Table 1.
A Summary of Experimentally Identified Results on the Interactions of Gas-phase Metal Species with CO2
Citation:
Yun-Zhu LIU, Xiao-Na LI, Sheng-Gui HE. Activation of Carbon Dioxide by Gas-phase Metal Species[J]. Chinese Journal of Structural Chemistry,
2021, 40(10): 1385-1403.
doi:
10.14102/j.cnki.0254–5861.2011–3081

Activation of Carbon Dioxide by Gas-phase Metal Species
English
Activation of Carbon Dioxide by Gas-phase Metal Species
Abstract:
Catalytic conversion of carbon dioxide (CO2) into value-added chemicals is an important and active field in both of the condensed-phase and gas-phase studies. This mini-review summarizes a variety of experimentally identified reactions in the activation and transformation of CO2 by metal species in the gas phase. The use of advanced mass spectrometric instrumentation in conjunction with quantum chemistry calculations can uncover the mechanistic details and determine the vital factors that control the activation of CO2. This review focuses mainly on three topics: the activation of CO2 by (1) bare metal ions and metal oxide species, (2) metal hydrides, and (3) other gas-phase metal species. Emphasis is placed on the latest advances in the hydrogenation of CO2 mediated with metal hydrides. A potential prospect toward the future effort in the activation and transformation of CO2 in gas phase has also been discussed.
-
1. INTRODUCTION
Recycle carbon dioxide (CO2) from industrial emission has long been an important subject because CO2 contributes greatly to the global warming[1-4]. CO2 is an abundant resource, less toxic than most of the chemicals, and can be used as potential C1 building block in chemical reactions. Catalytic conversion of CO2 into value-added products is an environmentally benign and sustainable way to recycle CO2. However, the inherent thermochemical stability and the kinetic inertness pose significant challenges to scientists to use CO2 as feedstock. Though nature has solved this problem by using CO2 in the photosynthesis process, it is still difficult for industry to use CO2 effectively due to the poor understanding of related chemistry. Transition metal catalysts play paramount roles in a wide range of reactions, including catalytic conversion of CO2 under relatively mild conditions[1], but the molecular-level mechanisms on how catalysts function are far from clear. It is hard to capture and then characterize the nature of the active sites on condensed-phase catalysts because of their complex and out-of-control chemistry environment, and then hinder the rational design of advanced catalysts.
Gas-phase ion-molecule experiments can be performed under isolated, controlled, and reproducible conditions[5-14]. With the aid of advanced mass spectrometry experiments in conjunction with theoretical calculations, the highly reactive species can be captured and characterized under unperturbed environment in which the difficult-to-control effects can be excluded and then the fundamental requirements necessary for bond activation and formation can be obtained at a strictly molecular level. Note that condensed-phase studies can guide the synthesis of catalysts that can be used directly in a wide range of catalytic reactions, while gas-phase studies are of great importance to parallel related condensed-phase studies to uncover the underlying mechanisms that govern the observed experimental results. Significant findings, such as the existence of complementary active site[15] and the discovery of spin conservation phenomenon[16] have been revealed by studying the reactions of gas-phase clusters with small molecules. For the activation of CO2, the reactions of different kinds of gas-phase metal species with CO2 have been extensively investigated and some excellent results have been summarized by Schwarz in a recent review[17]. The focus of this mini review is to permeate the latest advances in CO2 activation and transformation mediated with gas-phase metal species. The following topics are discussed: the reactions of (1) bare metal ions and metal oxide species, (2) metal hydrides, and (3) other gas-phase metal species, such as ScNH+[18], [MCN]+ (M = Fe and Co)[19], and Nb2BN2–[20] with CO2. Emphasis is placed on the fascinating reactivity of metal hydrides toward the hydrogenation of CO2. Structure characterization of CO2 adsorbed species using spectro- scopic techniques is shortly discussed. The experimentally identified results on the interactions of related gas-phase metal species with CO2 are summarized in Table 1.
Table 1

 DownLoad:
CSV
DownLoad:
CSV
Species Ion Products Neutral Products k1a Methodsb Remark Year Al+ AlO+ CO, C, O F 1992[82] Sc+ ScO+, ScO(CO2)1, 2+ CO 7.4 × 10−11 A 2006[21] Ti+ TiO+, TiO(CO2)+ CO 4.1 × 10−11 A 2006[21] V+ VO+, VCO+, VO2+ CO, O, C F 1995[83] V2+ V2O+ CO 3.8 × 10−12 D, theory 2020[23] Crn+ CrnO+ CO F n = 1~18 1998[84] Fen+ FenO+ CO F n = 1~18 1997[85] Fe2− Fe2O−, Fe2O2− CO (1.8 ± 0.4) × 10−10 C, theory 2020[54] Nix− Nix(CO2)y−, Nix(CO2)yOz− CO (0.63–1.09) × 10−10 A x = 3~5 1995[86] Cu+ CuO+, CuCO+ CO, O F 1999[87] Y+ YO+, YO(CO2)+ CO 5.9 × 10−10 A 2006[21] Y+ YO+, YCO+ CO, O F 1999[88] Zr+ ZrO+, ZrO(CO2)1, 2+ CO 2.5 × 10−10 A 2006[21] Zr+ ZrO+,
ZrCO+, ZrO2+CO, O, C F 1999[89] Nb+ NbO+,
NbO(CO2)+, NbO2(CO2)0−3+CO 1.8 × 10−10 A 2006[21] Nb+ NbO+, NbCO+, NbO2+ CO, O, C F 1998[90] Mo+ MoO+, MoCO+, MoO2+ CO, O, C F 1998[91] Pdx− Pdx(CO2)y− (0.05–3.4) × 10−10 A x = 3~8 1995[86] La+ LaO+, LaO(CO2)+ CO 4.2 × 10−10 A 2006[21] La+ LaO+, LaO(CO2)+ CO 4.4 × 10−10 A 2006[22] Ce+ CeO+, CeO(CO2)+ CO 4.6 × 10−10 A 2006[22] Ce+ CeO+ CO D 1997[92] Pr+ PrO+, PrO(CO2)+ CO 1.6 × 10−10 A 2006[22] Nd+ NdO+, NdO(CO2)1−2+ CO 3.7 × 10−11 A 2006[22] Nd+ NdO+ CO D 1997[92] Sm+ SmO+ CO F, theory 2017[93] Gd+ GdO+, GdCO+, GdO2+ CO, O, C F, theory 2018[94] Gd+ GdO+, GdO(CO2)1, 2+ CO 3.4 × 10−10 A 2006[22] Tb+ TbO+, TbO(CO2)1−4+ CO 3.8 × 10−11 A 2006[22] Lu+ LuO+, LuO(CO2)1−5+ CO 3.3 × 10−11 A 2006[22] Hf+ HfO+, HfO(CO2)1, 2+ CO 2.5 × 10−10 A 2006[21] Hf2+ HfO2+, HfO+, CO+ CO 6.3 × 10−10 D, theory 2012[95] Ta+ TaO+, TaO2(CO2)0−4+ CO 2.4 × 10−10 A 2006[21] Ta+ TaO+ CO D 1995[96] Ta2+ TaO2+, Ta+, TaO+, CO2+, CO+ CO 6.5 × 10−10 D, theory 2012[97] W+ WO+, WO2(CO2)0−3+ CO 4.2 × 10−11 A 2006[21] W+ WO+ CO 6.0 × 10−11 D 1991[98] Pt+ PtO+, PtCO+, CO2+ CO, O, Pt F, theory 2003[99] Ptx− Ptx(CO2)y− (0.06−1.71) × 10−10 A x = 3~7 1995[86] Th+ ThO+ CO D 2002[100] Th+ ThO+ CO D 1997[92] U+ UO+ CO D 2002[100] U+ UO+ CO D 1997[92] U+ UO+, UO2+ CO, C F 1980[101] Np+ NpO+ CO D 2002[100] Pu+ PuO+ CO D 2002[100] YO+ YO2+, Y+ CO, O + CO2 F 1999[88] ZrO+ ZrO2+, ZrCO2+ CO, O F 1999[89] NbO+ NbO2+, NbCO2+ CO, O F 1998[90] MoO+ MoO2+, MoCO2+, Mo+ CO, O, O+CO2 F 1998[91] HfO22+ HfO2+, CO2+ 3.2 × 10−10 D, theory 2012[95] TaO+ TaO2+ CO D 1995[96] TaO2+ TaO+, CO2+ 1.3 × 10−10 D, theory 2012[97] TaO22+ TaO2+, CO2+ 7.2 × 10−10 D, theory 2012[97] WO+ WO2+ CO 2.0 × 10−11 D 1991[98] [ReO2]− [ReO3]− CO (1.9 ± 0.1) × 10−12 C, theory 2016[102] PtO+ Pt+, PtO2+, PtCO2+ O + CO2, O2 + CO, CO, O F, theory 2003[103] UO+ UO2+ CO D 2002[100] UO+ UO2+ CO F 1980[101] V2O+ V2O2+ CO 8.8 × 10−10 D, theory 2020[23] MoxOy− MoxOy+1−, MoxOy+1CO− CO A, theory x = 2, 3; y = 2~5 2010[104] WxOy− WxOy+1− CO A, theory x = 2, 3; y = 2~6 2010[104] MoxWyOz− MoxWyOz+1−, MoxWyOz+2C− CO A, theory x + y = 3, z = 2~7 2010[104] RhVO3− RhVO3CO2− (295 K),
RhVO4− (600 K)CO (600 K) (5.2 ± 1.5) × 10−11 (295 K) C, theory 2019[62] Rh2VO− Rh2VO2− CO (9.9 ± 2.0) × 10−10 C, theory 2020[61] Rh2VO2− Rh2VO2CO2−, Rh2VO3− CO (10.3 ± 2.2) × 10−10 C, theory 2020[61] [Cp2TiH]+ [Cp2TiHCO2]+ B, theory 2015[30] [OTiH]+ [OTiOH]+ CO B, theory 2015[30] [HTaO] +/[TaOH]+ [OTaOH]+ CO B, theory 2016[106] FeH− HCO2−, HFeO−, FeO2H− Fe, CO (6.2 ± 1.2) × 10−10 C, theory 2017[33] Fe2H− FeHCO2−, Fe2HO−, Fe2HO2− Fe, CO (2.4 ± 0.5) × 10−10 C, theory 2020[54] Fe2H2− FeH2CO2−, Fe2H2O− Fe, CO (1.5 ± 0.3) × 10−10 C, theory 2020[54] Fe2H3− Fe2H3O− CO (6.4 ± 1.3) × 10−11 C, theory 2020[54] CoH− HCO2−, OCoH−, O2CoH− Co, CO (8.0 ± 1.6) × 10−10 C, theory 2020[34] NiH− NiOH−, NiO2H− CO (4.4 ± 0.9) × 10−10 C, theory 2020[34] CuH− HCO2−, CuHCO2− Cu (2.7 ± 0.5) × 10−12 C, theory 2020[34] CuH2− CuH2CO2− (2.8 ± 0.4) × 10−13 C, theory 2017[31] Cu2H2− Cu2H2(CO2)1, 2−, CuH2CO2− Cu 1.7 × 10−12 (298 K) C, theory 2018[53] Cu2H3− Cu2H3CO2− (1.3 ± 0.1) × 10−13 C, theory 2017[31] PtHn− H2Pt(HCO2)− B, G, theory n = 0~3, 5 2016[32] PdCuH4− PdCuH2− HCOOH B, G, theory 2020[57] TaCH2+ [TaCH2O]+ CO 6.9 × 10−10 D 1995[96] [TaCH2O]+ TaO2+ C2H2O 1.3 × 10−10 D 1995[96] ScS+ ScO+, Sc+ COS, S+CO2 F, D, theory 2000[107] TiS+ TiO+, TiOS+, Ti+ COS, CO, S + CO, S + CO2 F, D, theory 2000[107] VS+ VO+, VOS+, V+ COS, CO, S + CO, S + CO2 F, D, theory 1998[108] YS+ YO+, YOS+, Y+ COS, CO, S + CO2 F, D, theory 2006[109] ZrS+ ZrO+, ZrOS+, Zr+ COS, CO, S + CO2 F, D, theory 2006[109] NbS+ NbOS+, NbO+, Nb+ CO, CO + S, S + CO2 F, D, theory 2006[109] [ClMg]− [ClMgCO2]− B, theory 2018[110] [ClMgCO2]− [ClMgC2O4]−, [ClMgCO3]− CO B, theory 2018[110] ScNH+ ScO+ HNCO (8.5 ± 1.7) × 10−11 C, theory 2019[18] [FeCN]+ [NCCO2Fe]+ B, theory 2018[19] [CoCN]+ [NCCO2Co]+ B, theory 2018[19] [ClZn]− [ClZnCO2]− B, theory 2018[110] [ClZnCO2]− [ClZnC2O4]−, [ClZn]− B, theory 2018[110] [YC6D5]+ [YC7D5O2]+, [YC6D5O]+ CO B, theory 2016[111] [Re(CO)2]+ [Re(CO)2O]+ CO 3.9 × 10−11 D, theory 2017[112] NUOCl2− UO2(NCO)Cl2− C, theory 2016[113] CuB+ Cu+, CuOB+, CuCO+ CBO2, CO, BO (1.2 ± 0.2) × 10−9 C, theory 2018[63] Nb2BN2− Nb2BN2O− CO (5.3 ± 1.1) × 10−10 C, theory 2020[20] Nb2BN2O− Nb2BN2O2− CO (2.7 ± 0.5) × 10−10 C, theory 2020[20] Nb2BN2O2− Nb2BN2O3− CO (8.9 ± 1.8) × 10−11 C, theory 2020[20] Nb2BN2O3− Nb2BN2O4− CO (1.3 ± 0.3) × 10−11 C, theory 2020[20] Nb2N2− Nb2N2O− CO (8.8 ± 1.8) × 10−11 C, theory 2020[20] Nb2N2O− Nb2N2O2− CO (4.0 ± 0.8) × 10−11 C, theory 2020[20] Nb2B− Nb2BO− CO (1.6 ± 0.3) × 10−11 C, theory 2020[20] Nb2BO− Nb2BO2− CO (8.0 ± 1.7) × 10−12 C, theory 2020[20] Nb2BO2− Nb2BO3− CO (6.0 ± 1.3) × 10−13 C, theory 2020[20] CuBCH3+ Cu+, CuOBH+, CuCO+ C2H3BO2, C2H2O, CH3BO (5.3 ± 1.1) × 10−11 C, theory 2018[63] RhVO3CH4− RhVO3CH4CO2− (295 K),
RhVO5CH− (600 K),
RhVO4CH2− (600 K),
RhVO4C− (600 K),
RhVO4CH4− (600 K)CH3•, CH2O, CH3OH, CO C, theory 2019[62] Mg(CO2)n+ E, theory 2003[114] Al(CO2)n+ E, theory n = 1~11 2003[115] Si(CO2)n+ E, theory 2004[116] Ti(CO2)n+ E, theory n = 3~7 2013[117] V(CO2)n+ E, theory 2013[118] V(CO2)n+ E 2004[119] Fe(CO2)n+ E 2002[120] Fe(CO2)n+ E 2001[121] Co(CO2)n+ E, theory n = 2~6 2019[122] Co(CO2)n+ E, theory n = 2~15 2017[123] Ni(CO2)n+ E 2004[124] Ni(CO2)n+ E 2003[125] Cu(CO2)n+ E, theory n = 3~8 2017[126] Rh(CO2)n+ E, theory n = 2~15 2017[123] Ag(CO2)n+ E, theory n = 3~8 2017[126] Ir(CO2)n+ E, theory n = 2~15 2017[123] NiO2(CO2)n+ E 2003[125] YO(CO2)n+ E, theory n = 2~11 2018[66] NbO2(CO2)n+ E, theory n = 3~9 2020[127] NbO2(CO2)n+ E, theory n = 1~7 2019[128] TaO2(CO2)n+ E, theory n = 1~7 2019[128] Mn(CO2)n− E, theory n = 2~10 2017[129] Fe(CO2)n− E, theory n = 3~7 2017[130] Co(CO2)n− E, theory n = 3~11 2014[131] Ni(CO2)n− E, theory n = 2~8 2014[132] Cu(CO2)n− E, theory n = 2~9 2014[133] Ag(CO2)n− E, theory n = 2~11 2013[134] Au(CO2)n− E, theory n = 2~13 2012[135] Ni(CO2)− G, theory 2019[64] Cu(CO2)− G, theory 2015[65] Pd(CO2)− G, theory 2019[64] Ag(CO2)− G, theory 2015[65] Pt(CO2)− G, theory 2019[64] Au(CO2)− G, theory 2015[65] TiOx(CO2)y− E, theory x = 1~3; y > 1 2018[136] [ClMgCO2]− E, theory 2014[137] [Co(Pyridine)(CO2)]− G, theory 2015[138] Sc OScCO, OCScCO3 H, theory 2016[69] Ti OTiCO+, OTiOC+ OTiCO, O2Ti(CO)2,
O2Ti(CO), OTi(CO)2H, theory 1999[80] V OVCO+, OVOC+ OVCO,
O2V(CO)2, OV(CO)2H, theory 1999[80] Cr OCrCO−,
Cr(CO2)+, Cr(CO2)2+OCrCO, O2Cr(CO)2,
Cr(CO2), Cr(CO2)2,
CrOCrCO, OCCrCO3H, theory 2014[70] Cr OCrCO,
O2Cr(CO)2, O2CrCO,
CrO, CrCO,
CrO2, OCr(CO)2, CrCO2H, theory 1997[81] Mn OMnCO−, OMnCO+ OMnCO, O2MnCO, O2Mn(CO)2,
Mn(O2)CO, MnO, MnO2H, theory 1999[79] Fe OFeCO−, FeOCO+ OFeCO, O2FeCO,
Fe(O2)CO, FeO2H, theory 1999[79] Co OCoCO−, CoCO2−, OCoCO+ OCoCO, O2CoCO,
OCo2CO, CoOH, theory 2007[73] Co OCoCO−, CoCO2− OCoCO, CoO H, theory 1999[79] Ni ONiCO−, NiCO2− ONiCO H, theory 1999[79] Cu OCuCO−, CuCO2− H, theory 1999[79] Zr (ZrO) +CO OZrCO, ZrO H, theory 2000[76] Mo OMoCO, O2Mo(CO)2,
O2MoCO, MoCOH, theory 1997[81] Ru ORuCO− ORuCO, O2RuCO,
OCRu(O2)CO, RuCOH, theory 2002[74] Rh ORhCO− ORhCO, O2RhCO, RhO H, theory 2007[73] Ta OTaCO−, O2Ta(CO)2− OTaCO, O2Ta(CO)2 H, theory 2000[77] W OWCO, O2W(CO)2,
O2WCO, WO,
WCO, OW(CO)2WOH, theory 1997[81] Re OReCO−, ORe(CO)2− OReCO, O2ReCO,
ORe(CO)2, O2Re(CO)2H, theory 2002[[75] Os OOsCO− OOsCO, O2OsCO, O2Os(CO)2 H, theory 2002[74] Th OThCO+, O2Th(CO)2− OThCO, O2Th(CO)2, ThO H, theory 2000[78] U OUCO+, O2U(CO)2− OUCO, O2U(CO)2, O2UCO
UO, UO2H, theory 2000[78] ScO ScCO3 H, theory 2016[69] TiO TiO2(CO) H, theory 2012[71] NbO NbO2(CO) H, theory 2011[72] TiO2 OTiCO3 H, theory 2012[71] NbO2 NbO2(CO2)1, 2 H, theory 2011[72] a k1: in cm3 molecule−1 s−1.
b The experimental methods are labelled as A−H: A: fast flow reactor-mass spectrometry (MS), B: collision cell-MS, C: linear ion trap reactor-MS, D: ion cyclotron resonance cell-MS, E: IR-PD spectroscopy, F: guided ion beam-MS, G: photoelectron spectroscopy, and H: matrix isolation infrared spectroscop2. REACTIONS OF BARE METAL CATIONS AND METAL OXIDE SPECIES WITH CO2
Metal and metal oxide catalysts have been extensively used in the field of catalytic conversion of CO2 into more useful chemical materials. The studies on the interactions of bare metal cations and metal oxide species with CO2 are helpful to provide insights into the nature of M–O bond formation and define the fundamental requirement to activate CO2. The reactions of CO2 with 46 atomic metal cations have been studied systematically in the gas phase[21]. Only early transition metal cations exhibit oxygen atom transfer (OAT) reactivity, two in the first (Sc+ and Ti+), three in the second (Y+, Zr+, and Nb+), and four in the third row (La+, Hf+, Ta+, and W+). Note that all the nine atomic metal cations involved in the OAT reactions have O-atom affinities > 6.9 eV, which is much larger than the enthalpy of O–CO bond (5.5 eV) in CO2. This phenomenon shows that these OAT reactions are thermodynamically controlled. There is no indication for the formation of VO+ and AsO+, though the O-atom affinities for V+ and As+ are larger than that of O–CO bond in CO2. This absence of exothermic OAT from CO2 to V+ and As+ can be ascribed to the spin forbidden nature, for example, from 5V+ to 3VO+. In contrast, on the interactions of atomic lanthanide cations with CO2, the reactions are kinetically controlled[22]. Available experimental and theoretical results indicate that eleven out of fourteen cations have O-atom affinities higher than that of O–CO bond in CO2, while only seven of these were observed to react with CO2 through OAT chemistry. It has been proposed that a kinetic barrier was required for OAT and this barrier was found to correlate with the energy required to achieve two unpaired non-f valence electrons in 5d-orbital, that is the energy to excite the Ln+ cations from the ground state (4fn5d06s1) to the excited state (4fn-15d26s0), the electronic structure of which favors efficient bonding with oxygen atom in LnO+.
Comparing with the inert nature of V+ ion toward CO2 in OAT, a newly reported result demonstrated that the diatomic V2+ cation can abstract an oxygen atom from CO2 to generate V2O+ (Fig. 1a), which serves as a short-lived intermediate and it is surprisingly more reactive than V2+ in the reaction with CO2 to give rise to V2O2+ (Fig. 1b)[23]. Though the spin forbidden process (4V2+ → 2V2O+) also exists on the pathway for V2+ + CO2 (Fig. 1c), all the calculated energies for intermediates and transition states are well below the separate reactants. Thus, product V2O+ can be formed in the experiment (Fig. 1a). For the V2O+/CO2 couple, one reason that leads to the much higher reactivity of V2O+ may be due to the spin situation (Fig. 1d). The whole OAT process is confined to the doublet state and then the barriers for CO2 activation and dissociation (TS3 and TS4) can be more easily suppressed with respect to the V2+/CO2 couple. Moreover, the "prepared" structure of V2O+ due to the presence of the oxygen bridge was emphasized to account for its enhanced reactivity. The active V atom that is responsible to anchor CO2 in V2O+ is more positively charged than that in V2+. However, the oxygen bridge can overcompensate this unfavorable charge situation by elongating the V–V bond (251 pm) in V2O+, which can accept the oxygen atom from CO2 more easily to overcome TS3. The oxygen bridge can also raise the πdxz/dxz orbital energy from –10.5 eV in V2+ to –9.5 eV in V2O+, and this doubly occupied orbital is already delocalized extensively to the C atom of CO2 in TS3. This behavior greatly eases the transfer of d-electrons into the empty π*-orbital of CO2 and then favors the subsequent activation of CO2. Metal oxide catalysts are superior to metal catalysts due to their higher selectivity, durability, and stability in the conversion of CO2, however, it is challenging to define the exact structure of active sites. These gas-phase studies on the reactions of metal ions and metal oxide clusters with CO2 are pivotal to understand the underlying mechanisms behind their remarkably different reactivity.
Figure 1
 Figure 1. The reactions of V2+ and V2O+ with C18O2 inside the ion trap of the Fourier-transform ion cyclotron resonance mass spectrometer. The pressure of C18O2 in (a) is about 1.0×10−7 mbar and in (b) is 4.0×10−9 mbar. Calculated potential energy surfaces for the reactions of (c) V2+ and (d) V2O+ with CO2. Key structures with selected geometric parameters are provided. The relative energies are in unit of eV. Reprinted with permission from reference 23
Figure 1. The reactions of V2+ and V2O+ with C18O2 inside the ion trap of the Fourier-transform ion cyclotron resonance mass spectrometer. The pressure of C18O2 in (a) is about 1.0×10−7 mbar and in (b) is 4.0×10−9 mbar. Calculated potential energy surfaces for the reactions of (c) V2+ and (d) V2O+ with CO2. Key structures with selected geometric parameters are provided. The relative energies are in unit of eV. Reprinted with permission from reference 233. REACTIONS OF METAL HYDRIDES WITH CO2
The hydrogenation of CO2 serves as one of the most promising pathways to remove CO2 emission and produce value-added chemicals, such as methanol and formic acid[24, 25]. In addition to the extraordinary activity of noble-metal catalysts, 3d-metal (such as Fe, Co, Ni, and Cu) based catalysts has been demonstrated to have comparable or even improved reactivity with noble-metal catalysts in recent years[25-27]. However, the nature in the hydrogenation of CO2 remains far from clear due to the lack of experimental evidence of C–H bond formation, the key step to transform CO2 into methanol or formic acid. Metal hydrides[28, 29] are crucial intermediates to induce the hydrogenation of CO2, and studies on the reactions of metal hydrides with CO2 are of great importance to provide fundamental insights into this elementary step and then the guidelines can be obtained to optimize catalysts. In the field of gas phase, the studies on the reactions of metal hydrides with CO2 have been relatively scarcely reported and the potential formation of C–H bond has been proposed. For example, the formation of attached HCO2– moiety on the reactive system was suggested through the insertion of CO2 into the M–H bond of Cp2TiH+[30], CuH2–[31], and PtH3–[32]. Recently, we provided solid evidence that HCO2– can be formed directly as a product in the thermal reaction of CO2 with a diatomic metal hydride species FeH–, conforming the formation of C–H bond[33], as shown in Fig. 2b. Note that the reduction of CO2 into CO occupies a pivotal state during catalytic hydrogenation[4], and this channel can usually compete with the hydrogenation products. In this case, the scientists prefer to sacrifice the reactivity in order to pursue a high selectivity for a desired product. For the FeH–/CO2 couple, the reduction of CO2 into CO also serves as the main channel (Fig. 2b). Thus, the reactions of other late transition metal anions MH– with CO2 have also been investigated[34]. The results show that the CoH– anion exhibits similar reactivity to FeH– (Fig. 2d), in contrast, NiH– can selectively convert CO2 into CO (Fig. 2f) and CuH– gives rise to the hydrogenation product HCO2– selectively (Fig. 2h). These results highlight that the product selectivity is highly dependent on the nature of 3d-metal. To have a fundamental understanding on the factors that lead to different distributions of products, for example, the calculated pathway for FeH– + CO2 is shown (Fig. 3).
Figure 2
 Figure 2. Time-of-flight (TOF) mass spectra for the reactions of mass-selected FeH−, CoH−, NiH−, and CuH− (a, c, e, and g) with CO2 (b, d, f, and h). The reactant pressures are shown in mPa (= 10−3 Pa). Reprinted with permission from references 33 and 34
Figure 2. Time-of-flight (TOF) mass spectra for the reactions of mass-selected FeH−, CoH−, NiH−, and CuH− (a, c, e, and g) with CO2 (b, d, f, and h). The reactant pressures are shown in mPa (= 10−3 Pa). Reprinted with permission from references 33 and 34Figure 3
 Figure 3. Theoretical calculated potential energy profiles for the reaction of FeH− with CO2 in the quintet state. The relative energies are given in eV. The bond lengths are given in pm. Reprinted with permission from reference 33
Figure 3. Theoretical calculated potential energy profiles for the reaction of FeH− with CO2 in the quintet state. The relative energies are given in eV. The bond lengths are given in pm. Reprinted with permission from reference 33CO2 can be capture directly by the H atom of FeH– with a binding energy of –0.61 eV (I7). The next step is accompanied by complete rupture of Fe–H bond in a barrier-free process (I7 → TS5 → I8) to generate HCO2–, and then the nearly neutral Fe atom can be released from Fe(HCO2)– (I8). Moreover, the Fe atom in FeH– functions as a more preferred site to trap CO2 tightly (I9, ∆H0 = –1.43 eV), and a 0.46 eV barrier has to be surpassed to cleave C–O bond to produce CO and HFeO–. Several mechanisms for the hydrogenation of CO2 mediated with metal hydride have been proposed. The ƞ2-coordination of electrophilic CO2 to the metal center followed by migration of a H atom from metal to CO2 is the most generally accepted process to give rise to HCO2–[30, 32, 35, 36]. This mechanism can account for the hydrogenation of CO2 by Cp2TiH+[30] and PtH3–[32]. While the direct approach of CO2 to the terminally bonded H atom and subsequent H atom transfer to the C atom of CO2 has less been reported[37, 38]. The diatomic FeH– anion serves as an ideal model to explore the mechanistic details of CO2 hydrogenation and it demonstrates unambiguously that the direct H transfer from metal site to CO2 is an energetically more favorable pathway. This result parallels well the condensed-phase hydrogenation of CO2 by iron-based catalysts[37]. This mechanism was further evidenced by studying the reactions of CoH–, NiH–, and CuH– anions[34] with CO2 that the direct H atom transfer represents the most favorable pathway to form C–H bond, indicating that this chemistry may be universal during the hydrogenation of CO2 mediated with transition metal based catalysts. The much weaker Co–H/Ni–H bonds (2.34 eV/2.62 eV) than that of Co–O/Ni–O bonds (3.99 eV/3.96 eV) indicates that in principle, the channels to produce HCO2– and CO should both be observed for the two systems. The significantly different selectivity for CoH– + CO2 (Fig. 2d) and NiH– + CO2 (Fig. 2f) lies in the subsequent transformation of the crucial intermediates after CO2 dissociation, as shown in Fig. 4. The electronegativity of Ni (1.91) is slightly larger than that of Co (1.88)[39]. The leading result is that the H atom in intermediate B is more electrostatic attractive toward the terminal O atom, thus, the energy more stable products of NiOH– and CO with respect to that of Ni and HCO2– can be generated. However, this O–H bond formation process from intermediate A is kinetically hindered, and the channels for the reduction and hydrogenation of CO2 are competitive. These analysis indicates that a subtle difference in electronic structure of 3d-metal is powerful enough to bring about a strikingly different result in the selectivity of the final product. In contrast, the reduction of CO2 into CO by CuH– is the thermodynamic impediment process because of the stronger Cu–H bond (2.88 eV) than that of Cu–O bond (2.79 eV). This fact gives CuH– the chance to transform CO2 into HCO2– exclusively.
Figure 4
 Figure 4. Natural charge (e) distribution on the crucial intermediates A (in reaction CoH− + CO2) and B (in reaction NiH− + CO2). Reprinted with permission from reference 34
Figure 4. Natural charge (e) distribution on the crucial intermediates A (in reaction CoH− + CO2) and B (in reaction NiH− + CO2). Reprinted with permission from reference 34The active metal site plays a central role in catalytic transformation because the activity and selectivity of CO2 hydrogenation have been demonstrated to be very sensitive to the well-defined structure of metal sites[40, 41]. On the reaction of diatomic metal hydride anion MH− (M = Fe, Co, Ni, Cu) with CO2, both the metal and the H sites in MH− can adsorb CO2 and induce the activation of CO2. Further analysis indicates that the larger binding energies of CO2 facilitate the C−O bond cleavage and make the CO release process preferentially from the metal site, while the HCO2− product is formed through direct hydride transfer with a negligible barrier from the H site. Moreover, the hydride affinities of metal species have been calculated to be closely related to the insertion barrier of CO2 into the M−H bond, and a smaller hydride affinity corresponds to a lower barrier[30]. For the reaction of FeH− and CO2, the much weaker Fe−H bond (1.63 eV)[42] compared to the Pt−H bond (3.44 eV)[43] and Ti−H bond (2.12 eV)[44] may account for the barrier-free pathway for the insertion of CO2 into Fe−H bond. Thus, the nature of 3d-metals as well as the strengths of M−O and M−H bonds are all important factors for the product selectivity of different metal hydride species toward CO2.
The selectivity for CO2 catalytic hydrogenation mediated with a particular catalyst is even more essential than the reactivity in industrial applications to avoid the complex process of products separation[45]. The formate anion HCO2− was generated directly as a product on the reaction of MH− (M = Fe, Co) with CO2[33, 34], while the reduction of CO2 into CO was a more competitive channel (branching ratio of 60% for FeH− + CO2 and 77% for CoH− + CO2, respectively). In sharp contrast, CuH− can selectively transform CO2 into hydrogenation product HCO2− (branching ratio of 86%) while NiH− can only reduce CO2 into CO (branching ratio of 100%). The current results parallel the condensed-phase experiments that Ni-based catalysts can transform CO2 into CO effectively[46, 47], in contrast, Cu-based catalysts can selectively promote the generation of formate products[48-50].
Available studies have shown that hydrogen ligands can affect the electronic nature of metal hydrides and then modify their reactivity in the hydrogenation of CO2[51, 52]. For example, the Cu2H2– anion can adsorb CO2 strongly at room temperature and the channel responsible for the possible formation of the attached HCO2– moiety in CuH2CO2– is relatively weak (Fig. 5b)[53]. The generation of the HCO2– moiety on product CuH(HCO2)– was further identified with the increase of temperature (Fig. 5c), while the Cu2H3– anion reacts slowly with CO2 to generate adsorption product Cu2H3(CO2)–[31]. A recent study demonstrated that for the gas-phase reactions of the Fe2Hn– (n = 0~3) anions with CO2, the reduction of CO2 into CO dominants the channels, whereas only Fe2H– and Fe2H2– can induce the hydrogenation of CO2 effectively to produce Fe(HCO2)– (Fig. 5e) and HFe(HCO2)– (Fig. 5g), respectively[54]. Previous studies demonstrated that during C–H bond formation, the electron that can be used to reduce CO2 comes dominantly from the terminal H atom and the nearby metal atom[33, 34, 53]. As shown in Fig. 6, natural bond orbital analysis indicates that the hydrogen ligands in the Fe2H1~3– anions can remarkably modify the charge state of the Fe atom (marked as Fe1) that is directly connected with the terminal H atom. The more positively charged Fe1 atoms in Fe2H2– and Fe2H3– indicate that it is more difficult for these systems to transfer an electron to the coming CO2 molecule. A charge transfer model [(FeH)δ– + CO2 → Feδ+ + (HCO2)–] was proposed to account for the hydrogenation of CO2 mediated with Fe2Hn–. This study highlights the fact that only an appropriate number of hydrogen ligands on Fe2– can modify its electronic structures reasonably and then induce the effective hydrogenation of CO2.
Figure 5
 Figure 5. TOF mass spectra for the reactions of mass-selected Cu2H2− (a), Fe2H− (d), and Fe2H2− (f) with CO2 at different temperatures (b, c, e, and g). Reprinted with permission from references 53 and 54
Figure 5. TOF mass spectra for the reactions of mass-selected Cu2H2− (a), Fe2H− (d), and Fe2H2− (f) with CO2 at different temperatures (b, c, e, and g). Reprinted with permission from references 53 and 54Figure 6
 Figure 6. Natural charge (e) distribution on the Fe atoms in Fe2Hn− (n = 0~3). Cited with permission from reference 54
Figure 6. Natural charge (e) distribution on the Fe atoms in Fe2Hn− (n = 0~3). Cited with permission from reference 54The insertion of CO2 into a metal-hydride bond is a key step that has been the subject of much recent interest. The energetics and rate of CO2 insertion have been shown to vary widely with different metals and ligand environments. Several possible mechanisms for this process have been discussed[35]. It is intriguing to study the effect of other ligands in metal species on CO2 activation, and the important roles of different ligands in modifying the reactivity of metal clusters can be obtained. Metal carbide species are considered as the active sites on real-life catalysts to investigate the mechanism of CO2 reduction due to the fact that the surfaces of transition metal carbides can bind CO2 well and are highly active for the cleavage of C−O bonds[55, 56]. Our recent experimental results show that the CoCDn− (n = 0~3) species can reduce CO2 into CO while CoCD4− can only adsorb CO2 selectively under thermal collision conditions. The crucial roles of different number of D ligands in metal carbides for CO2 activation were rationalized by quantum chemical calculations and the results may provide fundamental understanding on the mechanisms that govern the catalytic reactions.
Bimetallic catalysts exhibit improved activity and selectivity over single-component ones in the catalytic conversion of CO2, as the active site of bimetallic catalysts can be well tailored and thus their performance can be regulated. The studies on the reactions of bimetallic hydrides with CO2 are of great importance to elucidate how interplay alters electronic structures, charge-transfer property, and then modify the catalytic properties. Very recently, for the first time, the Bowen group successfully generated and identified the reactivity of bimetallic palladium-copper hydride cluster anion (PdCuH4–) to catalytically convert CO2 into HCOOH in combination with mass spectrometric analysis, photoelectron spectroscopy, and theoretical calculations[57]. As shown in Fig. 7, the calculations show that structures A and B are two low-energy PdCuH4– isomers. In structure C, CO2 is inserted into the Cu–H bond of A, and D is obtained by the insertion of CO2 into the Pd–H bond in B. Both structures C and D have a HCO2– moiety and they can be potential intermediates to generate HCOOH. In structure D, the bridged H atom can transfer to the HCO2– moiety, forming structure E with a formic acid moiety. Subsequent dissociation of formic acid from E can generate PdCuH2–, which can react with H2 to regenerate PdCuH4–. Note that the structure C is a more stable intermediate, while a high energy (2.38 eV) is required for it to release HCOOH. Thus, it is unlikely that HCOOH can be formed via this process. In contrast, the reaction starting from B proceeds on a relatively smoother pathway, indicating that the metastable isomer B is the catalytic driving force, though both isomers can be identified in the experiment.
Figure 7
 Figure 7. Theoretical calculated relevant low-lying energy structures of PdCuH4− (A and B), PdCuCO2H4− (C, D, and E), and PdCuH2−. The relative energies for PdCuH4− (A and B) and PdCuCO2H4− (C, and D) are shown below each structure. Reprinted with permission from reference 57
Figure 7. Theoretical calculated relevant low-lying energy structures of PdCuH4− (A and B), PdCuCO2H4− (C, D, and E), and PdCuH2−. The relative energies for PdCuH4− (A and B) and PdCuCO2H4− (C, and D) are shown below each structure. Reprinted with permission from reference 574. REACTIONS OF OTHER METAL SPECIES WITH CO2
The studies on the reactions of CO2 with metal species coordinated by small ligands (CN–, Cl–, CO, OH–, and so on) are vital to have a more comprehensive understanding on the chemistry of CO2. For example, the reaction of ScNH+ with CO2 can generate neutral product HNCO, which is one of the first polyatomic molecules observed in the interstellar gas[18]. The reactions of M(CN)+ (M = Co and Fe) with CO2 produce addition product M(CN)(CO2)+, careful structure analysis of which can obtain more information about the knowledge in CO2 capture and reduction by related enzymes in nature[19]. Recently, the Ma group discovered a very interesting phenomenon that the Nb2BN2– cluster anion[20] can reduce four CO2 molecules consecutively to produce an oxygen-rich product Nb2BN2O4– (Fig. 8b~8d), while the species without B (Nb2N2–) or N (Nb2B–) atom can reduce two or three CO2 molecules, respectively. CO2 can be captured by the two Nb atoms in Nb2BN2– in the initial step. The presence of B atom in Nb2BN2– is vital to shorten the Nb–Nb bond (Fig. 8e). This can be reflected by the calculated lowest-lying structures for Nb2BN2– and Nb2N2– that the Nb–Nb bond in Nb2BN2– is greatly shorter (229 pm) than that in Nb2N2– (273 pm). Thus, more electrons can be stored in the Nb–Nb bond of Nb2BN2– and then the electrons can be used in the following CO2 reduction process. In the subsequent steps of CO2 activation and dissociation, the synergy between Nb atom and B atom was emphasized to drive electron transfer into the π*-orbitals of CO2 due to the similar d-orbital nature of the B p-orbital. This gas-phase study can be helpful to understand the nature of active site on BN substrate supported transition metal catalysts that exhibit superior reactivity for various catalytic reac- tions[58-60].
Figure 8
 Figure 8. TOF mass spectra for the reactions of mass-selected Nb2BN2− with Ar (a) and CO2 (b~d). The reactant pressures are shown in mPa (= 10−3 Pa). Calculated structures of Nb2BN2−, Nb2N2−, and Nb2B− (e). The relative energies are shown below each structure. Reprinted with permission from reference 20
Figure 8. TOF mass spectra for the reactions of mass-selected Nb2BN2− with Ar (a) and CO2 (b~d). The reactant pressures are shown in mPa (= 10−3 Pa). Calculated structures of Nb2BN2−, Nb2N2−, and Nb2B− (e). The relative energies are shown below each structure. Reprinted with permission from reference 20A more appealing and promising direction in CO2 utilization is to convert CO2 with CH4 directly into value-added chemicals but it is remarkably challenging due to the thermodynamic stability and kinetic inertness of both molecules. Design of better-performing catalysts for direct conversion of CO2 and CH4 under mild conditions requires fundamental understanding on the mechanistic details, however, related catalytic examples have been scarcely reported[61-63]. In gas-phase studies, it demonstrated that the establishment of catalytic cycles for CH4 conversion is challenging because it is difficult to design effective routes for the transformation of product clusters (for example, MxOyHzq and MxOyCH2q) to regenerate the reactant clusters. Recently, the examples for catalytic conversion of CO2 and CH4 by heteronuclear metal oxide clusters were reported[61, 62]. We take the conversion by RhVO3– as an example (Fig. 9). On the interaction of RhVO3– with CH4, only adsorption product RhVO3CH4– was observed at room temperature (Fig. 9b). Isotopic labeling experiment with CD4 as reactant gas indicated that the kinetic isotopic effect for this absorption channel was estimated to be 3.0 ± 0.6, implying that dissociative adsorption took place. When the reaction temperature was ramped up to 600 K, two new weak signals RhVO3CH2– (RhVO3CH4– → RhVO3CH2– + H2) and RhVO3C– (RhVO3CH4– → RhVO3C– + 2H2) appeared, convincing that CH4 was dissociated on RhVO3–. Note that RhVO3– can also reduce CO2 to CO, indicating that RhVO3– may be a promising candidate to induce co-conversion of CO2 and CH4. On the interaction of RhVO3CH4– with CO2 at 295 K, only adsorption product RhVO3CH4CO2– can be clearly observed. While the elevated temperature (Fig. 9d) brings about a series of stronger signals corresponding to the loss of neutral species of CH3•, CH2O, CH3OH, and CO from RhVO3CH4CO2–. These experiments indicate that the conversion of CO2 and CH4 can be mediated by RhVO3–. DFT calculations show that the exposed Rh site in RhVO3– (Fig. 9e) is nearly neutral, which is facile to dissociate CH4 in adsorption product A. Direct dehydrogenation from A is energetically demanding, consistent with the experimental result that a higher reaction temperature is required to drive the release of H2. In this case, CO2 has the chance to attach on RhVO3CH4– and then gives rise to a more stable intermediate B, from which different neutral products can be obtained. This fact emphasizes the crucial roles of the introduced CO2 that can switch the endothermic CH4 conversion to exothermic reactions and the partial oxidation of CH4 by RhVO3– can perform along the lower potential energy surface. Thus, the production of CH3OH and CH2O becomes thermodynamically and kinetically favorable. While the attached CO in structure C can be release only under the conditions of a higher temperature or collision-induced dissociation, and then RhVO3– can be regenerated to complete the catalytic cycle. The RhVO3– cluster represents the first example to convert CO2 and CH4 directly and a molecular-level mechanism on related catalytic conversion was obtained.
Figure 9
 Figure 9. TOF mass spectra for the reactions of mass-selected RhVO3− (a) with CH4 (b) seeded in the cooling gas (He) and then react with CO2 at 295K (c) and 600 K (d). The pressure of CH4 in panel b is about 1.4 Pa and the pressures of CO2 in panel c is about 0.2 Pa and in panel d about 0.7 Pa. Calculated structures for RhVO3−, RhVO3CH4−, RhVO3CH4CO2−, and RhVO4C− are shown (e). Reprinted with permission from reference 62
Figure 9. TOF mass spectra for the reactions of mass-selected RhVO3− (a) with CH4 (b) seeded in the cooling gas (He) and then react with CO2 at 295K (c) and 600 K (d). The pressure of CH4 in panel b is about 1.4 Pa and the pressures of CO2 in panel c is about 0.2 Pa and in panel d about 0.7 Pa. Calculated structures for RhVO3−, RhVO3CH4−, RhVO3CH4CO2−, and RhVO4C− are shown (e). Reprinted with permission from reference 625. SPECTROSCOPIC CHARACTERIZATION OF CO2 ADSORBED SPECIES M(CO2)n0/+/−
Adsorption induced CO2 fixation is an important field of scientific research. Understanding the nature of metal-CO2 interaction (chemisorption, physisorption, or the coexistence of both feasibility), is vital to provide insights into the structures of captured CO2. Powerful characterization techniques, such as infrared photodissociation spectroscopy, anion photoelectron spectroscopy, and matrix isolation spectroscopy, in combination with theoretical calculations play pivotal roles to investigate the structure of captured CO2. Early transition metal cations M+ have strong reducing power to reduce CO2 into CO[21], as mentioned in the second section, while the capability of late transition metal species in the reduction of CO2 is highly dependent on their positions in the periodic table and the charged state. For example, the neutral Ni, Pd, and Pt atoms are normally unable to activate CO2, while the addition of an electron in these systems can lead to the formation of chemisorbed anionic complexes M(CO2)− (M = Ni, Pd, and Pt)[64]. In contrast, anion photoelectron spectra investigation of M(CO2)− (M = Cu, Ag, and Au) species found that only physisorption in the case of Ag(CO2)−, only chemisorption in Cu(CO2)−, and both features for Au(CO2)−[65]. Moreover, spectroscopy characterization of M(CO2)n+/− or MO(CO2)n+/− complexes can understand the stepwise evolution of coordination environment with gradual addition of CO2[66]. Matrix isolation spectroscopic study has a number of advantages for investigating some transient intermediates. These extremely reactive and photolabile species can be trapped and accumulated over a long period of time in matrix isolation, so detection sensitivity can be enhanced and a broad spectral range can be easily explored in a short time[67, 68]. The reactions of transition metal atoms and simple oxide molecules with CO2 have been intensively studied in solid noble gas matrices[69-81], which indicate that CO2 not only can form complexes with transition metal centers but also can be reduced to CO via insertion reactions. The insertion intermediates and various coordination complexes in different coordination modes including η1−O, η1−C, η2−C, O, and η2−O, O fashions were characterized spectroscopically. These fundamental studies are vital to provide useful information for more effective CO2 fixation or separation, while this is not the main topic in this mini-review.
6. CONCLUSION
This review summarizes the latest process in the activation and transformation of CO2 by metal species in the gas phase. Hydrogenation of CO2 occupies a very important stage to catalytically convert CO2 into formic acid, which serves not only as an energy carrier and commodity but also a promising hydrogen storage material[24, 25]. Though some knowledge on C–H bond formation has been obtained, there is still a long way to go in this direction due to the following issues. The current studies are focused on homonuclear metal hydrides, while related study on heteronuclear species has been scarcely reported[57]. In this case, the pivotal synergy between different metal centers in the active site is elusive, thus, the strategy to engineer the reactivity and the selectivity of desirable product in practically used bimetallic catalysts is ambiguous. Secondly, only elementary steps have been emphasized and the catalysis in the hydrogenation of CO2 under mild conditions has been rarely established[57]. The fundamental chemistry and the key factors that control the rate-determining steps in the catalysis are still lack. Finally, the stepwise hydrogenation of CO2 to generate other C1 chemicals, such as CH3OH, has not also been experimentally identified in the gas phase. Gas-phase studies performed under isolated conditions, in principle, can never account for exactly the precise structure of active sites or the catalytic behaviors in the condensed phase, while related studies are of significant importance to uncover the underlying mechanisms behind the fascinating results, discover new species, and open new perspective for effectively catalytic conversion of CO2 in the future.
-
-
[1]
Franco, F.; Rettenmaier, C.; Jeon, H. S.; Roldan Cuenya, B. Transition metal-based catalysts for the electrochemical CO2 reduction: from atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884−6946. doi: 10.1039/D0CS00835D
-
[2]
Singh, G.; Lee, J.; Karakoti, A.; Bahadur, R.; Yi, J.; Zhao, D.; AlBahily, K.; Vinu, A. Emerging trends in porous materials for CO2 capture and conversion. Chem. Soc. Rev. 2020, 49, 4360−4404. doi: 10.1039/D0CS00075B
-
[3]
Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J. G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984−8034. doi: 10.1021/acs.chemrev.9b00723
-
[4]
Su, X.; Yang, X. F.; Huang, Y.; Liu, B.; Zhang, T. Single-atom catalysis toward efficient CO2 conversion to CO and formate products. Acc. Chem. Res. 2019, 52, 656−664. doi: 10.1021/acs.accounts.8b00478
-
[5]
Mou, L. H.; Jiang, G. D.; Li, Z. Y.; He, S. G. Activation of dinitrogen by gas-phase species. Chin. J. Chem. Phys. 2020, 33, 507−520. doi: 10.1063/1674-0068/cjcp2008141
-
[6]
Wang, L. N.; Li, X. N.; He, S. G. Recent research progress in the study of catalytic CO oxidation by gas phase atomic clusters. Sci. China Mater. 2020, 63, 892−902. doi: 10.1007/s40843-019-1206-2
-
[7]
Li, X. N.; Wang, L. N.; Mou, L. H.; He, S. G. Catalytic CO oxidation by gas-phase metal oxide clusters. J. Phys. Chem. A 2019, 123, 9257−9267. doi: 10.1021/acs.jpca.9b05185
-
[8]
Zhao, Y. X.; Li, Z. Y.; Yang, Y.; He, S. G. Methane activation by gas phase atomic clusters. Acc. Chem. Res. 2018, 51, 2603−2610. doi: 10.1021/acs.accounts.8b00403
-
[9]
Schwarz, H. Single-atom catalysis, mass spectrometry, and computational chemistry. Catal. Sci. Technol. 2017, 7, 4302−4314. doi: 10.1039/C6CY02658C
-
[10]
Chi, C.; Qu, H.; Meng, L.; Kong, F.; Luo, M.; Zhou, M. CO oxidation by group 3 metal monoxide cations supported on [Fe(CO)4]2−. Angew. Chem. Int. Ed. 2017, 56, 14096−14101. doi: 10.1002/anie.201707898
-
[11]
Zavras, A.; Khairallah, G. N.; Krstić, M.; Girod, M.; Daly, S.; Antoine, R.; Maitre, P.; Mulder, R. J.; Alexander, S. A.; Bonačić-Koutecký, V.; Dugourd, P.; O'Hair, R. A. J. Ligand-induced substrate steering and reshaping of [Ag2(H)]+ scaffold for selective CO2 extrusion from formic acid. Nat. Commun. 2016, 7, 11746−8. doi: 10.1038/ncomms11746
-
[12]
Harding, D. J.; Fielicke, A. Platinum group metal clusters: from gas-phase structures and reactivities towards model catalysts. Chem. Eur. J. 2014, 20, 3258−3267. doi: 10.1002/chem.201304586
-
[13]
Lang, S. M.; Bernhardt, T. M. Gas phase metal cluster model systems for heterogeneous catalysis. Phys. Chem. Chem. Phys. 2012, 14, 9255−9269. doi: 10.1039/c2cp40660h
-
[14]
Yin, S.; Bernstein, E. R. Gas phase chemistry of neutral metal clusters: distribution, reactivity and catalysis. Int. J. Mass Spectrom. 2012, 321–322, 49−65.
-
[15]
Roach, P. J.; Woodward, W. H.; Castleman, A. W.; Reber, A. C.; Khanna, S. N. Complementary active sites cause size-selective reactivity of aluminum cluster anions with water. Science 2009, 323, 492−495. doi: 10.1126/science.1165884
-
[16]
Burgert, R.; Schnöckel, H.; Grubisic, A.; Li, X.; Stokes, S. T.; Bowen, K. H.; Ganteför, G. F.; Kiran, B.; Jena, P. Spin conservation accounts for aluminum cluster anion reactivity pattern with O2. Science 2008, 319, 438−442. doi: 10.1126/science.1148643
-
[17]
Schwarz, H. Metal-mediated activation of carbon dioxide in the gas phase: mechanistic insight derived from a combined experimental/computational approach. Coord. Chem. Rev. 2017, 334, 112−123. doi: 10.1016/j.ccr.2016.03.009
-
[18]
Wang, M.; Sun, C.; Cui, J.; Zhang, Y.; Ma, J. Clean and efficient transformation of CO2 to isocyanic acid: the important role of triatomic cation ScNH+. J. Phys. Chem. A 2019, 123, 5762−5767. doi: 10.1021/acs.jpca.9b02133
-
[19]
Firouzbakht, M.; Rijs, N. J.; Schlangen, M.; Kaupp, M.; Schwarz, H. Ligand effects on the reactivity of [CoX]+ (X = CN, F, Cl, Br, O, OH) towards CO2: gas-phase generation of the elusive cyanoformate by [Co(CN)]+ and [Fe(CN)]+. Top. Catal. 2018, 61, 575−584. doi: 10.1007/s11244-018-0903-8
-
[20]
Zhou, H. Y.; Wang, M.; Ding, Y. Q.; Ma, J. B. Nb2BN2– cluster anions reduce four carbon dioxide molecules: reactivity enhancement by ligands. Dalton Trans. 2020, 49, 14081−14087. doi: 10.1039/D0DT02680H
-
[21]
Koyanagi, G. K.; Bohme, D. K. Gas-phase reactions of carbon dioxide with atomic transition-metal and main-group cations: room-temperature kinetics and periodicities in reactivity. J. Phys. Chem. A 2006, 110, 1232−1241. doi: 10.1021/jp0526602
-
[22]
Cheng, P.; Koyanagi, G. K.; Bohme, D. K. Gas-phase reactions of atomic lanthanide cations with CO2 and CS2: room-temperature kinetics and periodicities in reactivity. J. Phys. Chem. A 2006, 110, 12832−12838. doi: 10.1021/jp0637431
-
[23]
Li, J.; Geng, C.; Weiske, T.; Schwarz, H. Counter-intuitive gas-phase reactivities of [V2]+ and [V2O]+ towards CO2 reduction: insight from electronic structure calculations. Angew. Chem. Int. Ed. 2020, 59, 12308−12314. doi: 10.1002/anie.202001223
-
[24]
Goeppert, A.; Czaun, M.; Jones, J. P.; Surya Prakash, G. K.; Olah, G. A. Recycling of carbon dioxide to methanol and derived products-closing the loop. Chem. Soc. Rev. 2014, 43, 7995−8048. doi: 10.1039/C4CS00122B
-
[25]
Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703−3727. doi: 10.1039/c1cs15008a
-
[26]
Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. Rsc Adv. 2018, 8, 7651−7669. doi: 10.1039/C7RA13546G
-
[27]
Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A. V.; Wezendonk, T. A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 2017, 117, 9804−9838. doi: 10.1021/acs.chemrev.6b00816
-
[28]
Kato, S.; Matam, S. K.; Kerger, P.; Bernard, L.; Battaglia, C.; Vogel, D.; Rohwerder, M.; Züttel, A. The origin of the catalytic activity of a metal hydride in CO2 reduction. Angew. Chem. Int. Ed. 2016, 55, 6028−6032. doi: 10.1002/anie.201601402
-
[29]
Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551−12554. doi: 10.1002/anie.201105481
-
[30]
Tang, S. Y.; Rijs, N. J.; Li, J.; Schlangen, M.; Schwarz, H. Ligand-controlled CO2 activation mediated by cationic titanium hydride complexes, [LTiH]+ (L = Cp2, O). Chem. Eur. J. 2015, 21, 8483−8490. doi: 10.1002/chem.201500722
-
[31]
Zavras, A.; Ghari, H.; Ariafard, A.; Canty, A. J.; O'Hair, R. A. J. Gas-phase ion-molecule reactions of copper hydride anions [CuH2]− and [Cu2H3]−. Inorg. Chem. 2017, 56, 2387−2399. doi: 10.1021/acs.inorgchem.6b02145
-
[32]
Zhang, X.; Liu, G.; Meiwes-Broer, K. H.; Ganteför, G.; Bowen, K. CO2 activation and hydrogenation by PtHn− cluster anions. Angew. Chem. Int. Ed. 2016, 55, 9644−9647. doi: 10.1002/anie.201604308
-
[33]
Jiang, L. X.; Zhao, C.; Li, X. N.; Chen, H.; He, S. G. Formation of gas-phase formate in thermal reactions of carbon dioxide with diatomic iron hydride anions. Angew. Chem. Int. Ed. 2017, 56, 4187−4191. doi: 10.1002/anie.201611483
-
[34]
Jiang, L. X.; Li, X. N.; He, S. G. Metal-dependent selectivity on the reactions of carbon dioxide with diatomic hydride anions MH– (M = Co, Ni, and Cu). J. Phys. Chem. C 2020, 124, 5928−5933. doi: 10.1021/acs.jpcc.9b11619
-
[35]
Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W. A.; Kühn, F. E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011, 50, 8510−8537. doi: 10.1002/anie.201102010
-
[36]
Musashi, Y.; Sakaki, S. Theoretical study of ruthenium-catalyzed hydrogenation of carbon dioxide into formic acid. Reaction mechanism involving a new type of σ-bond metathesis. J. Am. Chem. Soc. 2000, 122, 3867−3877. doi: 10.1021/ja9938027
-
[37]
Langer, R.; Diskin-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Ben-David, Y.; Milstein, D. Low-pressure hydrogenation of carbon dioxide catalyzed by an iron pincer complex exhibiting noble metal activity. Angew. Chem. Int. Ed. 2011, 50, 9948−9952. doi: 10.1002/anie.201104542
-
[38]
Yang, X.; Hall, M. B. Monoiron hydrogenase catalysis: hydrogen activation with the formation of a dihydrogen, Fe−Hδ−···Hδ+−O, bond and methenyl-H4MPT+ triggered hydride transfer. J. Am. Chem. Soc. 2009, 131, 10901−10908. doi: 10.1021/ja902689n
-
[39]
Allred, A. L. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 1961, 17, 215−221. doi: 10.1016/0022-1902(61)80142-5
-
[40]
Li, S.; Xu, Y.; Chen, Y.; Li, W.; Lin, L.; Li, M.; Deng, Y.; Wang, X.; Ge, B.; Yang, C.; Yao, S.; Xie, J.; Li, Y.; Liu, X.; Ma, D. Tuning the selectivity of catalytic carbon dioxide hydrogenation over iridium/cerium oxide catalysts with a strong metal-support interaction. Angew. Chem. Int. Ed. 2017, 56, 10761−10765. doi: 10.1002/anie.201705002
-
[41]
Matsubu, J. C.; Yang, V. N.; Christopher, P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015, 137, 3076−3084. doi: 10.1021/ja5128133
-
[42]
Schultz, R. H.; Armentrout, P. B. The gas-phase thermochemistry of FeH. J. Chem. Phys. 1991, 94, 2262−2268. doi: 10.1063/1.459897
-
[43]
Mccarthy, M. C.; Field, R. W.; Engleman, R.; Bernath, P. F. Laser and Fourier transform spectroscopy of PtH and PtD. J. Mol. Spectrosc. 1993, 158, 208−236. doi: 10.1006/jmsp.1993.1067
-
[44]
Chen, Y. M.; Clemmer, D. E.; Armentrout, P. B. The gas-phase thermochemistry of TiH. J. Chem. Phys. 1991, 95, 1228−1233.
-
[45]
Kattel, S.; Liu, P.; Chen, J. G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739−9754. doi: 10.1021/jacs.7b05362
-
[46]
Zhao, B.; Yan, B.; Jiang, Z.; Yao, S.; Liu, Z.; Wu, Q.; Ran, R.; Senanayake, S. D.; Weng, D.; Chen, J. G. High selectivity of CO2 hydrogenation to CO by controlling the valence state of nickel using perovskite. Chem. Commun. 2018, 54, 7354−7357. doi: 10.1039/C8CC03829E
-
[47]
Yoo, C.; Kim, Y. E.; Lee, Y. Selective transformation of CO2 to CO at a single nickel center. Acc. Chem. Res. 2018, 51, 1144−1152. doi: 10.1021/acs.accounts.7b00634
-
[48]
Hicken, A.; White, A. J. P.; Crimmin, M. R. Selective reduction of CO2 to a formate equivalent with heterobimetallic gold···copper hydride complexes. Angew. Chem. Int. Ed. 2017, 56, 15127−15130. doi: 10.1002/anie.201709072
-
[49]
Sirijaraensre, J.; Limtrakul, J. Hydrogenation of CO2 to formic acid over a Cu-embedded graphene: a DFT study. Appl. Surf. Sci. 2016, 364, 241−248. doi: 10.1016/j.apsusc.2015.12.117
-
[50]
Zall, C. M.; Linehan, J. C.; Appel, A. M. A molecular copper catalyst for hydrogenation of CO2 to formate. ACS Catal. 2015, 5, 5301−5305. doi: 10.1021/acscatal.5b01646
-
[51]
Lee, Y.; Anderton, K. J.; Sloane, F. T.; Ermert, D. M.; Abboud, K. A.; García-Serres, R.; Murray, L. J. Reactivity of hydride bridges in high-spin [3M-3(μ-H)] clusters (M = FeII, CoII). J. Am. Chem. Soc. 2015, 137, 10610−10617. doi: 10.1021/jacs.5b05204
-
[52]
Yu, Y.; Sadique, A. R.; Smith, J. M.; Dugan, T. R.; Cowley, R. E.; Brennessel, W. W.; Flaschenriem, C. J.; Bill, E.; Cundari, T. R.; Holland, P. L. The reactivity patterns of low-coordinate iron-hydride complexes. J. Am. Chem. Soc. 2008, 130, 6624−6638. doi: 10.1021/ja710669w
-
[53]
Liu, Y. Z.; Jiang, L. X.; Li, X. N.; Wang, L. N.; Chen, J. J.; He, S. G. Gas-phase reactions of carbon dioxide with copper hydride anions Cu2H2–: temperature-dependent transformation. J. Phys. Chem. C 2018, 122, 19379−19384. doi: 10.1021/acs.jpcc.8b05216
-
[54]
Liu, Y. Z.; Li, X. N.; He, S. G. Reactivity of iron hydride anions Fe2Hn– (n = 0~3) with carbon dioxide. J. Phys. Chem. A 2020, 124, 8414−8420. doi: 10.1021/acs.jpca.0c06986
-
[55]
Kunkel, C.; Viñes, F.; Illas, F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 2016, 9, 141−144. doi: 10.1039/C5EE03649F
-
[56]
Posada-Pérez, S.; Viñes, F.; Ramirez, P. J.; Vidal, A. B.; Rodriguez, J. A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912−14921. doi: 10.1039/C4CP01943A
-
[57]
Liu, G.; Poths, P.; Zhang, X.; Zhu, Z.; Marshall, M.; Blankenhorn, M.; Alexandrova, A. N.; Bowen, K. H. CO2 hdrogenation to formate and formic acid by bimetallic palladium-copper hydride clusters. J. Am. Chem. Soc. 2020, 142, 7930−7936. doi: 10.1021/jacs.0c01855
-
[58]
Wu, J.; Wang, L.; Lv, B.; Chen, J. Facile fabrication of BCN nanosheet-encapsulated nano-iron as highly stable Fischer-Tropsch synthesis catalyst. ACS Appl. Mater. Interfaces 2017, 9, 14319−14327. doi: 10.1021/acsami.7b00561
-
[59]
Sun, W.; Meng, Y.; Fu, Q.; Wang, F.; Wang, G.; Gao, W.; Huang, X.; Lu, F. High-yield production of boron nitride nanosheets and its uses as a catalyst support for hydrogenation of nitroaromatics. ACS Appl. Mater. Interfaces 2016, 8, 9881−9888. doi: 10.1021/acsami.6b01008
-
[60]
Lin, S.; Ye, X.; Johnson, R. S.; Guo, H. First-principles investigations of metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) doped hexagonal boron nitride nanosheets: stability and catalysis of CO oxidation. J. Phys. Chem. C 2013, 117, 17319−17326. doi: 10.1021/jp4055445
-
[61]
Zhao, Y. X.; Yang, B.; Li, H. F.; Zhang, Y.; Yang, Y.; Liu, Q. Y.; Xu, H. G.; Zheng, W. J.; He, S. G. Photoassisted selective steam and dry reforming of methane to syngas catalyzed by rhodium-vanadium bimetallic oxide cluster anions at room temperature. Angew. Chem. Int. Ed. 2020, 59, 21216−21223. doi: 10.1002/anie.202010026
-
[62]
Yang, Y.; Yang, B.; Zhao, Y. X.; Jiang, L. X.; Li, Z. Y.; Ren, Y.; Xu, H. G.; Zheng, W. J.; He, S. G. Direct conversion of methane with carbon dioxide mediated by RhVO3− cluster anions. Angew. Chem. Int. Ed. 2019, 58, 17287−17292. doi: 10.1002/anie.201911195
-
[63]
Chen, Q.; Zhao, Y. X.; Jiang, L. X.; Chen, J. J.; He, S. G. Coupling of methane and carbon dioxide mediated by diatomic copper boride cations. Angew. Chem. Int. Ed. 2018, 57, 14134−14138. doi: 10.1002/anie.201808780
-
[64]
Liu, G.; Ciborowski, S. M.; Zhu, Z.; Chen, Y.; Zhang, X.; Bowen, K. H. The metallo-formate anions, M(CO2)−, M = Ni, Pd, Pt, formed by electron-induced CO2 activation. Phys. Chem. Chem. Phys. 2019, 21, 10955−10960. doi: 10.1039/C9CP01915D
-
[65]
Zhang, X.; Lim, E.; Kim, S. K.; Bowen, K. H. Photoelectron spectroscopic and computational study of (M−CO2)− anions, M = Cu, Ag, Au. J. Chem. Phys. 2015, 143, 174305−6. doi: 10.1063/1.4935061
-
[66]
Zhao, Z.; Kong, X.; Yuan, Q.; Xie, H.; Yang, D.; Zhao, J.; Fan, H.; Jiang, L. Coordination-induced CO2 fixation into carbonate by metal oxides. Phys. Chem. Chem. Phys. 2018, 20, 19314−19320. doi: 10.1039/C8CP02085J
-
[67]
Gong, Y.; Zhou, M. F.; Andrews, L. Spectroscopic and theoretical studies of transition metal oxides and dioxygen complexes. Chem. Rev. 2009, 109, 6765−6808. doi: 10.1021/cr900185x
-
[68]
Zhou, M. F.; Andrews, L.; Bauschlicher, C. W. Spectroscopic and theoretical investigations of vibrational frequencies in binary unsaturated transition-metal carbonyl cations, neutrals, and anions. Chem. Rev. 2001, 101, 1931−1961. doi: 10.1021/cr990102b
-
[69]
Zhang, Q.; Qu, H.; Chen, M.; Zhou, M. Carbon dioxide activation by scandium atoms and scandium monoxide molecules: formation and spectroscopic characterization of ScCO3 and OCScCO3 in solid neon. J. Phys. Chem. A 2016, 120, 425−432. doi: 10.1021/acs.jpca.5b11809
-
[70]
Zhang, Q.; Chen, M.; Zhou, M. Infrared spectra and structures of the neutral and charged CrCO2 and Cr(CO2)2 isomers in solid neon. J. Phys. Chem. A 2014, 118, 6009−6017. doi: 10.1021/jp505740j
-
[71]
Zhuang, J.; Li, Z. H.; Fan, K.; Zhou, M. Matrix isolation spectroscopic and theoretical study of carbon dioxide activation by titanium oxide molecules. J. Phys. Chem. A 2012, 116, 3388−3395. doi: 10.1021/jp301025n
-
[72]
Zhou, M.; Zhou, Z.; Zhuang, J.; Li, Z. H.; Fan, K.; Zhao, Y.; Zheng, X. Carbon dioxide coordination and activation by niobium oxide molecules. J. Phys. Chem. A 2011, 115, 14361−14369. doi: 10.1021/jp208291g
-
[73]
Jiang, L.; Teng, Y. L.; Xu, Q. Infrared spectroscopic and density functional theory study on the reactions of rhodium and cobalt atoms with carbon dioxide in rare-gas matrixes. J. Phys. Chem. A 2007, 111, 7793−7799. doi: 10.1021/jp0728095
-
[74]
Liang, B.; Andrews, L. Reactions of laser-ablated osmium and ruthenium atoms with carbon dioxide: matrix infrared spectra and density functional calculations on OMCO, O2MCO, OMCO− (M = Os, Ru), O2Os(CO)2, and OCRu(O2)CO. J. Phys. Chem. A 2002, 106, 4042−4053. doi: 10.1021/jp0200167
-
[75]
Liang, B. Y.; Andrews, L. Reactions of laser-ablated rhenium atoms with carbon dioxide: matrix infrared spectra and density functional calculations on OReCO, O2ReCO, ORe(CO)2, O2Re(CO)2, OReCO−, and ORe(CO)2−. J. Phys. Chem. A 2002, 106, 595−602. doi: 10.1021/jp013184s
-
[76]
Zhang, L. N.; Wang, X. F.; Chen, M. H.; Qin, Q. Z. Activation of CO2 by Zr atom. Matrix-isolation FTIR spectroscopy and density functional studies. Chem. Phys. 2000, 254, 231−238. doi: 10.1016/S0301-0104(00)00031-8
-
[77]
Wang, X. F.; Chen, M. H.; Zhang, L. N.; Qin, Q. Z. Spectroscopic and theoretical studies on the reactions of laser-ablated tantalum with carbon dioxide. J. Phys. Chem. A 2000, 104, 758−764. doi: 10.1021/jp9927808
-
[78]
Andrews, L.; Zhou, M. F.; Liang, B. Y.; Li, J.; Bursten, B. E. Reactions of laser-ablated U and Th with CO2: neon matrix infrared spectra and density functional calculations of OUCO, OThCO, and other products. J. Am. Chem. Soc. 2000, 122, 11440−11449. doi: 10.1021/ja0016699
-
[79]
Zhou, M. F.; Liang, B. Y.; Andrews, L. Infrared spectra of OMCO (M = Cr−Ni), OMCO− (M = Cr−Cu), and MCO2− (M = Co−Cu) in solid argon. J. Phys. Chem. A 1999, 103, 2013−2023. doi: 10.1021/jp984439d
-
[80]
Zhou, M. F.; Andrews, L. Infrared spectra and density functional calculations for OMCO, OM−(η2-CO), OMCO+, and OMOC+ (M = V, Ti) in solid argon. J. Phys. Chem. A 1999, 103, 2066−2075. doi: 10.1021/jp9844009
-
[81]
Souter, P. F.; Andrews, L. A spectroscopic and theoretical study of the reactions of group 6 metal atoms with carbon dioxide. J. Am. Chem. Soc. 1997, 119, 7350−7360. doi: 10.1021/ja971038n
-
[82]
Clemmer, D. E.; Weber, M. E.; Armentrout, P. B. Reactions of Al+(1S) with NO2, N2O, and CO2: thermochemistry of AlO and AlO+. J. Phys. Chem. 1992, 96, 10888−10893. doi: 10.1021/j100205a052
-
[83]
Sievers, M. R.; Armentrout, P. B. Potential energy surface for carbon-dioxide activation by V+: a guided ion beam study. J. Chem. Phys. 1995, 102, 754−762. doi: 10.1063/1.469188
-
[84]
Griffin, J. B.; Armentrout, P. B. Guided ion beam studies of the reactions of Crn+ (n = 1~18) with CO2: chromium cluster oxide bond energies. J. Chem. Phys. 1998, 108, 8075−8083. doi: 10.1063/1.476246
-
[85]
Griffin, J. B.; Armentrout, P. B. Guided ion-beam studies of the reactions of Fen+ (n = 1~18) with CO2: iron cluster oxide bond energies. J. Chem. Phys. 1997, 107, 5345−5355. doi: 10.1063/1.474244
-
[86]
Hintz, P. A.; Ervin, K. M. Chemisorption and oxidation reactions of nickel group cluster anions with N2, O2, CO2, and N2O. J. Chem. Phys. 1995, 103, 7897−7906. doi: 10.1063/1.470207
-
[87]
Rodgers, M. T.; Walker, B.; Armentrout, P. B. Reactions of Cu+ (1S and 3D) with O2, CO, CO2, N2, NO, N2O, and NO2 studied by guided ion beam mass spectrometry. Int. J. Mass Spectrom. 1999, 182/183, 99−120. doi: 10.1016/S1387-3806(98)14228-8
-
[88]
Sievers, M. R.; Armentrout, P. B. Activation of carbon dioxide: gas-phase reactions of Y+, YO+, and YO2+ with CO and CO2. Inorg. Chem. 1999, 38, 397−402. doi: 10.1021/ic981117f
-
[89]
Sievers, M. R.; Armentrout, P. B. Oxidation of CO and reduction of CO2 by gas phase Zr+, ZrO+, and ZrO2+. Int. J. Mass Spectrom. 1999, 185/186/187, 117−129.
-
[90]
Sievers, M. R.; Armentrout, P. B. Gas phase activation of carbon dioxide by niobium and niobium monoxide cations. Int. J. Mass Spectrom. 1998, 179/180, 103−115. doi: 10.1016/S1387-3806(98)14064-2
-
[91]
Sievers, M. R.; Armentrout, P. B. Reactions of CO and CO2 with gas-phase Mo+, MoO+, and MoO2+. J. Phys. Chem. A 1998, 102, 10754−10762. doi: 10.1021/jp983694v
-
[92]
Cornehl, H. H.; Wesendrup, R.; Diefenbach, M.; Schwarz, H. A comparative study of oxo-ligand effects in the gas-phase chemistry of atomic lanthanide and actinide cations. Chem. Eur. J. 1997, 3, 1083−1090. doi: 10.1002/chem.19970030716
-
[93]
Armentrout, P. B.; Cox, R. M. Potential energy surface for the reaction Sm+ + CO2 → SmO+ + CO: guided ion beam and theoretical studies. Phys. Chem. Chem. Phys. 2017, 19, 11075−11088. doi: 10.1039/C7CP00914C
-
[94]
Demireva, M.; Armentrout, P. B. Activation of CO2 by gadolinium cation (Gd+): energetics and mechanism from experiment and theory. Top. Catal. 2018, 61, 3−19. doi: 10.1007/s11244-017-0858-1
-
[95]
Lourenço, C.; Michelini, M. C.; Marçalo, J.; Gibson, J. K.; Oliveira, M. C. Gas-phase reaction studies of dipositive hafnium and hafnium oxide ions: generation of the peroxide HfO22+. J. Phys. Chem. A 2012, 116, 12399−12405. doi: 10.1021/jp3088385
-
[96]
Wesendrup, R.; Schwarz, H. Tantalum-mediated coupling of methane and carbon dioxide in the gas phase. Angew. Chem, Int. Ed. 1995, 34, 2033−2035. doi: 10.1002/anie.199520331
-
[97]
Santos, M.; Michelini, M. C.; Lourenço, C.; Marçalo, J.; Gibson, J. K.; Oliveira, M. C. Gas-phase oxidation reactions of Ta2+: synthesis and properties of TaO2+ and TaO22+. J. Phys. Chem. A 2012, 116, 3534−3540. doi: 10.1021/jp300294c
-
[98]
Irikura, K. K.; Beauchamp, J. L. Electronic structure considerations for methane activation by third-row transition-metal ions. J. Phys. Chem. 1991, 95, 8344−8351. doi: 10.1021/j100174a057
-
[99]
Zhang, X. G.; Armentrout, P. B. Activation of O2, CO, and CO2 by Pt+: the thermochemistry of PtO+. J. Phys. Chem. A 2003, 107, 8904−8914. doi: 10.1021/jp036014j
-
[100]
Santos, M.; Marçalo, J.; de Matos, A. P.; Gibson, J. K.; Haire, R. G. Gas-phase oxidation reactions of neptunium and plutonium ions investigated via Fourier transform ion cyclotron resonance mass spectrometry. J. Phys. Chem. A 2002, 106, 7190−7194. doi: 10.1021/jp025733f
-
[101]
Armentrout, P. B.; Beauchamp, J. L. Reactions of U+ and UO+ with O2, CO, CO2, COS, CS2 and D2O. Chem. Phys. 1980, 50, 27−36. doi: 10.1016/0301-0104(80)87022-4
-
[102]
Canale, V.; Robinson, R.; Zavras, A.; Khairallah, G. N.; d'Alessandro, N.; Yates, B. F.; O'Hair, R. A. J. Two spin-state reactivity in the activation and cleavage of CO2 by [ReO2]−. J. Phys. Chem. Lett. 2016, 7, 1934−1938. doi: 10.1021/acs.jpclett.6b00754
-
[103]
Zhang, X. G.; Armentrout, P. B. Activation of O2 and CO2 by PtO+: the thermochemistry of PtO2+. J. Phys. Chem. A 2003, 107, 8915−8922. doi: 10.1021/jp036015b
-
[104]
Hossain, E.; Rothgeb, D. W.; Jarrold, C. C. CO2 reduction by group 6 transition metal suboxide cluster anions. J. Chem. Phys. 2010, 133, 024305−10. doi: 10.1063/1.3455220
-
[105]
Rothgeb, D. W.; Hossain, E.; Mann, J. E.; Jarrold, C. C. Disparate product distributions observed in Mo(3-x)WxOy− (x = 0~3; y = 3~9) reactions with D2O and CO2. J. Chem. Phys. 2010, 132, 064302−10. doi: 10.1063/1.3313927
-
[106]
Firouzbakht, M.; Rijs, N. J.; González-Navarrete, P.; Schlangen, M.; Kaupp, M.; Schwarz, H. On the activation of methane and carbon dioxide by [HTaO]+ and [TaOH]+ in the gas phase: a mechanistic study. Chem. Eur. J. 2016, 22, 10581−10589. doi: 10.1002/chem.201601339
-
[107]
Kretzschmar, I.; Schröder, D.; Schwarz, H.; Rue, C.; Armentrout, P. B. Thermochemistry and reactivity of cationic scandium and titanium sulfide in the gas phase. J. Phys. Chem. A 2000, 104, 5046−5058. doi: 10.1021/jp994228o
-
[108]
Kretzschmar, I.; Schröder, D.; Schwarz, H.; Rue, C.; Armentrout, P. B. Experimental and theoretical studies of vanadium sulfide cation. J. Phys. Chem. A 1998, 102, 10060−10073. doi: 10.1021/jp982199w
-
[109]
Kretzschmar, I.; Schröder, D.; Schwarz, H.; Armentrout, P. B. Gas-phase thermochemistry of the early cationic transition-metal sulfides of the second row: YS+, ZrS+, and NbS+. Int. J. Mass Spectrom. 2006, 249−250, 263−278.
-
[110]
Miller, G. B. S.; Uggerud, E. C−C bond formation of Mg- and Zn-activated carbon dioxide. Chem. Eur. J. 2018, 24, 4710−4717. doi: 10.1002/chem.201706069
-
[111]
Firouzbakht, M.; Schlangen, M.; Kaupp, M.; Schwarz, H. Mechanistic aspects of CO2 activation mediated by phenyl yttrium cation: a combined experimental/theoretical study. J. Catal. 2016, 343, 68−74. doi: 10.1016/j.jcat.2015.09.012
-
[112]
Zhou, S. D.; Li, J. L.; Firouzbakht, M.; Schlangen, M.; Schwarz, H. Sequential gas-phase activation of carbon dioxide and methane by [Re(CO)2]+: the sequence of events matters! J. Am. Chem. Soc. 2017, 139, 6169−6176. doi: 10.1021/jacs.7b01255
-
[113]
Dau, P. D.; Armentrout, P. B.; Michelini, M. C.; Gibson, J. K. Activation of carbon dioxide by a terminal uranium-nitrogen bond in the gas-phase: a demonstration of the principle of microscopic reversibility. Phys. Chem. Chem. Phys. 2016, 18, 7334−7340. doi: 10.1039/C6CP00494F
-
[114]
Gregoire, G.; Brinkmann, N. R.; van Heijnsbergen, D.; Schaefer, H. F.; Duncan, M. A. Infrared photodissociation spectroscopy of Mg+(CO2)n and Mg+(CO2)nAr clusters. J. Phys. Chem. A 2003, 107, 218−227. doi: 10.1021/jp026373z
-
[115]
Walters, R. S.; Brinkmann, N. R.; Schaefer, H. F.; Duncan, M. A. Infrared photodissociation spectroscopy of mass-selected Al+(CO2)n and Al+(CO2)nAr clusters. J. Phys. Chem. A 2003, 107, 7396−7405.
-
[116]
Jaeger, J. B.; Jaeger, T. D.; Brinkmann, N. R.; Schaefer, H. F.; Duncan, M. A. Infrared photodissociation spectroscopy of Si+(CO2)n and Si+(CO2)nAr complexes − evidence for unanticipated intracluster reactions. Can. J. Chem. 2004, 82, 934−946. doi: 10.1139/v04-044
-
[117]
Xing, X. P.; Wang, G. J.; Wang, C. X.; Zhou, M. F. Infrared photodissociation spectroscopy of Ti+(CO2)2Ar and Ti+(CO2)n (n = 3~7) complexes. Chin. J. Chem. Phys. 2013, 26, 687−693. doi: 10.1063/1674-0068/26/06/687-693
-
[118]
Ricks, A. M.; Brathwaite, A. D.; Duncan, M. A. IR spectroscopy of gas phase V(CO2)n+ clusters: solvation-induced electron transfer and activation of CO2. J. Phys. Chem. A 2013, 117, 11490−11498.
-
[119]
Walker, N. R.; Walters, R. S.; Duncan, M. A. Infrared photodissociation spectroscopy of V+(CO2)n and V+(CO2)nAr complexes. J. Chem. Phys. 2004, 120, 10037−10045. doi: 10.1063/1.1730217
-
[120]
Gregoire, G.; Duncan, M. A. Infrared spectroscopy to probe structure and growth dynamics in Fe+-(CO2)n clusters. J. Chem. Phys. 2002, 117, 2120−2130. doi: 10.1063/1.1490600
-
[121]
Gregoire, G.; Velasquez, J.; Duncan, M. A. Infrared photodissociation spectroscopy of small Fe+-(CO2)n and Fe+-(CO2)nAr clusters. Chem. Phys. Lett. 2001, 349, 451−457. doi: 10.1016/S0009-2614(01)01247-7
-
[122]
Yang, D.; Su, M. Z.; Zheng, H. J.; Zhao, Z.; Li, G.; Kong, X. T.; Xie, H.; Fan, H. J.; Zhang, W. Q.; Jiang, L. Infrared photodissociation spectroscopic and theoretical study of Co(CO2)n+ clusters. Chin. J. Chem. Phys. 2019, 32, 223−228. doi: 10.1063/1674-0068/cjcp1902032
-
[123]
Iskra, A.; Gentleman, A. S.; Kartouzian, A.; Kent, M. J.; Sharp, A. P.; Mackenzie, S. R. Infrared spectroscopy of gas-phase M+(CO2)n (M = Co, Rh, Ir) ion-molecule complexes. J. Phys. Chem. A 2017, 121, 133−140. doi: 10.1021/acs.jpca.6b10902
-
[124]
Walker, N. R.; Walters, R. S.; Grieves, G. A.; Duncan, M. A. Growth dynamics and intracluster reactions in Ni+(CO2)n complexes via infrared spectroscopy. J. Chem. Phys. 2004, 121, 10498−10507. doi: 10.1063/1.1806821
-
[125]
Walker, N. R.; Grieves, G. A.; Walters, R. S.; Duncan, M. A. The metal coordination in Ni+(CO2)n and NiO2+(CO2)m complexes. Chem. Phys. Lett. 2003, 380, 230−236. doi: 10.1016/j.cplett.2003.08.107
-
[126]
Zhao, Z.; Kong, X.; Yang, D.; Yuan, Q.; Xie, H.; Fan, H.; Zhao, J.; Jiang, L. Reactions of copper and silver cations with carbon dioxide: an infrared photodissociation spectroscopic and theoretical study. J. Phys. Chem. A 2017, 121, 3220−3226. doi: 10.1021/acs.jpca.7b01320
-
[127]
Kong, X.; Shi, R.; Wang, C.; Zheng, H.; Wang, T.; Liang, X.; Yang, J.; Jing, Q.; Liu, Y.; Han, H.; Zhao, Z.; Fan, H.; Li, G.; Xie, H. Interaction between CO2 and NbO2+: infrared photodissociation spectroscopic and theoretical study. Chem. Phys. 2020, 534, 110755−7. doi: 10.1016/j.chemphys.2020.110755
-
[128]
Iskra, A.; Gentleman, A. S.; Cunningham, E. M.; Mackenzie, S. R. Carbon dioxide binding to metal oxides: infrared spectroscopy of NbO2+(CO2)n and TaO2+(CO2)n complexes. Int. J. Mass Spectrom. 2019, 435, 93−100. doi: 10.1016/j.ijms.2018.09.038
-
[129]
Thompson, M. C.; Ramsay, J.; Weber, J. M. Interaction of CO2 with atomic manganese in the presence of an excess negative charge probed by infrared spectroscopy of [Mn(CO2)n]− clusters. J. Phys. Chem. A 2017, 121, 7534−7542. doi: 10.1021/acs.jpca.7b06870
-
[130]
Thompson, M. C.; Dodson, L. G.; Weber, J. M. Structural motifs of [Fe(CO2)n]− clusters (n = 3~7). J. Phys. Chem. A 2017, 121, 4132−4138. doi: 10.1021/acs.jpca.7b02742
-
[131]
Knurr, B. J.; Weber, J. M. Infrared spectra and structures of anionic complexes of cobalt with carbon dioxide ligands. J. Phys. Chem. A 2014, 118, 4056−4062. doi: 10.1021/jp503194v
-
[132]
Knurr, B. J.; Weber, J. M. Interaction of nickel with carbon dioxide in [Ni(CO2)n]− clusters studied by infrared spectroscopy. J. Phys. Chem. A 2014, 118, 8753−8757. doi: 10.1021/jp507149u
-
[133]
Knurr, B. J.; Weber, J. M. Structural diversity of copper-CO2 complexes: infrared spectra and structures of [Cu(CO2)n]− clusters. J. Phys. Chem. A 2014, 118, 10246−10251. doi: 10.1021/jp508219y
-
[134]
Knurr, B. J.; Weber, J. M. Solvent-mediated reduction of carbon dioxide in anionic complexes with silver atoms. J. Phys. Chem. A 2013, 117, 10764−10771. doi: 10.1021/jp407646t
-
[135]
Knurr, B. J.; Weber, J. M. Solvent-driven reductive activation of carbon dioxide by gold anions. J. Am. Chem. Soc. 2012, 134, 18804−18808. doi: 10.1021/ja308991a
-
[136]
Dodson, L. G.; Thompson, M. C.; Weber, J. M. Interactions of molecular titanium oxides TiOx (x = 1~3) with carbon dioxide in cluster anions. J. Phys. Chem. A 2018, 122, 6909−6917. doi: 10.1021/acs.jpca.8b06229
-
[137]
Miller, G. B. S.; Esser, T. K.; Knorke, H.; Gewinner, S.; Schöllkopf, W.; Heine, N.; Asmis, K. R.; Uggerud, E. Spectroscopic identification of a bidentate binding motif in the anionic magnesium-CO2 complex ([ClMgCO2]−). Angew. Chem. Int. Ed. 2014, 53, 14407−14410. doi: 10.1002/anie.201409444
-
[138]
Graham, J. D.; Buytendyk, A. M.; Zhang, X.; Kim, S. K.; Bowen, K. H. Carbon dioxide is tightly bound in the [Co(Pyridine)(CO2)]− anionic complex. J. Chem. Phys. 2015, 143, 184315−4. doi: 10.1063/1.4935573
-
[1]
-
Figure 1 The reactions of V2+ and V2O+ with C18O2 inside the ion trap of the Fourier-transform ion cyclotron resonance mass spectrometer. The pressure of C18O2 in (a) is about 1.0×10−7 mbar and in (b) is 4.0×10−9 mbar. Calculated potential energy surfaces for the reactions of (c) V2+ and (d) V2O+ with CO2. Key structures with selected geometric parameters are provided. The relative energies are in unit of eV. Reprinted with permission from reference 23
Figure 2 Time-of-flight (TOF) mass spectra for the reactions of mass-selected FeH−, CoH−, NiH−, and CuH− (a, c, e, and g) with CO2 (b, d, f, and h). The reactant pressures are shown in mPa (= 10−3 Pa). Reprinted with permission from references 33 and 34
Figure 3 Theoretical calculated potential energy profiles for the reaction of FeH− with CO2 in the quintet state. The relative energies are given in eV. The bond lengths are given in pm. Reprinted with permission from reference 33
Figure 4 Natural charge (e) distribution on the crucial intermediates A (in reaction CoH− + CO2) and B (in reaction NiH− + CO2). Reprinted with permission from reference 34
Figure 5 TOF mass spectra for the reactions of mass-selected Cu2H2− (a), Fe2H− (d), and Fe2H2− (f) with CO2 at different temperatures (b, c, e, and g). Reprinted with permission from references 53 and 54
Figure 6 Natural charge (e) distribution on the Fe atoms in Fe2Hn− (n = 0~3). Cited with permission from reference 54
Figure 7 Theoretical calculated relevant low-lying energy structures of PdCuH4− (A and B), PdCuCO2H4− (C, D, and E), and PdCuH2−. The relative energies for PdCuH4− (A and B) and PdCuCO2H4− (C, and D) are shown below each structure. Reprinted with permission from reference 57
Figure 8 TOF mass spectra for the reactions of mass-selected Nb2BN2− with Ar (a) and CO2 (b~d). The reactant pressures are shown in mPa (= 10−3 Pa). Calculated structures of Nb2BN2−, Nb2N2−, and Nb2B− (e). The relative energies are shown below each structure. Reprinted with permission from reference 20
Figure 9 TOF mass spectra for the reactions of mass-selected RhVO3− (a) with CH4 (b) seeded in the cooling gas (He) and then react with CO2 at 295K (c) and 600 K (d). The pressure of CH4 in panel b is about 1.4 Pa and the pressures of CO2 in panel c is about 0.2 Pa and in panel d about 0.7 Pa. Calculated structures for RhVO3−, RhVO3CH4−, RhVO3CH4CO2−, and RhVO4C− are shown (e). Reprinted with permission from reference 62
Table 1. A Summary of Experimentally Identified Results on the Interactions of Gas-phase Metal Species with CO2
Species Ion Products Neutral Products k1a Methodsb Remark Year Al+ AlO+ CO, C, O F 1992[82] Sc+ ScO+, ScO(CO2)1, 2+ CO 7.4 × 10−11 A 2006[21] Ti+ TiO+, TiO(CO2)+ CO 4.1 × 10−11 A 2006[21] V+ VO+, VCO+, VO2+ CO, O, C F 1995[83] V2+ V2O+ CO 3.8 × 10−12 D, theory 2020[23] Crn+ CrnO+ CO F n = 1~18 1998[84] Fen+ FenO+ CO F n = 1~18 1997[85] Fe2− Fe2O−, Fe2O2− CO (1.8 ± 0.4) × 10−10 C, theory 2020[54] Nix− Nix(CO2)y−, Nix(CO2)yOz− CO (0.63–1.09) × 10−10 A x = 3~5 1995[86] Cu+ CuO+, CuCO+ CO, O F 1999[87] Y+ YO+, YO(CO2)+ CO 5.9 × 10−10 A 2006[21] Y+ YO+, YCO+ CO, O F 1999[88] Zr+ ZrO+, ZrO(CO2)1, 2+ CO 2.5 × 10−10 A 2006[21] Zr+ ZrO+,
ZrCO+, ZrO2+CO, O, C F 1999[89] Nb+ NbO+,
NbO(CO2)+, NbO2(CO2)0−3+CO 1.8 × 10−10 A 2006[21] Nb+ NbO+, NbCO+, NbO2+ CO, O, C F 1998[90] Mo+ MoO+, MoCO+, MoO2+ CO, O, C F 1998[91] Pdx− Pdx(CO2)y− (0.05–3.4) × 10−10 A x = 3~8 1995[86] La+ LaO+, LaO(CO2)+ CO 4.2 × 10−10 A 2006[21] La+ LaO+, LaO(CO2)+ CO 4.4 × 10−10 A 2006[22] Ce+ CeO+, CeO(CO2)+ CO 4.6 × 10−10 A 2006[22] Ce+ CeO+ CO D 1997[92] Pr+ PrO+, PrO(CO2)+ CO 1.6 × 10−10 A 2006[22] Nd+ NdO+, NdO(CO2)1−2+ CO 3.7 × 10−11 A 2006[22] Nd+ NdO+ CO D 1997[92] Sm+ SmO+ CO F, theory 2017[93] Gd+ GdO+, GdCO+, GdO2+ CO, O, C F, theory 2018[94] Gd+ GdO+, GdO(CO2)1, 2+ CO 3.4 × 10−10 A 2006[22] Tb+ TbO+, TbO(CO2)1−4+ CO 3.8 × 10−11 A 2006[22] Lu+ LuO+, LuO(CO2)1−5+ CO 3.3 × 10−11 A 2006[22] Hf+ HfO+, HfO(CO2)1, 2+ CO 2.5 × 10−10 A 2006[21] Hf2+ HfO2+, HfO+, CO+ CO 6.3 × 10−10 D, theory 2012[95] Ta+ TaO+, TaO2(CO2)0−4+ CO 2.4 × 10−10 A 2006[21] Ta+ TaO+ CO D 1995[96] Ta2+ TaO2+, Ta+, TaO+, CO2+, CO+ CO 6.5 × 10−10 D, theory 2012[97] W+ WO+, WO2(CO2)0−3+ CO 4.2 × 10−11 A 2006[21] W+ WO+ CO 6.0 × 10−11 D 1991[98] Pt+ PtO+, PtCO+, CO2+ CO, O, Pt F, theory 2003[99] Ptx− Ptx(CO2)y− (0.06−1.71) × 10−10 A x = 3~7 1995[86] Th+ ThO+ CO D 2002[100] Th+ ThO+ CO D 1997[92] U+ UO+ CO D 2002[100] U+ UO+ CO D 1997[92] U+ UO+, UO2+ CO, C F 1980[101] Np+ NpO+ CO D 2002[100] Pu+ PuO+ CO D 2002[100] YO+ YO2+, Y+ CO, O + CO2 F 1999[88] ZrO+ ZrO2+, ZrCO2+ CO, O F 1999[89] NbO+ NbO2+, NbCO2+ CO, O F 1998[90] MoO+ MoO2+, MoCO2+, Mo+ CO, O, O+CO2 F 1998[91] HfO22+ HfO2+, CO2+ 3.2 × 10−10 D, theory 2012[95] TaO+ TaO2+ CO D 1995[96] TaO2+ TaO+, CO2+ 1.3 × 10−10 D, theory 2012[97] TaO22+ TaO2+, CO2+ 7.2 × 10−10 D, theory 2012[97] WO+ WO2+ CO 2.0 × 10−11 D 1991[98] [ReO2]− [ReO3]− CO (1.9 ± 0.1) × 10−12 C, theory 2016[102] PtO+ Pt+, PtO2+, PtCO2+ O + CO2, O2 + CO, CO, O F, theory 2003[103] UO+ UO2+ CO D 2002[100] UO+ UO2+ CO F 1980[101] V2O+ V2O2+ CO 8.8 × 10−10 D, theory 2020[23] MoxOy− MoxOy+1−, MoxOy+1CO− CO A, theory x = 2, 3; y = 2~5 2010[104] WxOy− WxOy+1− CO A, theory x = 2, 3; y = 2~6 2010[104] MoxWyOz− MoxWyOz+1−, MoxWyOz+2C− CO A, theory x + y = 3, z = 2~7 2010[104] RhVO3− RhVO3CO2− (295 K),
RhVO4− (600 K)CO (600 K) (5.2 ± 1.5) × 10−11 (295 K) C, theory 2019[62] Rh2VO− Rh2VO2− CO (9.9 ± 2.0) × 10−10 C, theory 2020[61] Rh2VO2− Rh2VO2CO2−, Rh2VO3− CO (10.3 ± 2.2) × 10−10 C, theory 2020[61] [Cp2TiH]+ [Cp2TiHCO2]+ B, theory 2015[30] [OTiH]+ [OTiOH]+ CO B, theory 2015[30] [HTaO] +/[TaOH]+ [OTaOH]+ CO B, theory 2016[106] FeH− HCO2−, HFeO−, FeO2H− Fe, CO (6.2 ± 1.2) × 10−10 C, theory 2017[33] Fe2H− FeHCO2−, Fe2HO−, Fe2HO2− Fe, CO (2.4 ± 0.5) × 10−10 C, theory 2020[54] Fe2H2− FeH2CO2−, Fe2H2O− Fe, CO (1.5 ± 0.3) × 10−10 C, theory 2020[54] Fe2H3− Fe2H3O− CO (6.4 ± 1.3) × 10−11 C, theory 2020[54] CoH− HCO2−, OCoH−, O2CoH− Co, CO (8.0 ± 1.6) × 10−10 C, theory 2020[34] NiH− NiOH−, NiO2H− CO (4.4 ± 0.9) × 10−10 C, theory 2020[34] CuH− HCO2−, CuHCO2− Cu (2.7 ± 0.5) × 10−12 C, theory 2020[34] CuH2− CuH2CO2− (2.8 ± 0.4) × 10−13 C, theory 2017[31] Cu2H2− Cu2H2(CO2)1, 2−, CuH2CO2− Cu 1.7 × 10−12 (298 K) C, theory 2018[53] Cu2H3− Cu2H3CO2− (1.3 ± 0.1) × 10−13 C, theory 2017[31] PtHn− H2Pt(HCO2)− B, G, theory n = 0~3, 5 2016[32] PdCuH4− PdCuH2− HCOOH B, G, theory 2020[57] TaCH2+ [TaCH2O]+ CO 6.9 × 10−10 D 1995[96] [TaCH2O]+ TaO2+ C2H2O 1.3 × 10−10 D 1995[96] ScS+ ScO+, Sc+ COS, S+CO2 F, D, theory 2000[107] TiS+ TiO+, TiOS+, Ti+ COS, CO, S + CO, S + CO2 F, D, theory 2000[107] VS+ VO+, VOS+, V+ COS, CO, S + CO, S + CO2 F, D, theory 1998[108] YS+ YO+, YOS+, Y+ COS, CO, S + CO2 F, D, theory 2006[109] ZrS+ ZrO+, ZrOS+, Zr+ COS, CO, S + CO2 F, D, theory 2006[109] NbS+ NbOS+, NbO+, Nb+ CO, CO + S, S + CO2 F, D, theory 2006[109] [ClMg]− [ClMgCO2]− B, theory 2018[110] [ClMgCO2]− [ClMgC2O4]−, [ClMgCO3]− CO B, theory 2018[110] ScNH+ ScO+ HNCO (8.5 ± 1.7) × 10−11 C, theory 2019[18] [FeCN]+ [NCCO2Fe]+ B, theory 2018[19] [CoCN]+ [NCCO2Co]+ B, theory 2018[19] [ClZn]− [ClZnCO2]− B, theory 2018[110] [ClZnCO2]− [ClZnC2O4]−, [ClZn]− B, theory 2018[110] [YC6D5]+ [YC7D5O2]+, [YC6D5O]+ CO B, theory 2016[111] [Re(CO)2]+ [Re(CO)2O]+ CO 3.9 × 10−11 D, theory 2017[112] NUOCl2− UO2(NCO)Cl2− C, theory 2016[113] CuB+ Cu+, CuOB+, CuCO+ CBO2, CO, BO (1.2 ± 0.2) × 10−9 C, theory 2018[63] Nb2BN2− Nb2BN2O− CO (5.3 ± 1.1) × 10−10 C, theory 2020[20] Nb2BN2O− Nb2BN2O2− CO (2.7 ± 0.5) × 10−10 C, theory 2020[20] Nb2BN2O2− Nb2BN2O3− CO (8.9 ± 1.8) × 10−11 C, theory 2020[20] Nb2BN2O3− Nb2BN2O4− CO (1.3 ± 0.3) × 10−11 C, theory 2020[20] Nb2N2− Nb2N2O− CO (8.8 ± 1.8) × 10−11 C, theory 2020[20] Nb2N2O− Nb2N2O2− CO (4.0 ± 0.8) × 10−11 C, theory 2020[20] Nb2B− Nb2BO− CO (1.6 ± 0.3) × 10−11 C, theory 2020[20] Nb2BO− Nb2BO2− CO (8.0 ± 1.7) × 10−12 C, theory 2020[20] Nb2BO2− Nb2BO3− CO (6.0 ± 1.3) × 10−13 C, theory 2020[20] CuBCH3+ Cu+, CuOBH+, CuCO+ C2H3BO2, C2H2O, CH3BO (5.3 ± 1.1) × 10−11 C, theory 2018[63] RhVO3CH4− RhVO3CH4CO2− (295 K),
RhVO5CH− (600 K),
RhVO4CH2− (600 K),
RhVO4C− (600 K),
RhVO4CH4− (600 K)CH3•, CH2O, CH3OH, CO C, theory 2019[62] Mg(CO2)n+ E, theory 2003[114] Al(CO2)n+ E, theory n = 1~11 2003[115] Si(CO2)n+ E, theory 2004[116] Ti(CO2)n+ E, theory n = 3~7 2013[117] V(CO2)n+ E, theory 2013[118] V(CO2)n+ E 2004[119] Fe(CO2)n+ E 2002[120] Fe(CO2)n+ E 2001[121] Co(CO2)n+ E, theory n = 2~6 2019[122] Co(CO2)n+ E, theory n = 2~15 2017[123] Ni(CO2)n+ E 2004[124] Ni(CO2)n+ E 2003[125] Cu(CO2)n+ E, theory n = 3~8 2017[126] Rh(CO2)n+ E, theory n = 2~15 2017[123] Ag(CO2)n+ E, theory n = 3~8 2017[126] Ir(CO2)n+ E, theory n = 2~15 2017[123] NiO2(CO2)n+ E 2003[125] YO(CO2)n+ E, theory n = 2~11 2018[66] NbO2(CO2)n+ E, theory n = 3~9 2020[127] NbO2(CO2)n+ E, theory n = 1~7 2019[128] TaO2(CO2)n+ E, theory n = 1~7 2019[128] Mn(CO2)n− E, theory n = 2~10 2017[129] Fe(CO2)n− E, theory n = 3~7 2017[130] Co(CO2)n− E, theory n = 3~11 2014[131] Ni(CO2)n− E, theory n = 2~8 2014[132] Cu(CO2)n− E, theory n = 2~9 2014[133] Ag(CO2)n− E, theory n = 2~11 2013[134] Au(CO2)n− E, theory n = 2~13 2012[135] Ni(CO2)− G, theory 2019[64] Cu(CO2)− G, theory 2015[65] Pd(CO2)− G, theory 2019[64] Ag(CO2)− G, theory 2015[65] Pt(CO2)− G, theory 2019[64] Au(CO2)− G, theory 2015[65] TiOx(CO2)y− E, theory x = 1~3; y > 1 2018[136] [ClMgCO2]− E, theory 2014[137] [Co(Pyridine)(CO2)]− G, theory 2015[138] Sc OScCO, OCScCO3 H, theory 2016[69] Ti OTiCO+, OTiOC+ OTiCO, O2Ti(CO)2,
O2Ti(CO), OTi(CO)2H, theory 1999[80] V OVCO+, OVOC+ OVCO,
O2V(CO)2, OV(CO)2H, theory 1999[80] Cr OCrCO−,
Cr(CO2)+, Cr(CO2)2+OCrCO, O2Cr(CO)2,
Cr(CO2), Cr(CO2)2,
CrOCrCO, OCCrCO3H, theory 2014[70] Cr OCrCO,
O2Cr(CO)2, O2CrCO,
CrO, CrCO,
CrO2, OCr(CO)2, CrCO2H, theory 1997[81] Mn OMnCO−, OMnCO+ OMnCO, O2MnCO, O2Mn(CO)2,
Mn(O2)CO, MnO, MnO2H, theory 1999[79] Fe OFeCO−, FeOCO+ OFeCO, O2FeCO,
Fe(O2)CO, FeO2H, theory 1999[79] Co OCoCO−, CoCO2−, OCoCO+ OCoCO, O2CoCO,
OCo2CO, CoOH, theory 2007[73] Co OCoCO−, CoCO2− OCoCO, CoO H, theory 1999[79] Ni ONiCO−, NiCO2− ONiCO H, theory 1999[79] Cu OCuCO−, CuCO2− H, theory 1999[79] Zr (ZrO) +CO OZrCO, ZrO H, theory 2000[76] Mo OMoCO, O2Mo(CO)2,
O2MoCO, MoCOH, theory 1997[81] Ru ORuCO− ORuCO, O2RuCO,
OCRu(O2)CO, RuCOH, theory 2002[74] Rh ORhCO− ORhCO, O2RhCO, RhO H, theory 2007[73] Ta OTaCO−, O2Ta(CO)2− OTaCO, O2Ta(CO)2 H, theory 2000[77] W OWCO, O2W(CO)2,
O2WCO, WO,
WCO, OW(CO)2WOH, theory 1997[81] Re OReCO−, ORe(CO)2− OReCO, O2ReCO,
ORe(CO)2, O2Re(CO)2H, theory 2002[[75] Os OOsCO− OOsCO, O2OsCO, O2Os(CO)2 H, theory 2002[74] Th OThCO+, O2Th(CO)2− OThCO, O2Th(CO)2, ThO H, theory 2000[78] U OUCO+, O2U(CO)2− OUCO, O2U(CO)2, O2UCO
UO, UO2H, theory 2000[78] ScO ScCO3 H, theory 2016[69] TiO TiO2(CO) H, theory 2012[71] NbO NbO2(CO) H, theory 2011[72] TiO2 OTiCO3 H, theory 2012[71] NbO2 NbO2(CO2)1, 2 H, theory 2011[72] a k1: in cm3 molecule−1 s−1.
b The experimental methods are labelled as A−H: A: fast flow reactor-mass spectrometry (MS), B: collision cell-MS, C: linear ion trap reactor-MS, D: ion cyclotron resonance cell-MS, E: IR-PD spectroscopy, F: guided ion beam-MS, G: photoelectron spectroscopy, and H: matrix isolation infrared spectroscop 下载: 导出CSV
下载: 导出CSV
-

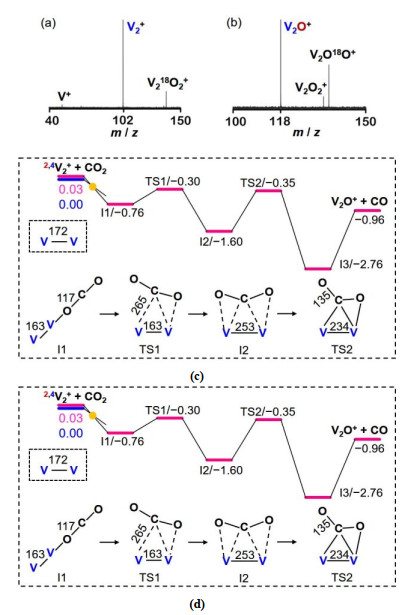
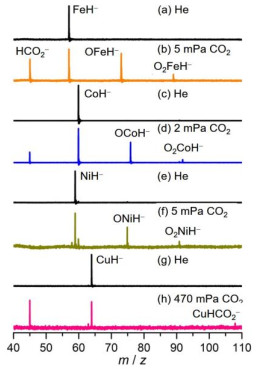
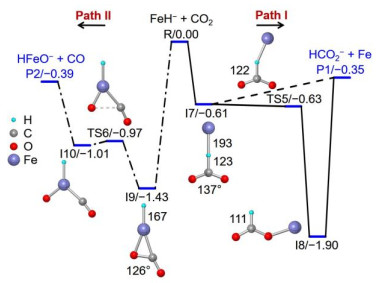
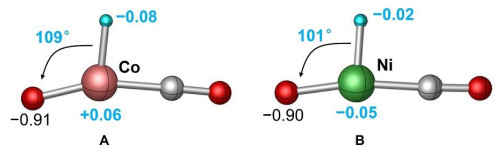
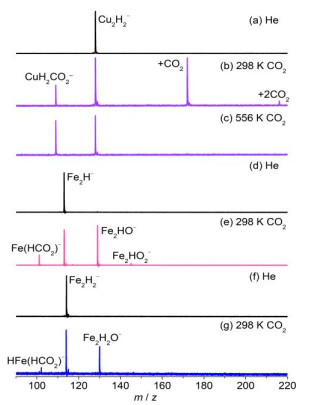
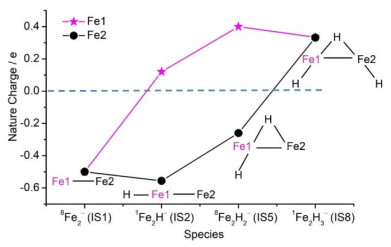
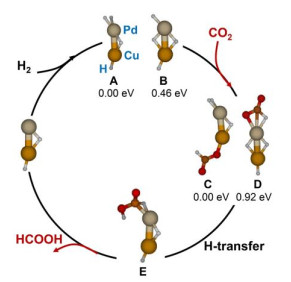
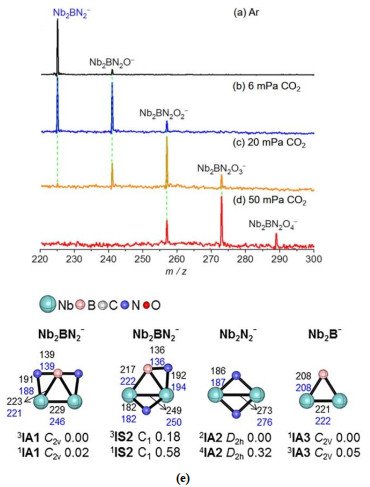
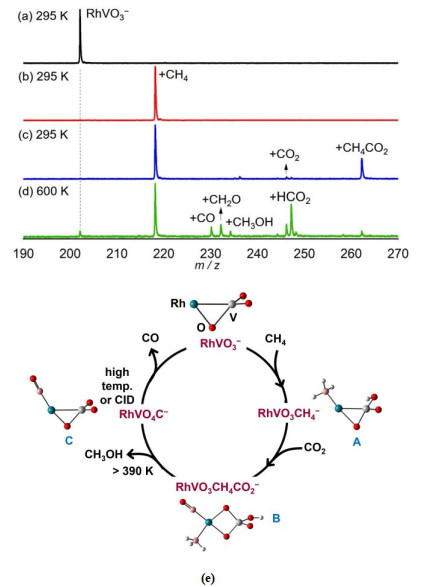









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 618
- HTML全文浏览量: 6
 下载:
下载:
