 图 图式 1
冬凌草甲素卟啉壳聚糖微球合成路线
Figure 图式 1.
Synthetic route of Ori-BHP-CS
图 图式 1
冬凌草甲素卟啉壳聚糖微球合成路线
Figure 图式 1.
Synthetic route of Ori-BHP-CS
引用本文:
何洁, 曹泳, 袁强. 冬凌草甲素卟啉壳聚糖微球的制备及体外光动力抗肿瘤活性研究[J]. 有机化学,
2017, 37(3): 759-766.
doi:
10.6023/cjocx201607022

Citation: He Jie, Cao Yong, Yuan Qiang. Preparation and Photocytotoxicity in vitro of Oridonin-porphyrinchitosan Microspheres[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 759-766. doi: 10.6023/cjocx201607022
Citation: He Jie, Cao Yong, Yuan Qiang. Preparation and Photocytotoxicity in vitro of Oridonin-porphyrinchitosan Microspheres[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 759-766. doi: 10.6023/cjocx201607022
冬凌草甲素卟啉壳聚糖微球的制备及体外光动力抗肿瘤活性研究
摘要:
基于光-化联合抗肿瘤的目的,通过共价偶联将5-对(6-溴已氨基苯基)-10,15,20-三苯基卟啉(BHP)引入壳聚糖侧基制备复合物,卟啉枝接率平均达30.80%;采用乳化交联法制备了冬凌草甲素卟啉壳聚糖微球,经高效液相色谱(HPLC)测得微球载药量为12.41%,包封率为8.72%,48 h体外释放量达81.74%;采用噻唑蓝(MTT)法考察合成的系列化合物对人乳腺癌细胞MCF-7的光毒性.实验结果表明:浓度为25,50和100 μmol/mL的冬凌草甲素光敏微球光照30 min后对MCF-7细胞的光动力杀伤率的平均值分别达(31.55±1.70)%,(71.03±0.76)%和(82.74±0.38)%,光动力杀伤效果显著.
English
Preparation and Photocytotoxicity in vitro of Oridonin-porphyrinchitosan Microspheres
Abstract:
A emulsion crosslinking method to prepare oridonin-porphyrin-chitosan microspheres with oridonin and 5-p-(6-bromohexylaminophenyl)-10, 15, 20-triphenylporphyrin-chitosan (BHP-CS) is reported, which in turn was synthesized from 5-p-(6-bromo-hexylaminophenyl)-10, 15, 20-triphenylporphyrin (BHP) and chitosan. The porphyrin grafting rate of BHP-CS reached 30.80% on average, the drug loading rate and entrapment rate of the prepared microspheres reached 12.41% and 8.72% measured by HPLC, and the accumulated release rate was 81.74% in 48 h in vitro. The thiazolyl blue tetrazolium bromide (MTT) method was used to evaluate the photocytotoxicities of these derivatives against MCF-7 cells. The results revealed that oridonin-porphyrin-chitosan microspheres showed high photocytotoxicity in concentrations of 25, 50 and 100 μmol/mL against MCF-7 cells, and the inhibit ratio of MCF-7 cells under light irradiation for 30 min were (31.55±1.70)%, (71.03±0.76)% and (82.74±0.38)%, respectively.
-
Key words:
- oridonin
- / porphyrin
- / microspheres
- / photocytotoxicity
- / antitumor
-
癌症是全球的主要疾病负担, 化学疗法是除手术外最重要的治疗手段, 但肿瘤化学药物却存在靶向性差、毒副作用大等问题[1, 2].中国拥有沿用了几千年的巨大中药宝库, 因此, 开发研制具有靶向性的中西医结合抗肿瘤新药具有极高的科学价值和现实意义.冬凌草是一种传统的中草药, 味甘苦, 性彻寒, 具有清热解毒、消炎止痛和健胃活血等作用, 近年来研究发现它有独特的抗肿瘤活性.冬凌草甲素 (Ori) 是从冬凌草中提取的贝壳杉烯二萜类天然有机物, 是其主要抗癌活性成分[3~7], 但其水溶性差, 生物利用度低, 严重影响临床应用.研究表明, 冬凌草甲素制备成微/纳米给药系统后, 在增加体循环时间、提高生物利用度、增强抗肿瘤活性等方面都有明显的作用[8~10].
光动力疗法 (PDT) 是利用光敏剂在光照下, 通过O2的参与, 造成肿瘤组织的定向损伤.它具有组织选择性好, 对正常组织损伤小, 抗癌谱广等优点, 是除手术、放疗、化疗之外的第四种成熟的癌症治疗方法[11, 12].卟啉类化合物因其独特的大π共轭平面构型对光具有很好的灵敏性[13], 是极好的光敏剂[14~17].本文采用具有光动力抗肿瘤活性的卟啉环作为侧基引入壳聚糖制成光敏新材料, 包埋冬凌草甲素制成微球.冬凌草甲素通过扩散作用从用戊二醛交联的壳聚糖微球的孔隙中释放出来, 达到缓释药物的目的; 微/纳米给药系统在水中良好的分散性, 可克服冬凌草甲素水溶性差、生物利用度低等问题.本文设计并合成具有缓释功能的中药药效物质, 将中药与光动力杀伤相结合, 光-化联合抗肿瘤, 达到高效低毒的目的.合成路线见Scheme 1.
图 图式 1
冬凌草甲素卟啉壳聚糖微球合成路线
Figure 图式 1.
Synthetic route of Ori-BHP-CS
1 结果与讨论
1.1 卟啉壳聚糖复合物的合成
以合成的卟啉5-对 (6-溴已氨基苯基)-10, 15, 20-三苯基卟啉 (BHP) 和壳聚糖为原料, 利用壳聚糖上的游离氨基与卟啉上的卤代烃部分共价偶连, 考虑到卟啉和壳聚糖分别处于有机相和无机相, 采用相转移催化剂以提高产率, 将合成的BHP、复合物 (BHP-CS)、微球 (Ori-BHP-CS) 进行紫外表征 (表 1, 图 2), 与底物冬凌草甲素 (Ori) 和壳聚糖 (CS) 比较, 卟啉Soret带和Q带典型峰的出现, 表明共价偶连的成功, 复合物、微球中引入了卟啉分子.
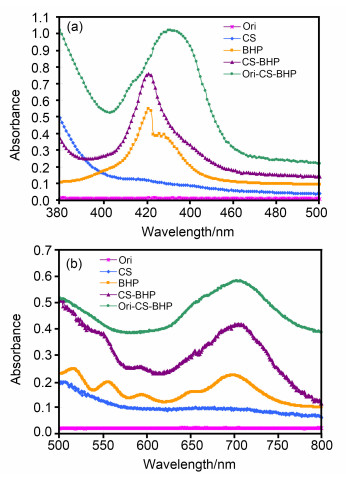 图 2
壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外谱图
Figure 2.
UV-Vis spectrogram of CS, BHP, BHP-CS and Ori-BHP-CS
图 2
壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外谱图
Figure 2.
UV-Vis spectrogram of CS, BHP, BHP-CS and Ori-BHP-CS
 表 1
冬凌草甲素、壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外数据a
Table 1.
UV-Vis spectral data of Ori, CS, BHP, BHP-CS and Ori-BHP-CS
表 1
冬凌草甲素、壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外数据a
Table 1.
UV-Vis spectral data of Ori, CS, BHP, BHP-CS and Ori-BHP-CS
Compd. Soret band/nm Q band/nm Ori — — CS — — BHP 421.0 517.0, 556.0, 594.0, 698.0 BHP-CS 421.0, 465.0, 472.0 705.0 Ori-BHP-CS 430.0, 480.0, 494.0 703.0, 792.0 a V(CH3COOH):V(CHCl3)=1:2, 25 ℃. 表 1 冬凌草甲素、壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外数据a
Table 1. UV-Vis spectral data of Ori, CS, BHP, BHP-CS and Ori-BHP-CSOri由于结构中仅有一个羰基和烯键共扼, 缺少大的生色团, 羟基由于位置的原因, 助色团效应体现也不明显, 故在可见区没有明显的吸收.然而卟啉就不同了, BHP在421 nm的强峰称为卟啉的Soret带, 在517.0, 556.0, 594.0, 698.0 nm的四个小峰称为Q带, 两者均由π-π*跃迁产生, 是表征卟啉环的最有力证明.由图可见, 在380~500 nm区域, BHP-CS、Ori-BHP-CS均出现了卟啉的Soret带, 并且相对于BHP有不同程度的谱带变宽和红移现象, 三者的峰宽依次增大, 微球尤其突出, 它的Soret带比BHP红移了9 nm.而在380~400 nm的区域, BHP-CS、Ori-BHP-CS也同时体现了壳聚糖在此区域的吸收特征, 证明了壳聚糖与卟啉的成功链接. BHP在Q带的四个特征峰, 在形成复合物和微球后依然存在, 其中复合物较明显, 而微球体现相对较弱.但698 nm峰表现强势, 在复合物和微球中分别有7和5 nm的红移, 且强度增大, 峰宽变宽, 这恰恰是我们的兴趣所在, 我们所致力于的光敏剂要能有效地吸收长波长的光, 以致得到尽可能深入的光动力治疗效果.因为人体组织的最佳透射波段是在620~900 nm, 在光动力治疗肿瘤疾病中, 如果光敏剂的吸收波长越长, 穿透则越深, 治疗深度也就越深.我们制备的微球在703 nm有强吸收, 可以推知其具备作为光动力治疗光敏剂的潜质.
采用共价偶联法将BHP与壳聚糖进行枝接时, 为了增加有机功能材料的光敏性, 需要提高卟啉的枝接率.参照常规的卟啉合成法, 以氯仿为溶剂, 首先控制温度40~70 ℃, 加入四丁基溴化胺为相转移催化剂 (Entries 1~4), 发现温度到70 ℃后, 枝接率反而下降, 在反应体系中发现焦油状物体, 分析可能是卟啉环被破坏, 故温度不能太高.第二考察无机碱 (Entries 5, 6), 结果表明经干燥处理的K2CO3明显优于NaOH.第三方面考察溶剂, 由于壳聚糖和无机碱在氯仿溶液中不溶, 故在溶剂中加入适量水, 考察对卟啉枝接率是否有贡献 (Entries 7~10), 实验结果发现V(H2O):V(CHCl3)=1/3时枝接率比单纯以氯仿作溶剂高.第四考察相转移催化剂与壳聚糖中游离氨基的当量比 (Entries 11~14), 考虑到不可能所有壳聚糖游离氨基都参与反应, 故催化剂当量无需太高, 实验发现催化剂与游离氨基比例为1:1时, 枝接率最高.最后考察反应时间, 数据表明反应5.5 h卟啉枝接率最高.故最后优化的最佳实验条件为:反应温度60 ℃, 反应时间5.5 h, 混合溶剂比1:3, 相转移催化剂与壳聚糖游离氨基摩尔比1:1.根据最优条件合成的复合物平均卟啉枝接率为30.80%.
1.2 冬凌草甲素光敏微球的制备
1.3 体外释放曲线测定
为了考察制备的冬凌草甲素光敏微球的缓控释药性能, 我们对其进行了体外释放性能研究, 以高效液相色谱 (HPLC) 测定药物浓度, 结果如图 5所示.
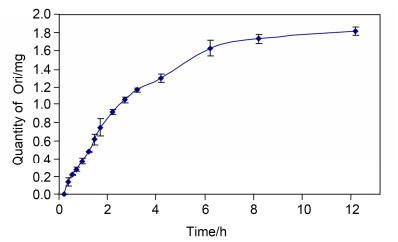 图 5
冬凌草甲素卟啉壳聚糖微球的体外释放曲线
Figure 5.
In vitro release curve of oridonin-porphyrin-chitosan microspheres
图 5
冬凌草甲素卟啉壳聚糖微球的体外释放曲线
Figure 5.
In vitro release curve of oridonin-porphyrin-chitosan microspheres
由图 5可见, 在释放的前2 h内, 冬凌草甲素释放较快, 累计释放达41.59%; 2~8 h内释放放缓, 截至8 h累计释放76.31%; 8 h以后释放平缓, 在释药12 h后累计Ori质量达到80.04%;之后随着时间增加Ori质量基本保持不变, 可以认为壳聚糖载药微球已最大程度释放出所载药物.根据投药量与载体质量比为2.2:1时包封率和载药量情况计算, 其体外释放48 h累计释放平均达81.74%.上述数据说明, 冬凌草甲素光敏微球体外释药具有一定的缓释制剂特征, 24 h内均能持续释药, 且有效避免了药物突释现象.但光敏微球前4 h释药过快的现象还是说明微球的稳定性不够, 制备工艺有待改进, 项目组正在考虑在微球表面包裹一层脂质材料, 以提高制剂稳定性、减缓药物释放.
1.4 体外光动力抗肿瘤活性研究
以人乳腺癌细胞MCF-7为肿瘤细胞模型, 考察制备的冬凌草甲素光敏微球的体外抗肿瘤活性.将实验分组为冬凌草甲素 (1)、空白壳聚糖微球 (2)、冬凌草甲素壳聚糖微球 (3)、空白卟啉壳聚糖微球 (4)、冬凌草甲素卟啉壳聚糖微球 (5), 将上述1~5号化合物分别作为光动力抗肿瘤药物 (normal组指不加药的正常组), 分别设置了25, 50, 100 μmol/mL等三个浓度, 同时设置避光组和光照组进行对比, 冬凌草甲素卟啉壳聚糖微球对人乳腺癌细胞MCF-7的光动力杀伤力数据见表 3, 化合物1~5的肿瘤细胞存活率情况见图 6.
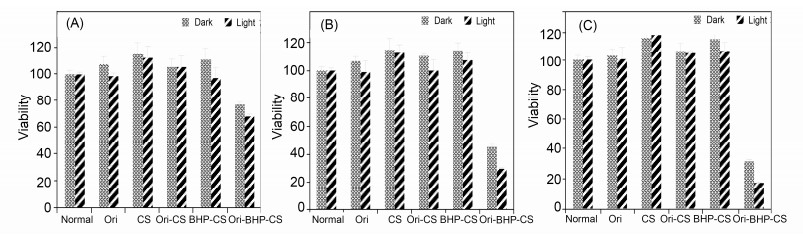 图 6
MCF-7细胞在不同浓度药物光动力作用下的存活率情况
Figure 6.
Viability of MCF-7 cells under different concentrations of drugs in photodynamic therapy
表 3
冬凌草甲素卟啉壳聚糖微球对MCF-7细胞的体外光动力杀伤力数据
Table 3.
In vitro photocytotoxicity datas of oridonin-porphyrin-chitosan microspheres against MCF-7 cells
图 6
MCF-7细胞在不同浓度药物光动力作用下的存活率情况
Figure 6.
Viability of MCF-7 cells under different concentrations of drugs in photodynamic therapy
表 3
冬凌草甲素卟啉壳聚糖微球对MCF-7细胞的体外光动力杀伤力数据
Table 3.
In vitro photocytotoxicity datas of oridonin-porphyrin-chitosan microspheres against MCF-7 cells
Cytotoxity ratio [(%)±SD] of MCF-7 cells 100 μmol/L 50 μmol/L 25 μmol/L Light 82.74±0.38 71.03±0.76 31.55±1.70 Dark 68.50±1.16 55.49±1.85 22.59±1.31 表 3 冬凌草甲素卟啉壳聚糖微球对MCF-7细胞的体外光动力杀伤力数据
Table 3. In vitro photocytotoxicity datas of oridonin-porphyrin-chitosan microspheres against MCF-7 cells由图 6可知:化合物1~4的抗肿瘤活性均不理想.首先是化合物2, 作为壳聚糖空白微球, 既无冬凌草甲素也不含卟啉, 因此细胞存活率数据最高, 完全没有表现出抗肿瘤活性, 但这也从另一个侧面证明了该微球囊材没有细胞毒性, 是安全的; 其次是含冬凌草甲素的化合物1和3, 显示了较弱的抗肿瘤活性; 而含有卟啉的化合物4表现出了显著的光敏活性, 即光照组杀伤力明显高于避光组; 最可喜的是化合物5, 即冬凌草甲素光敏微球, 表现出了卓越的抗肿瘤活性. 表 3可见3个浓度的冬凌草甲素光敏微球光照组对人乳腺癌细胞MCF-7的光动力杀伤率的平均值分别达 (31.55±1.70)%、(71.03±0.76)%、(82.74±0.38)%, 药物的活性随着浓度的增高而显著提升, 并明显高于相应浓度的避光组, 这说明光敏微球延承了卟啉的光敏活性, 故发挥了光动力杀伤作用的光照组疗效优于避光组.同时, 光敏微球的光毒性优于Ori, 提示我们光敏微球的剂型改进改善了冬凌草甲素的溶解度问题, 并发挥了缓释持续给药的作用, 从而提高了疗效.最后, 化合物5的疗效既优于1也高于4, 这说明冬凌草甲素光敏微球切实发挥了中药抗肿瘤和光动力杀伤的协同作用, 才使得活性增强显著.
1.2.1 扫描电镜表征
以冬凌草甲素为药物模型, 上述合成的卟啉壳聚糖复合物为载体, 戊二醛为交联剂, 乳化交联制备微球法实则是戊二醛的醛基与壳聚糖的氨基通过生成席夫碱而发生交联.严格控制水相/油相比例、搅拌速度, 优化后的制备工艺制得的冬凌草甲素光敏微球扫描电镜图如图 3所示, 由图可见, 制得的微球球形完整、表面光滑、结团率低, 经测定粒径分布为2~8 μm.
 图 3
冬凌草甲素卟啉壳聚糖微球的扫描电镜图
Figure 3.
The SEM of oridonin-porphyrin-chitosan microspheres
图 3
冬凌草甲素卟啉壳聚糖微球的扫描电镜图
Figure 3.
The SEM of oridonin-porphyrin-chitosan microspheres
1.2.2 载药量及包封率的测定
为了提高微球的包封率和载药量, 在上述优化的微球制备条件的基础上, 进一步考察药物与载体投料比例对包封率和载药量的影响.按投药量与载体 (载体为33.0 mg) 的质量比为0.1:1, 0.2:1, 0.3:1, 0.4:1, 0.5:1, 0.6:1, 1.5:1, 2:1, 2.2:1, 2.5:1, 3:1, 3.5:1制得的冬凌草甲素壳聚糖光敏微球的载药量和包封率情况如图 4.
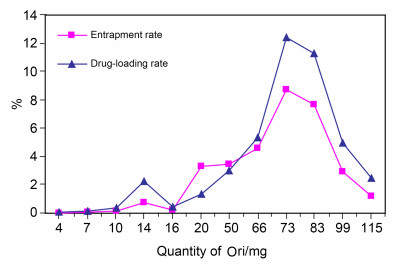 图 4
冬凌草甲素投药量对其微球的包封率及载药量的影响
Figure 4.
Effect of dosage of Ori on the entrapment rate and drug loading rate
表 2
BHP-CS复合物合成条件优化
Table 2.
Optimization of the reaction conditions for the formation of BHP-CS
图 4
冬凌草甲素投药量对其微球的包封率及载药量的影响
Figure 4.
Effect of dosage of Ori on the entrapment rate and drug loading rate
表 2
BHP-CS复合物合成条件优化
Table 2.
Optimization of the reaction conditions for the formation of BHP-CS
Entry Time/h Temp./℃ V(H2O):V(CHCl3) Base Cat./mol% Absorbance Grafting ratio/% 1 4.0 40 0:1 K2CO3 0.1 0.858 16.25 2 4.0 50 0:1 K2CO3 0.1 0.749 24.76 3 4.0 60 0:1 K2CO3 0.1 0.701 28.51 4 4.0 70 0:1 K2CO3 0.1 0.759 23.98 5 5.5 60 0:1 K2CO3 0.1 0.603 36.16 6 5.5 60 0:1 NaOH 0.1 0.830 18.44 7 5.5 60 0:1 K2CO3 1.0 0.723 26.79 8 5.5 60 1:2 K2CO3 1.0 0.717 27.26 9 5.5 60 1:3 K2CO3 1.0 0.663 31.48 10 5.5 60 1:4 K2CO3 1.0 0.694 29.06 11 5.5 60 1:3 K2CO3 0.1 0.663 31.48 12 5.5 60 1:3 K2CO3 0.5 0.633 33.82 13 5.5 60 1:3 K2CO3 1.0 0.593 36.94 14 5.5 60 1:3 K2CO3 3.0 0.610 35.61 15 5.0 60 1:3 K2CO3 1.0 0.684 29.84 16 5.5 60 1:3 K2CO3 1.0 0.643 33.04 17 6.0 60 1:3 K2CO3 1.0 0.669 31.01 18 6.5 60 1:3 K2CO3 1.0 0.661 31.63 表 2 BHP-CS复合物合成条件优化
Table 2. Optimization of the reaction conditions for the formation of BHP-CS由图 4可知, 包封率和载药量的总趋势随着冬凌草甲素的投药量增加而增大, 当投药量与载体的质量比为2.2:1时达到最高, 但当比例为2.5:1后开始下降.分析其原因有三: (1) 一般情况下, 药物 (即冬凌草甲素) 的投入量越多, 微球在形成过程中能纳入的药物也就越多, 包封率和载药量就越大; 但微球可容纳药物的内部空间有限, 故微球包埋的药物量应该有一个极值, 超过此极值, 过多的药物无法包埋入球. (2) 剩余药物可吸附在微球表面, 但由于吸附力有限, 在微球制备的多次离心、洗涤中被除去, 故不可被计算在内.而当投药量过多时, 溶液中、微球表面均可见大量药物结晶, 这些结晶由于颗粒太大, 反而影响了微球对其的包埋, 故使得包封率和载药量数值出现下降. (3) 在包封率的计算方法中, 分母为投入的总药量, 因此投药量过多, 分母变大, 而入球药物量达到极值, 故包封率数据的下降明显快于载药量.经测定, 冬凌草甲素投药量与载体的质量比为2.2:1时, 载药量为12.41%, 包封率为8.72%, 达到最高值.
2 结论
本文基于光-化联合抗肿瘤的考虑, 将卟啉环作为侧基引入壳聚糖制成光敏材料, 卟啉平均接枝率达到30.80%, 用其包埋冬凌草甲素制得微球, 赋予其光敏特性.制得的微球产率达90%以上, 载药量为12.41%, 包封率为8.72%.体外光动力抗肿瘤实验发现:在采用8 W光源、含药培养4 h、光照30 min的情况下, 浓度为25, 50, 100 μmol/mL的冬凌草甲素光敏微球对人乳腺癌细胞MCF-7的光动力杀伤率的平均值分别达 (31.55±1.70)%, (71.03±0.76)%, (82.74±0.38)%, 光照组活性明显高于避光组, 表现出了显著的光动力杀伤力.光敏微球的缓释新剂型, 可为进一步开发通过实体瘤的高通透性和滞留效应 (EPR) 实现被动靶向的中西医结合抗肿瘤光敏纳米药物的研制提供一种新方法.
3 实验部分
3.1 仪器与试剂
UV-Vis由UT-1900型双光束紫外可见分光光度计 (北京普析通用仪器有限责任公司) 测定; FTIR由Spectrum BX型傅立叶变换红外光谱仪 (珀金埃尔默仪器有限公司) 测定; 1H NMR由Mercury-500型核磁共振仪 (美国Varian公司) 测定; ESI-MS由LCQ Advantage质谱仪 (美国Thermofisher公司) 测定; ESI-HRMS由6210 LC/TOF质谱仪 (美国Agilent公司) 测定; 药物浓度由UltiMate-3000(美国戴安公司) 高效液相色谱仪测定; 光动力实验光源Philips Tornado 8 W.
壳聚糖购自阿拉丁试剂有限公司 (脱乙酰度≥95%, 粘度100~200 mPa•s); MCF-7购自中国医学科学院肿瘤医院肿瘤研究所; DMEM (Dulbecco’s modified Eagle’s medium), 胎牛血清 (FBS) 购自Gibico公司; 噻唑蓝 (MTT) 购自Sigma公司; 其余所用试剂均为分析纯.
3.2 卟啉衍生物的合成
3.3 合成卟啉壳聚糖复合物
将50 mg壳聚糖加入三颈烧瓶中, 加入150 mg K2CO3, 滴入2.0 mL氯仿40 ℃密封搅拌0.5 h; 加入20 mg四丁基溴化铵, 再将50 mg BHP溶解在15 mg氯仿中, 逐滴加入烧瓶中, 60 ℃回流6 h.反应停止, 冷却至室温.抽滤, 用氯仿洗至滤液无色, 再用水洗3次, 产物真空干燥.采用茚三酮法测定复合物的卟啉枝接率.
3.4 制备冬凌草甲素光敏微球
3.5 体外释放度实验
取2.00 mL超纯水于透析袋中, 加入20.0 mg冬凌草甲素光敏微球, 再将透析袋置于离心管中, 加入适量超纯水将透析袋浸没, 设置温度, 于10, 20, 30, 45 min和1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48 h精密吸取1.0 mL液体用过滤器过滤到液相进样瓶中, 同时补加1.0 mL超纯水, 由HPLC测其含量, 计算药物累计释放量.
3.6 体外光动力抗肿瘤活性实验
实验分组:冬凌草甲素 (1)、壳聚糖空白微球 (2)、冬凌草甲素壳聚糖微球 (3)、空白壳聚糖光敏微球 (4)、冬凌草甲素光敏微球 (5).
取对数生长期的细胞, 以每孔5000的密度种于96孔板中, 培养24 h.种两板, 一板为光照组, 一板为不光照组.弃去培养液, 加入含药的培养液.每板均设置正常组和加药组.化合物1~5的浓度为25, 50, 100 μmol/mL.避光培养4 h.将光照组的板光照0.5 h, 板与灯泡之间的距离为10 cm.不光照组则继续避光培养.光照组和不光照组均弃掉培养液, PBS洗一遍, 加入不含药的新鲜培养液, 培养24 h.每孔吸出培养液后, 加入培养液-MTT混合溶液 (培养液: MTT溶液5 mg/mL V:V=9:1) 100 μL, 孵育4 h.吸弃培养液, 每孔加入100 μL的DMSO, 振荡10 min, 580 nm酶标仪测定.实验重复3次.
辅助材料 (Supporting Information)化合物TPP, NTPP, ATPP, BHP的核磁、质谱及高分辨质谱的谱图, 化合物Ori, BHP-CS, Ori-BHP-CS的HPLC谱图.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.2.2 5-对硝基苯基-10, 15, 20-三苯基卟啉 (NTPP) 的合成
将200 mg四苯基卟啉、30 mL二氯甲烷搅拌溶解后分四次加入65%硝酸, 反应时间40 min, 薄层色谱 (TCL) 检测反应完全, 加入氨水中和, 终止反应.分液后, 下层有机相用水洗三次, 无水Na2SO4固体干燥.减压蒸馏得到的固体经真空干燥得到粗产物.用硅胶G柱色谱分离, 以V(石油醚):V(氯仿)=1:1的混合液为展开剂, 收集第二条色带得到紫色固体NTPP 178 mg, 产率82.8%. m.p.>300 ℃; UV-Vis (CHCl3) λ: 420.0, 516.5, 553.0, 592, 646.0 nm; 1H NMR (CDCl3) δ: 8.71~8.88 (m, 6H, pyrrole β-CH), 8.60 (d, J=4.5 Hz, 2H, pyrrole β-CH), 8.59 (d, J=8.4 Hz, 2H, o-C6H5NO2), 8.37 (d, J=8.4 Hz, 2H, m-C6H5NO2), 8.19~8.21 (m, 6H, o-C6H5), 7.72~7.79 (m, 9H, m, p-C6H5), -2.77 (s, 2H, porphine ring NH); MS (ESI) m/z: 660.2 (M+H)+; HRMS (ESI) calcd for C44H29N5O2(M+H)+ 660.2394, found 660.2374.
3.2.1 四苯基卟啉 (TPP) 的合成
将0.84 g间硝基苯甲酸溶解于16 mL二甲苯溶液中加热回流, 在剧烈搅拌下, 滴加6 mL溶解了0.7 mL吡咯和1 mL苯甲醛的二甲苯溶液.反应持续2~3 h, 停止加热, 冷却至120 ℃时, 加入25 mL甲醇.冷却至室温后, 将反应器转移到冰箱中放置过夜.混合液抽滤, 用甲醇反复洗涤滤饼, 直至洗出液基本无色为止, 收集固体, 真空干燥.得紫色固体TPP[18] 3.6 g, 产率60%. m.p.>300 ℃; UV-Vis (CHCl3) λ: 419.0, 515.5, 550.0, 590.0, 645.5 nm; 1H NMR (CDCl3) δ: 8.85 (s, 8H, pyrrole β-CH), 8.22 (d, J=6.8 Hz, 8H, o-C6H5), 7.72~7.79 (m, 12H, m, p-C6H5), -2.78 (s, 2H, porphine ring NH); MS (ESI) m/z: 615.3 (M+H)+; HRMS (ESI) calcd for C44H31N4 (M+H)+ 615.2543, found 615.2562.
3.2.4 5-对 (6-溴已氨基苯基)-10, 15, 20-三苯基卟啉 (BHP) 的合成
将150 mg ATPP、218 mg K2CO3、四丁基溴化铵10 mg混合, 搅拌, 加热至60 ℃, 分三次滴加二溴己烷, 每次0.4 mL, 反应每30 min加一次.反应120 min, 停止加热, 加入30 mL石油醚, 抽滤, 减压蒸馏得到粗产品.粗产品用CH2Cl2溶解, 上硅胶G色谱柱, 先用石油醚洗下残余的二溴己烷, 再用V(石油醚)︰V(CH2Cl2)= 1︰1为展开剂, 洗脱得紫色固体[19]BHP 90 mg, 产率60%. m.p.>300 ℃. UV-Vis (CHCl3)λ: 420.50, 517.50, 556.00, 591.50, 650.00 nm; 1H NMR (CDCl3) δ: 8.95 (d, J=4.0 Hz, 2H, pyrrole β-CH), 8.82~8.86 (m, 6H, pyr-role β-CH), 8.21 (d, J=5.5 Hz, 6H, o-C6H5), 8.00 (d, J=8.0 Hz, 2H, m-C6H5NH), 7.73~7.75 (m, 9H, m, p-C6H5), 6.94 (d, J=7.5 Hz, 2H, o-C6H5NH), 4.05 (t, J=6.5 Hz, 1H, NH), 3.45 (t, J=6.5 Hz, 2H, CH2Br), 3.33~3.40 (m, 2H, CH2NH), 1.94 (t, J=6.5 Hz, 2H, CH2CH2Br), 1.78~1.81 (m, 2H, CH2CH2NH), 1.44~1.57 (m, 4H, (CH2)2(CH2)2Br), -2.73 (s, 2H, porphine ring NH); IR (KBr) ν: 3416.11, 3319.43, 2927.31, 1609.20, 1519.65, 1469.49, 1350.56, 1183.27, 801.23, 731.56, 701.78 cm-1; MS (ESI) m/z: 792.2 (M+H)+; HRMS (ESI) calcd for C50H43BrN5 (M+H)+ 792.2696, found 792.2696.
3.2.3 5-对氨基苯基-10, 15, 20-三苯基卟啉 (ATPP) 的合成
将200 mg NTPP溶于浓盐酸中, 加入0.8 g氯化亚锡, 均匀搅拌并加热到65 ℃, 反应1 h.反应结束后, 将产物移入砂芯漏斗中, 抽去过量的浓盐酸.依次用浓氨水、蒸馏水、饱和NaCl溶液洗涤三次, 分出有机层, 无水Na2SO4干燥, 蒸干, 得紫色固体ATPP 185 mg, 产率97%. m.p.>300 ℃; UV-vis (CHCl3)λ: 421.0, 517.5, 554.0, 591.5, 648.0 nm; 1H NMR (CDCl3) δ: 8.93 (d, J=4.5 Hz, 2H, pyrrole β-CH), 8.83~8.84 (m, 6H, pyrrole β-CH), 8.21 (d, J=6.5 Hz, 6H, o-C6H5), 7.97 (d, J=8.2 Hz, 2H, m-C6H5NH2), 7.71~7.78 (m, 9H, m, p-C6H5), 7.00 (d, J=8.2 Hz, 2H, o-C6H5NH2), 3.93 (s, 2H), -2.75 (s, 2H, porphine ring NH); MS (ESI) m/z: 630.3 (M+H)+; HRMS (ESI) calcd for C44H32N5 (M+H)+ 630.2652, found 630.2657.
3.4.1 制备方法
精密称取33.0 mg卟啉壳聚糖复合物于50 mL离心管中, 加入1.1 mL 5%的醋酸溶液中, 超声10 min.加入0.2 mL丙酮以及适量冬凌草甲素, 超声10 min, 备用.塑料烧杯中加入50.0 mL液体石蜡, 1000 r/min, 缓慢加入1.0 mL Span-80, 搅拌20 min, 将离心管中液体 加入上述油相中, 搅拌乳化30 min后, 调转速450 r/min, 加入0.3 mL戊二醛和1.0 mL 1, 4-二氧六环的混合溶液, 搅拌90 min.溶液离心2~3次, 转速2500 r/min, 5 min, 抽滤, 用3%亚硫酸氢钠溶液洗1次, 离心, 抽滤, 再依次用3%亚硫酸氢钠溶液洗1次, 水洗2次、异丙醇洗3次, 产物真空干燥.
3.4.2 微球的粒径及形态
用扫描电镜观察微球表面形态, 计算粒径.
3.4.3 载药量及包封率的测定
精密称取10.0 mg冬凌草甲素卟啉壳聚糖微球于25 mL圆底烧瓶, 加入10.0 mL 3%盐酸, 60 ℃加热搅拌反应4 h, 反应结束后将反应液全部转移到25 mL容量瓶内, 用0.1000 g/mL NaOH溶液调节pH至4左右, 用超纯水定容, 经0.45 μm微孔滤膜过滤, 滤液收集到50 mL容量瓶中, 用甲醇定容, 备用.
取10 μL供试品溶液进样.用回归方程求出浓度, 按下列公式计算药物载药量和包封率:
包封率=(微球中所含药物的重量/投入的总药量)×100%
载药量=(微球中所含药物的重量/微球的总重量)×100%
-
-
[1]
Pinzani, V.; Bressolle, F.; Haug, I. J.; Galtier, M.; Blayac, J. P.; Balmes, P. Cancer Chemother. Pharmacol. 1994, 35, 1. doi: 10.1007/BF00686277
-
[2]
Gottesman, M. M. Annu. Rev. Med. 2002, 53, 615. doi: 10.1146/annurev.med.53.082901.103929
-
[3]
邓志成, 陈胜, 刘双海, 中国肿瘤外科杂志, 2013, 5, 50.Deng, Z. C.; Chen, S.; Liu, S. H. Chin. J. Surg. Oncol. 2013, 5, 50 (in Chinese).
-
[4]
Liu, Y.; Liu, J. H.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. J. Pharm. Pharmacol. 2013, 65, 1622. doi: 10.1111/jphp.2013.65.issue-11
-
[5]
Jin, S.; Shen, J.; Wang, J.; Huang, G.; Zhou, J. G. Cancer Biol. Ther. 2007, 6, 261 doi: 10.4161/cbt.6.2.3621
-
[6]
Cheng, Y.; Qiu, F.; Ye, Y. C.; Guo, Z. M.; Tashiro, S.; Onodera, S.; Ikejima, T. FEBS J. 2009, 276, 1291. doi: 10.1111/j.1742-4658.2008.06864.x
-
[7]
Cui, Q.; Tashiro, S.; Onodera, S.; Minami, M.; Ikejima, T. Biol. Pharm. Bull. 2007, 30, 859. doi: 10.1248/bpb.30.859
-
[8]
Feng, N.; Wu, P.; Li, Q.; Mei, Y.; Shi, S.; Yu, J.; Xu, J.; Liu, Y.; Wang, Y. J. Drug Targeting 2008, 16, 479. doi: 10.1080/10611860802102282
-
[9]
Duan, C.; Gao, J.; Zhang, D.; Jia, L.; Liu, Y.; Zheng, D.; Liu, G.; Tian, X.; Wang, F.; Zhang, Q. Biomacromolecules 2011, 12, 4335. doi: 10.1021/bm201270m
-
[10]
Liu, Y.; Zhang, P.; Feng, N.; Zhang, X.; Wu, S.; Zhao, J. H. Int. J. Pharm. 2009, 365, 136. doi: 10.1016/j.ijpharm.2008.08.009
-
[11]
Antoni, P. M.; Naik, A.; Albert, I.; Rubbiani, R.; Gupta, S.; Ruiz-Sanchez, P.; Munikorn, P.; Mateos, J. M.; Luginbuehl, V.; Thamyongkit, P.; Ziegler, U.; Gasser, G.; Jeschke, G.; Spingler, B. Chem. Eur. J. 2015, 21, 1179. doi: 10.1002/chem.201405470
-
[12]
Yano, S.; Hirohara, S.; Obata, M.; Hagiya, Y.; Ogura, S.; Ikeda, A.; Kataoka, H.; Tanaka, M.; Joh, T. J. Photochem. Photobiol. C 2011, 12, 46. doi: 10.1016/j.jphotochemrev.2011.06.001
-
[13]
顾承志, 孟书献, 冯亚青, 有机化学, 2015, 35, 1229. doi: 10.6023/cjoc201412018Gu, C. Z.; Meng, S. X.; Feng, Y. Q. Chin. J. Org. Chem. 2015, 35, 1229 (in Chinese). doi: 10.6023/cjoc201412018
-
[14]
Joana, F. B. B.; Alicia, Z.; Graça, P. M. S. N.; Amparo, F. F.; Augusto, C. T.; José, A. S. C.; Beate, R.; Ángeles, J.; Francisco, S. R. Eur. J. Med. Chem. 2015, 92, 135 doi: 10.1016/j.ejmech.2014.12.025
-
[15]
Zhao, Z.; Wen, J. Y.; Lv, B. B.; Li, X.; Ying, X.; Wang, Y. J.; Zhang, H. T.; Wang, H.; Liu, H. Y.; Chang, C. K. Appl. Organomet. Chem. 2016, 30, 132. doi: 10.1002/aoc.v30.3
-
[16]
Naik, A.; Rubbiani, R.; Gasser, G.; Spingler, B. Angew. Chem., Int. Ed. 2014, 53, 6938. doi: 10.1002/anie.201400533
-
[17]
Gianferrara, T.; Spagnul, C.; Alberto, R.; Gasser, G.; Ferrari, S.; Pierroz V.; Bergamo, A.; Alessio, E. Chem. Med. Chem. 2014, 9, 1231. doi: 10.1002/cmdc.201300501
-
[18]
何洁, 罗伟丰, 宋雪妍, 毛丽园, 化学通报, 2013, 76, 179.He, J.; Luo, W. F.; Song, X. Y.; Mao, L. Y. Chemistry 2013, 76, 179 (in Chinese).
-
[19]
何洁, 刘文洪, 何利民, 陈丽园, 高等学校化学学报, 2013, 34, 2308.He, J.; Liu, W. H.; He, L. M.; Chen, L. Y. Chem. J. Chin. Univ. 2013, 34, 2308 (in Chinese).
-
[1]
-

图 2 壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外谱图
Figure 2 UV-Vis spectrogram of CS, BHP, BHP-CS and Ori-BHP-CS

图 4 冬凌草甲素投药量对其微球的包封率及载药量的影响
Figure 4 Effect of dosage of Ori on the entrapment rate and drug loading rate

图 5 冬凌草甲素卟啉壳聚糖微球的体外释放曲线
Figure 5 In vitro release curve of oridonin-porphyrin-chitosan microspheres

图 6 MCF-7细胞在不同浓度药物光动力作用下的存活率情况
Figure 6 Viability of MCF-7 cells under different concentrations of drugs in photodynamic therapy
(A) cdrug=25 μmol/mL; (B) cdrug=50 μmol/mL; (C) cdrug=100 μmol/mL
表 1 冬凌草甲素、壳聚糖、卟啉、复合物及冬凌草甲素卟啉壳聚糖微球的紫外数据a
Table 1. UV-Vis spectral data of Ori, CS, BHP, BHP-CS and Ori-BHP-CS
Compd. Soret band/nm Q band/nm Ori — — CS — — BHP 421.0 517.0, 556.0, 594.0, 698.0 BHP-CS 421.0, 465.0, 472.0 705.0 Ori-BHP-CS 430.0, 480.0, 494.0 703.0, 792.0 a V(CH3COOH):V(CHCl3)=1:2, 25 ℃.  下载: 导出CSV
下载: 导出CSV
表 2 BHP-CS复合物合成条件优化
Table 2. Optimization of the reaction conditions for the formation of BHP-CS
Entry Time/h Temp./℃ V(H2O):V(CHCl3) Base Cat./mol% Absorbance Grafting ratio/% 1 4.0 40 0:1 K2CO3 0.1 0.858 16.25 2 4.0 50 0:1 K2CO3 0.1 0.749 24.76 3 4.0 60 0:1 K2CO3 0.1 0.701 28.51 4 4.0 70 0:1 K2CO3 0.1 0.759 23.98 5 5.5 60 0:1 K2CO3 0.1 0.603 36.16 6 5.5 60 0:1 NaOH 0.1 0.830 18.44 7 5.5 60 0:1 K2CO3 1.0 0.723 26.79 8 5.5 60 1:2 K2CO3 1.0 0.717 27.26 9 5.5 60 1:3 K2CO3 1.0 0.663 31.48 10 5.5 60 1:4 K2CO3 1.0 0.694 29.06 11 5.5 60 1:3 K2CO3 0.1 0.663 31.48 12 5.5 60 1:3 K2CO3 0.5 0.633 33.82 13 5.5 60 1:3 K2CO3 1.0 0.593 36.94 14 5.5 60 1:3 K2CO3 3.0 0.610 35.61 15 5.0 60 1:3 K2CO3 1.0 0.684 29.84 16 5.5 60 1:3 K2CO3 1.0 0.643 33.04 17 6.0 60 1:3 K2CO3 1.0 0.669 31.01 18 6.5 60 1:3 K2CO3 1.0 0.661 31.63
下载: 导出CSV
表 3 冬凌草甲素卟啉壳聚糖微球对MCF-7细胞的体外光动力杀伤力数据
Table 3. In vitro photocytotoxicity datas of oridonin-porphyrin-chitosan microspheres against MCF-7 cells
Cytotoxity ratio [(%)±SD] of MCF-7 cells 100 μmol/L 50 μmol/L 25 μmol/L Light 82.74±0.38 71.03±0.76 31.55±1.70 Dark 68.50±1.16 55.49±1.85 22.59±1.31
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 2168
- HTML全文浏览量: 290
 下载:
下载:
