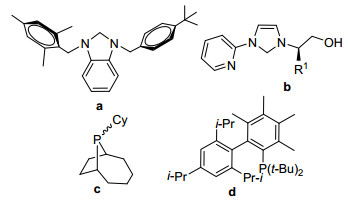
Figure 1.
Some typical ligands
Pd/1, 3-Bis(diphenylphosphino)propane Catalyzed Arylation of Benzoxazoles at C-2 Position with Aryl Bromides
diandian WangYang , Xiaojun Yu , Haiyan Fu , Xueli Zheng , Hua Chen , Ruixiang Li

Heteroaromatics are not only useful structural units in organic synthesis, [1] but 2-aryl substituted benzoxazole and its derivatives are also found in many natural products and anti-inflammatory[2] or anticancer pharmaceuticals.[3] Due to their excellent biological activity, 2-aryl substituted benzoxazoles and derivatives have drawn numerous attentions to explore the efficiently synthetic approaches to them. Regarded as one of the practical means to form C—C bond, transition-metal-catalyzed direct arylation of C—H bond is widely used.[4] PdCl2(PPh3)2-catalyzed direct 2-arylation of benzoxazoles by C—C coupling between organic halides and organotin compounds was firstly reported in 1986.[5] Doucet and co-workers[6] used PdCl(dppb)(C3H5) as catalyst to achieve the same reaction, but different substituents required different loadings of catalysts (0.2~5 mol%), high reaction temperature of 100~150 ℃ and long reaction time of 20 h had to be needed. Pd(OAc)2/xantphos catalytic system, being developed by Walsh and co-workers, [7] could catalyze this reaction at room temperature, but 5 mol% Pd(OAc)2 and excess base must be used. In order to address the high loading of catalyst, co-catalyst strategy has been employed. Pd-Cu co-catalytic system[8] was one of the most frequently-used approaches. For example, Pd(OAc)2-Cu(Ⅱ)/PPh3 carried out the reaction in a loading of 1.0 mol%.[8a] Furthermore, dichlorobis(chloro-di-tert-butyl-phosphine)Pd-Cu(xantphos)Ⅰ system could achieve this transformation with a low amount of 0.25 mol%, but an excessive Cs2CO3 (2.5 equiv.) was necessary.[9] In addition, palladium-catalyzed silver-assisted direct C-5-H arylation of 3-substituted 1, 2, 4-oxadiazoles was accomplished at 150 ℃ under microwave irradiation in the presence of 3.0 equiv. AgOAc.[10] Similarly, the co-catalytic system of palladium-nickel binary nanocluster required 2.5 mol% PdCl2, 2.5 mol% NiCl2 and 10 mol% TBAF for ortho-heterocycle-tethered sterically hindered aryl bromides.[11] Besides the previous co-catalytic systems, many new ligands and their complexes were designed and synthesized to achieve the highly efficient transformation. Arslan's group[12a] and Guo's group[12b] designed two different N-heterocyclic carbenes (Figures 1, a, b) and prepared their palladium complexes to catalyze the direct arylation of benzoxazoles with bromobenzenes. For this system, a high reaction temperature of 130 ℃, a high catalyst loading of 1.5 mol% and a long reaction time of 48 h were needed to carry out a high conversion. Furthermore, even if some complicated phosphine ligands were synthesized and applied in the reaction, the reaction conditions were still very serious. Such as Bergman et al.[13a] used a phosphine (Figure 1, c), a high catalyst loading of 5.0 mol%, high reaction temperature of 250 ℃ and microwave irradiation. Using phosphine d (Figure 1, d), Wilson and co-workers[13b] performed this reaction in the presence of a high catalyst loading of 5.0 mol% and used PivOH as additive. Recently, some phosphine-free or ligand-free synthetic methods were developed, but some co-catalyst and additive had to be introduced into the reaction system.[14]
Based on our continuous interests in C—C coupling reactions catalyzed by transition metal complexes bearing polydentate phosphines, [15] a simple catalytic system was discovered which was combined of commercially available PdCl2 and dppp showed the highly catalytic activity with the low catalyst loading of 0.01% without any co-catalyst.
Firstly, the direct arylation of benzoxazole with bromobenzene was chosen as the model reaction to investigate the effects of a variety of reaction parameters, such as ligands, Pd precursors, bases and solvents, on the reaction, and the results were summarized in Table 1.
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | Catalyst | Ligand | Base | Solvent | Yieldd/% |
| 1 | PdCl2 | dppe | LiOtBu | DME | 10 |
| 2 | PdCl2 | dppp | LiOtBu | DME | 98 |
| 3 | PdCl2 | dppb | LiOtBu | DME | 94 |
| 4 | PdCl2 | xantphos | LiOtBu | DME | 93 |
| 5 | PdCl2 | dppf | LiOtBu | DME | 92 |
| 6 | PdCl2(CNMe)2 | dppp | LiOtBu | DME | 21 |
| 7 | [PdCl(C3H5)]2 | dppp | LiOtBu | DME | 50 |
| 8 | Pd2(dba)3 | dppp | LiOtBu | DME | 32 |
| 9 | PdCl2 | dppp | NaOH | DME | 69 |
| 10 | PdCl2 | dppp | KOH | DME | N.R. |
| 11 | PdCl2 | dppp | Cs2CO3 | DME | 10 |
| 12 | PdCl2 | dppp | KOtBu | DME | N.R |
| 13 | PdCl2 | dppp | NaOtBu | DME | 72 |
| 14 | PdCl2 | dppp | LiOtBu | DME | 98 |
| 15 | PdCl2 | dppp | LiOtBu | DMF | 78 |
| 16 | PdCl2 | dppp | LiOtBu | Toluene | 85 |
| 17 | PdCl2 | dppp | LiOtBu | DMA | 53 |
| 18 | PdCl2 | dppp | LiOtBu | Dioxane | 80 |
| 19b | PdCl2 | dppp | LiOtBu | DME | 87 |
| 20c | PdCl2 | dppp | LiOtBu | DME | N.R. |
| a PhBr (1.0 mmol), benzoxazole (1.2 mmol), base (1.0 equiv.), Pd (0.10 mol%), ligand (0.10 mol%), solvent (4.0 mL), temperature 90 ℃, time 4 h. b PhBr (1.0 mmol), benzoxazole (1.2 mmol), LiOtBu (1.0 equiv, ) PdCl2 (0.10 mol%), dppp (0.050 mol%), DME (4.0 mL), temperature 90 ℃, time 4 h. c PdCl2 (0.10 mol%), dppp (0.20 mol%), DME (4.0 mL), temperature 90 ℃, time 4 h. d GC yield. |
|||||
The catalytic performance of the combination of PdCl2 with some simple and commercially available bidentate phosphines 1, 2-bis(diphenylphosphino)ethane (dppe), 1, 3-bis(diphenylphosphino)propane (dppp), 1, 4-bis(diphenyl-phosphino) (dppb), 4, 5-bis(diphenylphosphino)-9, 9-dimethylxanthene (xantphos) and 1, 1'-bis(diphenypho-sphino)ferrocene (dppf) were firstly examined for the direct arylation of benzoxazole with bromobenzene (Table 1, Entries 1~5). When the reaction time was 4 h and the loading of palladium was 0.1 mol%, the combination of palladium with dppe gave only a low yield of 10%. Good yields could be achieved with dppp, dppb, dppf and xantphos, respectively. Especially, PdCl2/dppp system gave the highest yield. Subsequently, the effect of palladium precursors on the reaction was examined (Table 1, Entries 2, 6~8). It was found that both PdCl(CNMe)2 and Pd2(dba)3 gave the low yields of 21% and 32%, respectively, [PdCl(C3H5)]2 as palladium precursor gave the yield of 50%. PdCl2 showed the hightest activity and gave a high yield of 98%. Hence, in the following experiments PdCl2 was used as palladium precursor. The effect of bases on the reaction was also evaluated (Table 1, Entries 9~14). The low yields were obtained if the reaction was performed in the presence of NaOH or NaOtBu, while the reaction did not occur in Cs2CO3, KOtBu and KOH, respectively. When LiOtBu was used as base, the highest yield of 98% was given (Table 1, Entry 14). The effect of solvents on the reaction (Table 1, Entries 14~18) showed that yields of the target products were not improved in N, N-dimethylform-amide (DMF), toluene and dioxane, and only moderate yield was observed in N, N-dimethylacetamide (DMA). For the effect of the ratio of palladium to dppp on the reaction, it was found that 1: 1 molar ratio of dppp to palladium gave the highest yield. The high molar ratio of palladium to dppp (2: 1) caused the yield to be reduced to 87% (Table 1, Entry 19). On the contrary, the low molar ratio of palladium to dppp (1: 2) led to the reaction was almost completely inhibited (Table 1, Entry 20). Considering the high conversions of some inactive substrates, we decided to prolong the reaction time from 4 h to 6 h in the scope test of substrates. Finally, the optimum composition of this catalyst system was 0.10 mol% PdCl2, 0.10 mol% dppp and 1.0 equiv. LiOtBu, and the reaction was performed in 1, 2-dimethoxy- ethane (DME) at 90 ℃ for 6 h.
With the optimized reaction conditions in hand, the substrate scope for the direct C-2 arylation of benzoxazole and derivatives was further examined and the results were summarized in Table 2. The reaction was not sensitive to the electronic factor of substituents on bromobenzene except of steric factor. For all substrates bearing substituents at 3- and 4-positions of bromobenzene, whatever it was electron-withdrawing group or electron-donating group on aryl bromide, the high yields could be obtained (Table 2, 3ab~3ax). For aryl bromides with substituent in 2-position, all of them gave relatively low yields due to steric hindrance (Table 2, 3ad, 3ag, 3al). For the bulky 1-bromonaphthalene and 9-bromoanthracene, this system also afforded corresponding products in good to excellent yields (Table 2, 3ba~3bb). For heterocyclic compounds, they are generally difficult to carry out this reaction due to the strong coordination ability of heteroatoms with palladium. However, for some typical heteroaryl bromides, such as 2-bromopyridine or 3-bromopyridine, 5-bromopyrimidine, 2-bromofuran and 3-bromothiophene, this system was still suitable and provided the corresponding products with good to excellent yields (Table 2, 3ca~3ce).
下载:
导出CSV
 |
 |
 |
| a ArBr (1.0 equiv.), benzoxazole (1.2 equiv.), LiOtBu (1.0 equiv.), PdCl2 (0.10 mol%), dppp (0.10 mol%), DME (4.0 mL), temperature 90 ℃, time 6 h. Isolated yields.b PdCl2 (0.01 mol%), time 24 h. Isolated yields in parenthesis. c PdCl2 (0.01 mol%), time 48 h. Isolated yields in parenthesis. d PdCl2 (0.50 mol%), time 24 h. Isolated yields. |
On account of the excellent catalytic activity of this system, we tried to decrease the catalyst loading to 0.01 mol% and further evaluated its catalytic performance. Good to excellent yields were still obtained in the presence of electron-withdrawing groups (Table 2, 3ah, 3aj). For electron-donating groups, satisfying yields could be achieved if reaction time was prolonged to 48 h (Table 2, 3ab, 3ae). In addition, benzoxazole derivatives were also suitable for this reaction. However, benzoxazoles with electron-donating groups (Table 2, 3da~3dc) gave higher yields than electron-withdrawing groups (Table 2, 3dd~3de), which was agreed with the reported results.[16] Heterocycles, such as benzothiazole, were quite hard to transform under the standard condition. The amount of catalyst was increased to 0.50 mol% and the time was extended to 24 h. The product yield of 45% was obtained (Table 2, 3df). Compared with the reported results, this catalytic system was suitable for the coupling of benzoxazoles with a wide range of aryl bromides and the yields of desired products were as high as 95%~99% for electron-withdrawing or electron-donating aryl bromides, which were obviously higher than 70%~88% of the reported results.[8a, 8b, 17]
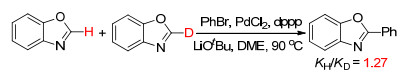
In order to explore the reaction mechanism, we used the mixture (50: 50) of deuterium-labelled-benzoxazole and benzoxazole to perform this reaction and detected the consumption of substrates with NMR (Scheme 1). A kinetic isotope effect (KIE) value (KH/KD) of 1.27 was obtained. The result indicated that the cleavage of C—H bond was not the rate-limiting step of the reaction.[18]
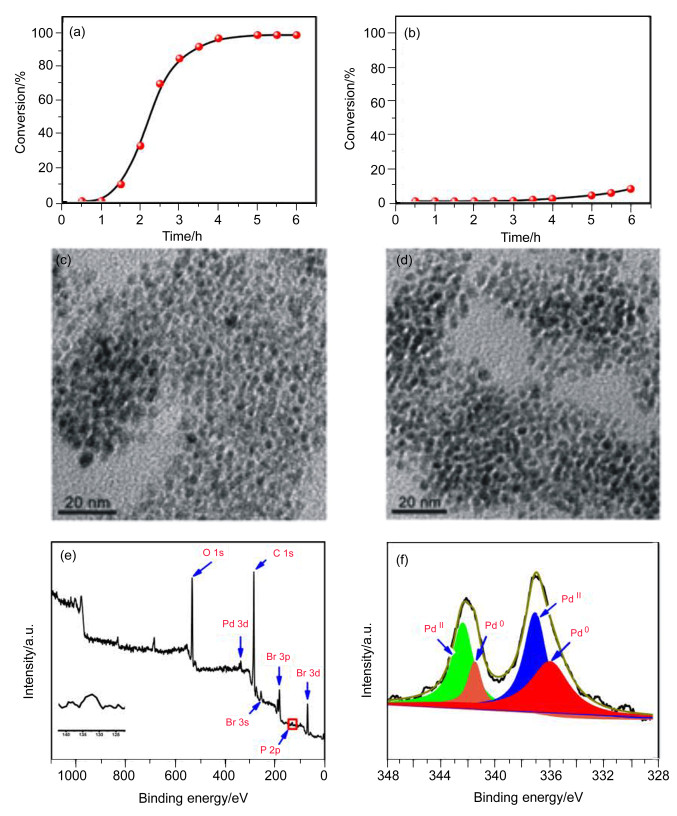
To clarify the oxidation state of palladium species, the relationship between the conversion and reaction time was investigated. Figure 2a depicted the reaction progress with time in the presence of dppp. The curve was a typical S form and the reaction had an induction period of about one hour. At the same time, the reaction could be completely inhibited in the presence of mercury. These were the typically catalytic characters of palladium nanoparticles. Analysis of transmission electron microscopy (TEM) in Figure 2c showed that palladium was uniform nanoparticles with spherical shape in the average diameter of about 3.0 nm after the reaction progressed for 6 h. When the reaction time was extended to 14 h, the size of nanoparticles was almost maintained (Figure 2d). Compared with result in Figure 2a, the conversion of aryl bromide in the absence of ligand (Figure 2b) was very low even if the reaction time prolonged to 6 h. And TEM showed that the size of palladium particles without ligand was much larger than that with dppp. Combined with the HAADF-STEM (high-angle annular dark field-scanning transmission electron microscopy) image of the catalyst at the end of reaction, these results revealed that dppp played a key role in stabilizing palladium nanoparticles. The isolated Pd nanoparticles were analyzed with X-ray photoelectron spectroscopy (XPS) to find the existence of oxygen, bromide and phosphorus (red box, inserted graph) besides palladium (Figure 2e). The result indicated that the surface of nanoparticles absorbed dppp by the coordination interaction. High-resolution XPS spectra of the sample in Figure 2f showed that palladium nanoparticles existed as Pd(Ⅱ) and Pd(0).[19] Because XPS analysis was performed in air, it was suggested that the formation of Pd(Ⅱ) resulted from the oxidation of air to Pd(0). To further verify the state of catalytic active species, a half of reaction mixture solution was immediately filtered into another preheated Schlenk tube[20] containing a stir bar after the reaction was performed for 2 h. At this time, GC conversion of aryl bromide was 25%, and then both examples continued reacting for 4 h. As a result, the original example and the filtrate gave GC conversions of 97% and 77%, respectively. These results indicated that the catalytic active species should be soluble Pd(0) species dissociated from palladium nanoparticals.
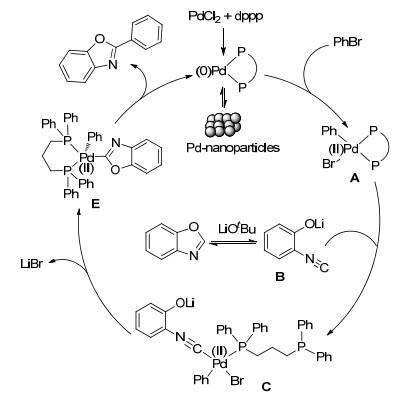
Subsequently, we tried to obtain some information about the composition or structure of intermediates in the process of the reaction. According to the result with deuterium- labelled-benzoxazole and benzoxazole, the cleavage of C—H bond was not rate-determining step. It has been reported that benzoxazoles in the presence of bases underwent a ring-opening pathway to form isocyanophenolate derivatives[21] and they were good ligands for Pd(Ⅱ).[22] In order to verify the ring-opening pathway of benzoxazoles in this system, PdCl2, dppp, LiOtBu and benzoxazole were added in Schlenk tube at 90 ℃ and reacted for 1 h, and then the mixture solution was filtered immediately and inspected with 31P NMR. The result showed that a strong singlet was found at δ 3.65. It suggested that this system existed a major species containing phosphine.
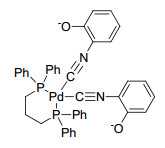
Furthermore, HRMS (SHIMADZU-LCMS-IT-TOF) analysis found the signal of (dppp)Pd(C6H5ONC)2 (m/z 757.1367, calcd for [M+H]+ 757.1371) in the filtrate. The HRMS signal was in good consistent with the molecular weight of palladium complex bearing isocyanophenolate and dppp (Figure 3). Besides, 3-chloro-1-propene was applied to capture phenolic hydroxyl group to further prove the open ring of oxazole rings. PdCl2, dppp, LiOtBu, 3-chloro-1-propene and benzoxazole were added in Schlenk tube and reacted at 90 ℃ for 6 h. Some products were separated by thin-layer chromatography and characterized by 1H NMR. Although we did not separate pure product, the signal of C6H4O(C3H5)NC (m/z 160.0757, calcd for [M+H]+ 160.0751) was found in HRMS and some matched signals were observed in 1H NMR spectrum.
The palladium particles were removed by hot filtration after the reaction was performed for 2 h. It was found that the reaction continued progressing, HRMS of the filtrate showed the signal of m/z 674.9909, which was corresponding the molecular weight of PdBr(C6H5)dppp (calcd for [M+H]+ m/z 675.0197; A in Figure 4). At the same time, the signal of PdBr(C6H5)dppp(C6H4ONC) (m/z 794.0547, calcd for [M+H]+ 794.0574, C in Figure 4) was also found in the filtrate. The results further proved that the really active species were soluble Pd(0) and Pd(Ⅱ) species rather than palladium nanoparticles. This is the first time to obtain the direct evidence of active species with HRMS in the arylation of benzoxazole, while the reported mechanism information was from density functional theory (DFT) calculation.[21]
Finally, a possible mechanism of the direct arylation of benzoxazole was proposed as Figure 4. The catalytic cycle firstly involves the reduction of Pd(Ⅱ) to Pd(0) during the induction period of the reaction, followed by the oxidative-addition of PhBr with Pd(0) to form species (A). Through the ring-opening pathway, 2-isocyanophenolate (B) entered to the catalytic cycle to form Pd-isocyanide (C). An intramolecular nucleophilic attack led to giving species E. E processed reductive-elimination to generate the desired product 2-phenylbenzoxazole and Pd(0) species was regenerated to start a new recycling.
In conclusion, a convenient effective Pd-dppp system to achieve C—H functionalization of benzoxazoles with aryl bromides was developed in good to excellent yields. This catalytic system could tolerate to wide functional groups. dppp played a key role to stabilize active Pd species. Based on TEM, XPS and HRMS results, the reaction mechanism was proposed.
All materials and reagents were commercially available and used without further purification.
1.2 mmol of benzoxazole (143 mg), 1.0 mmol of LiOtBu (80 mg), 0.1 mol% PdCl2 (0.2 mg) and 0.1 mol% dppp (0.5 mg) were added into a Schlenk tube under N2 atmoshere, and then 1.0 mmol of bromobenzene (105 μL) and 4.0 mL of dry DME were added to the reaction tube with syringe. The reaction tube was stirred at 363 K for 6 h. At the end of reaction, the mixture was cooled to room temperature and added with 3.0 mL ether. The solution was filtered with silica gel and the solvent in filtrate was removed under reduced pressure. The residue was purified by silica gel column chromatography with 1: 100 ratio of ethyl acetate (EA) to petroleum ether (PE) to isolate corresponding product.
2-Phenylbenzoxazole (3aa): White powder, m.p. 101~102 ℃ (lit.[23] m.p. 102~104 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.31~8.23 (m, 2H), 7.81~7.76 (m, 1H), 7.62~7.57 (m, 1H), 7.57~7.50 (m, 3H), 7.39~7.34 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 163.16, 150.84, 131.70, 129.06, 127.76, 125.27, 124.74, 120.10, 110.74.
2-p-Tolylbenzoxazole (3ab): Pale-yellow powder, m.p. 112~113 ℃ (lit.[23] m.p. 112~114 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.08~8.06 (m, 2H), 7.69~7.67 (m, 1H), 7.50~7.48 (m, 1H), 7.27~7.18 (m, 4H), 2.36 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.41, 150.79, 142.19, 129.77, 127.71, 124.99, 124.54 (d, J=11.3 Hz), 119.94, 110.61, 21.77.
2-m-Tolylbenzoxazole (3ac): White powder, m.p. 77~79 ℃ (lit.[24] m.p. 81.3~82.6 ℃); 1H NMR (CDCl3, 400.1 MHz) δ: 8.10 (s, 1H), 8.05 (d, J=7.6 Hz, 1H), 7.79~7.11 (m, 1H), 7.60~7.58 (m, 1H), 7.40 (t, J=7.6 Hz, 1H), 7.38~7.34 (m, 3H), 2.46 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.39, 150.86, 142.21, 138.89, 132.52, 128.96, 128.33, 127.13, 125.17, 124.90, 124.68, 120.08, 110.70, 21.49.
2-o-Tolylbenzoxazole (3ad): White powder, m.p. 62~64 ℃ (lit.[25] m.p. 63~65 ℃); 1H NMR (CDCl3, 400.1 MHz) δ: 8.17 (d, J=7.2 Hz, 1H), 7.85~7.79 (m, 1H), 7.61~7.59 (m, 1H), 7.42~7.26 (m, 5H), 2.82 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.52, 150.41, 142.24, 138.96, 131.91, 131.02, 130.06, 126.26, 125.12, 124.48, 120.25, 110.60, 22.34.
2-(4-Methoxyphenyl)benzoxazole (3ae): White powder, m.p. 96~97 ℃ (lit.[24] m.p. 96.3~97.2 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.21 (d, J=8.7 Hz, 2H), 7.74 (d, J=9.1 Hz, 1H), 7.56 (d, J=9.2 Hz, 1H), 7.36~7.29 (m, 2H), 7.03 (d, J=8.8 Hz, 2H), 3.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.31, 162.47, 150.80, 142.38, 129.53, 124.65, 119.78, 114.49, 110.52, 55.59.
2-(3-Methoxyphenyl)benzoxazole (3af): White powder, m.p. 72~74 ℃ (lit.[23] m.p. 70~72 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.89~7.83 (m, 1H), 7.82~7.76 (m, 2H), 7.62~7.56 (m, 1H), 7.43 (t, J=8.0 Hz, 1H), 7.39~7.33 (m, 2H), 7.10~7.08 (m, 1H), 3.92 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.08, 160.07, 150.86, 142.13, 130.13, 128.43, 125.30, 124.73, 120.18, 118.49, 112.02, 110.73, 55.65.
2-(2-Methoxyphenyl)benzoxazole (3ag): White powder, m.p. 55~57 ℃ (lit.[26] m.p. 55~57 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.14 (dd, J=7.6, 1.6 Hz, 1H), 7.85~7.83 (m, 1H), 7.60~7.58 (m, 1H), 7.52~7.50 (m, 1H), 7.36~7.26 (m, 2H), 7.11 (t, J=8.0 Hz, 2H), 4.03 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 161.66, 158.58, 150.42, 142.23, 132.92, 131.41, 125.07, 124.41, 120.84, 120.35, 116.24, 112.17, 110.57, 56.34.
2-(4-Fluorophenyl)benzoxazole (3ah): White powder, m.p. 98~99 ℃ (lit.[23] m.p. 102~104 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.20~8.14 (m, 2H), 7.74~7.65 (m, 1H), 7.50~7.48 (m, 1H), 7.27 (ddd, J=3.7, 3.2, 1.9 Hz, 2H), 7.15~7.11 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 166.20, 163.69, 162.28, 150.89, 142.14, 129.92, 125.2, 124.80, 120.10, 116.20, 110.69.
2-(2-Fluorophenyl)benzoxazole (3ai): Orange powder, m.p. 93~95 ℃ (lit.[27] m.p. 91~93 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.16 (td, J=7.6, 1.7 Hz, 1H), 7.80~7.73 (m, 1H), 7.58~7.51 (m, 1H), 7.48~7.40 (m, 1H), 7.34~7.27 (m, 2H), 7.26~7.21 (m, 1H), 7.21~7.18 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 162.22, 159.68, 150.58, 141.87, 133.19, 130.65, 125.61, 124.81, 124.60, 120.49, 117.11, 115.63, 110.82.
2-(4-(Trifluoromethyl)phenyl)benzoxazole (3aj): White powder, m.p. 140~141 ℃ (lit.[23] m.p. 148~150 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.38~8.24 (m, 2H), 7.81~7.67 (m, 3H), 7.59~7.49 (m, 1H), 7.32 (dd, J=5.9, 2.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 161.61, 151.00, 142.03, 130.56, 128.00, 126.08, 125.95, 125.23, 125.08, 120.54, 110.94.
2-(3-(Trifluoromethyl)phenyl)benzoxazole (3ak): White powder, m.p. 119~120 ℃ (lit.[28] m.p. 119~120 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H), 8.34 (d, J=7.8 Hz, 1H), 7.74~7.66 (m, 2H), 7.53 (ddd, J=10.2, 6.5, 4.7 Hz, 2H), 7.33~7.25 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 161.65, 146.49, 131.94, 131.61, 130.76, 129.69, 128.20, 128.09, 125.87, 125.08, 124.67, 122.49, 120.47, 110.92.
2-(2-(Trifluoromethyl)phenyl)benzoxazole (3al): Pale- orange powder, m.p. 119~121 ℃ (lit.[28] m.p. 119~121 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.07 (dd, J=4.2, 3.5 Hz, 1H), 7.85~7.73 (m, 2H), 7.68~7.51 (m, 3H), 7.37~7.29 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 161.30, 151.37, 141.74, 132.37, 132.05, 131.18, 129.54, 127.26, 126.31, 125.83, 124.91, 122.22, 120.69, 111.05.
2-(4-Isopropylphenyl)benzoxazole (3am): White powder, m.p. 80~82 ℃ (lit.[29] m.p. 80~82 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.19 (d, J=8.3 Hz, 2H), 7.82~7.73 (m, 1H), 7.62~7.54 (m, 1H), 7.43~7.31 (m, 4H), 3.06~2.93 (m, 1H), 1.31 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ: 163.40, 153.02, 150.81, 142.27, 127.86, 127.19, 124.90, 124.60, 119.96, 110.63, 34.37, 23.87.
2-(4-(tert-Butyl)phenyl)benzoxazole (3an): White powder, m.p. 107~108 ℃ (lit.[30] m.p. 107.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.34~8.09 (m, 2H), 7.83~7.72 (m, 1H), 7.72~7.45 (m, 3H), 7.44~7.27 (m, 2H), 1.37 (d, J=7.0 Hz, 9H); 13C NMR (101 MHz, CDCl3) δ: 163.35, 155.28, 150.81, 142.23, 127.60, 126.04, 125.00, 124.61, 124.40, 119.96, 110.64, 35.19, 31.27.
2-(3, 5-Dimethylphenyl)benzoxazole (3ao): White powder, m.p. 119~121 ℃ (lit.[24] m.p. 109.4~113 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.90 (s, 2H), 7.80~7.73 (m, 1H), 7.64~7.52 (m, 1H), 7.35 (ddd, J=5.5, 3.7, 1.0 Hz, 2H), 7.18 (s, 1H), 2.42 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 163.58, 150.80, 141.99, 138.78, 133.55, 126.87, 125.56, 125.15, 124.70, 119.98, 110.66, 21.36.
2-(2, 5-Dimethylphenyl)benzoxazole (3ap): White powder, m.p. 40~42 ℃ (lit.[31] m.p. 40~42 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.93 (s, 1H), 7.79~7.65 (m, 1H), 7.58~7.44 (m, 1H), 7.35~7.22 (m, 2H), 7.17~7.14 (m, 2H), 2.68 (s, 3H), 2.34 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.84, 150.48, 142.18, 135.81, 131.90, 130.55, 126.06, 125.06, 124.48, 120.20, 110.58, 21.80, 20.98.
2-(2, 4-Dimethylphenyl)benzoxazole (3aq): White powder, m.p. 87~88 ℃ (lit.[32] m.p. 87~88 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.09 (d, J=8.6 Hz, 1H), 7.83~7.76 (m, 1H), 7.61~7.55 (m, 1H), 7.38~7.31 (m, 2H), 7.16 (d, J=7.4 Hz, 2H), 2.79 (s, 3H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.82, 150.37, 142.19, 141.50, 138.90, 132.74, 130.14, 127.04, 124.97, 124.46, 120.09, 110.55, 22.28, 21.55.
2-(4-Fluoro-3-methylphenyl)benzoxazole (3ar): White powder, m.p. 116~118 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.02 (dd, J=27.6, 4.4 Hz, 2H), 7.72~7.65 (m, 1H), 7.53~7.46 (m, 1H), 7.31~7.23 (m, 2H), 7.08 (d, J=9.0 Hz, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 164.84, 162.56, 150.88, 142.19, 131.24, 127.29, 125.96, 125.16, 124.74, 123.20, 120.03, 116.00, 110.65, 14.54.
2-(3-Fluoro-4-methylphenyl)benzoxazole (3as): White powder, m.p. 129~131 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.84 (ddd, J=11.6, 9.1, 1.4 Hz, 2H), 7.72~7.66 (m, 1H), 7.49 (d, J=0.5 Hz, 1H), 7.31~7.23 (m, 3H), 2.29 (d, J=1.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.09, 150.96, 144.40, 142.34, 140.15, 129.10, 128.24 (d, J=3.4 Hz), 127.72, 127.32, 126.10, 125.26, 124.77, 120.14, 110.74.
2-(3, 5-Bis(trifluoromethyl)phenyl)benzoxazole (3at): White powder, m.p. 121~123 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.64 (s, 2H), 7.95 (s, 1H), 7.80~7.70 (m, 1H), 7.60~7.51 (m, 1H), 7.43~7.29 (m, 2H), 7.19 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 160.13, 151.06, 141.83, 133.02, 129.51, 127.66, 124.78, 124.44, 121.73, 120.80, 111.12.
4-(benzoxazol-2-yl)benzonitrile (3au): White powder, m.p. 207~208 ℃ (lit.[23] m.p. 209~210 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.36~8.21 (m, 2H), 7.82~7.67 (m, 3H), 7.54 (s, 1H), 7.34 (dd, J=6.5, 2.5 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 161.04, 151.02, 141.98, 132.82, 128.09, 126.29, 125.26, 120.69, 118.29, 114.86, 111.00.
2-(4-(Benzoxazol-2-yl)phenyl)acetonitrile (3av): White powder, m.p. 169~170 ℃ (lit.[34] m.p. 169~170 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.30~8.28 (m, 2H), 7.76~7.72 (m, 2H), 7.56~7.54 (m, 2H), 7.35~7.33 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 160.75, 150.95, 141.89, 134.53, 131.52, 131.12, 130.01, 126.11, 125.20, 120.58, 118.02, 113.64, 110.97.
4-(Benzoxazol-2-yl)benzaldehyde (3aw): White powder, m.p. 112~114 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.03 (s, 1H), 8.35 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.2 Hz, 2H), 7.77~7.71 (m, 1H), 7.53 (d, J=0.6 Hz, 1H), 7.36~7.28 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 191.58, 161.71, 151.03, 142.08, 138.10, 132.39, 131.67, 130.23, 128.20, 126.10, 125.13, 120.60, 110.96.
2-([1, 1'-Biphenyl]-4-yl)benzoxazole (3ax): White powder, m.p. 139~140 ℃ (lit.[24] m.p. 135~136.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=8.5 Hz, 2H), 7.77~7.64 (m, 3H), 7.59 (d, J=7.4 Hz, 2H), 7.55~7.49 (m, 1H), 7.41 (t, J=7.5 Hz, 2H), 7.36~7.22 (m, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.09, 150.96, 144.40, 142.34, 140.15, 129.10, 128.24 (d, J=3.4 Hz), 127.72, 127.32, 126.10, 125.26, 124.77, 120.14, 110.74.
2-(Naphthalen-1-yl)benzoxazole (3ba): Pale-yellow powder, m.p. 106~107 ℃ (lit.[24] m.p. 103.6~109.2 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.39 (d, J=8.7 Hz, 1H), 8.35 (dd, J=7.3, 1.2 Hz, 1H), 7.94 (d, J=8.2 Hz, 1H), 7.89~7.76 (m, 2H), 7.63~7.60 (m, 1H), 7.58~7.47 (m, 3H), 7.32~7.30 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 162.96, 146.40, 134.11, 132.45, 130.85, 129.47, 128.80, 128.05, 126.59, 126.43, 125.24, 124.64, 123.77, 120.42, 110.65.
2-(Anthracen-9-yl)benzoxazole (3bb): Pale-yellow powder, m.p. 164~166 ℃ (lit.[31] m.p. 164~166 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.58 (s, 1H), 8.03~7.96 (m, 4H), 7.92~7.90 (m, 1H), 7.64~7.61 (m, 1H), 7.45~7.40 (m, 6H); 13C NMR (101 MHz, CDCl3) δ: 162.19, 146.41, 131.49, 131.21, 130.97, 128.82, 127.51, 125.71, 125.62, 124.82, 121.42, 120.69, 111.07.
2-(Pyridin-2-yl)benzoxazole (3ca): Yellow powder, m.p. 108~109 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.81~8.71 (m, 1H), 8.31~8.28 (m, 1H), 7.85~7.80 (m, 1H), 7.77~7.75 (m, 1H), 7.61~7.59 (m, 1H), 7.35~7.32 (m, 3H); 13C NMR (101 MHz, CDCl3) δ: 161.54, 151.16, 150.40, 146.17, 141.88, 137.22, 126.16, 125.68, 125.05, 123.55, 120.75, 111.35.
2-(Pyridin-3-yl)benzoxazole (3cb): Pale-yellow powder, m.p. 113~114 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.41 (d, J=1.4 Hz, 1H), 8.69 (dd, J=4.8, 1.6 Hz, 1H), 8.46~8.43 (m, 1H), 7.74~7.71 (m, 1H), 7.54~7.53 (m, 1H), 7.42~7.39 (m, 1H), 7.35~7.31 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 160.74, 152.05, 150.86, 148.78, 141.88, 134.91, 125.85, 125.05, 123.76 (d, J=15.0 Hz), 120.40, 110.90.
2-(Pyrimidin-5-yl)benzoxazole (3cc): White powder, m.p. 166~168 ℃ (lit.[36] m.p. 166~168 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.46 (dd, J=6.3, 4.4 Hz, 2H), 9.35~9.23 (m, 1H), 7.74 (d, J=6.8 Hz, 1H), 7.56 (t, J=6.0 Hz, 1H), 7.41~7.28 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 160.36, 158.25, 155.67, 150.95, 141.72, 126.54, 125.49, 122.34, 120.81, 111.16.
2-(Furan-2-yl)benzoxazole (3cd): White powder, m.p. 83~85 ℃ (lit.[26] m.p. 83~85 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.67 (s, 1H), 7.60 (dd, J=1.7, 0.6 Hz, 1H), 7.48 (s, 1H), 7.31~7.25 (m, 2H), 7.21 (dd, J=3.5, 0.6 Hz, 1H), 6.54 (dd, J=3.5, 1.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ: 155.41, 150.26, 145.87, 142.72, 141.74, 125.42, 124.98, 120.26, 114.42, 112.40, 110.70.
2-(Thiophen-3-yl)benzoxazole (3ce): White powder, m.p. 136~139 ℃ (lit.[23] m.p. 147~149 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.13 (dd, J=2.9, 1.0 Hz, 1H), 7.72 (dd, J=5.1, 0.9 Hz, 1H), 7.70~7.65 (m, 1H), 7.51~7.45 (m, 1H), 7.38 (dd, J=5.1, 3.0 Hz, 1H), 7.30~7.25 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 159.83, 150.45, 142.01, 129.35, 128.22, 127.13, 126.73, 125.14, 124.71, 120.00, 110.59.
5-Methyl-2-phenylbenzoxazole (3da): White powder, m.p. 102~103 ℃ (lit.[27] m.p. 99~101 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.22~8.11 (m, 2H), 7.48 (s, 1H), 7.46~7.41 (m, 3H), 7.37 (d, J=8.3 Hz, 1H), 7.08 (dd, J=8.3, 1.1 Hz, 1H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.25, 149.12, 142.38, 134.53, 131.52, 129.00, 127.68, 127.43, 126.36, 120.03, 110.07, 21.66.
5-Methoxy-2-phenylbenzoxazole (3db): White powder, m.p. 60~61 ℃ (lit.[25] m.p. 60~61 ℃); 1H NMR (CDCl3, 400 MHz) δ: 8.24~8.21 (m, 2H), 7.4~7.6 (m, 4H), 7.26~7.25 (m, 1H), 6.96 (dd, J=8.8 Hz, 1H, 2.4 Hz), 3.88 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.93, 157.55, 153.39, 145.54, 142.97, 131.58, 129.04, 127.63, 113.89, 110.87, 102.97, 56.08.
5-(tert-Butyl)-2-phenylbenzoxazole (3dc): White powder, m.p. 83~84 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.16 (dd, J=6.6, 3.0 Hz, 2H), 7.72 (d, J=1.4 Hz, 1H), 7.46~7.38 (m, 4H), 7.33 (dd, J=8.6, 1.8 Hz, 1H), 1.30 (d, J=5.6 Hz, 9H); 13C NMR (101 MHz, CDCl3) δ: 163.29, 152.77, 148.90, 148.26, 142.10, 131.50, 129.02, 127.65, 123.00, 116.65, 109.84, 35.08, 31.91.
2-Phenyl-5-(trifluoromethyl)benzoxazole (3dd): White powder, m.p. 86~87 ℃ (lit.[37] m.p. 86~87 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.19 (dd, J=8.0, 1.6 Hz, 2H), 7.97 (d, J=0.7 Hz, 1H), 7.64~7.53 (m, 2H), 7.49 (dd, J=6.0, 4.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 164.86, 152.60, 142.37, 132.30, 129.18, 128.00, 127.54, 127.34, 126.56, 122.47, 117.75, 111.15.
5-Fluoro-2-phenylbenzoxazole (3de): White powder, m.p. 93~97 ℃ (lit.[25] m.p. 93~97 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.16 (dd, J=7.7, 1.9 Hz, 2H), 7.48~7.41 (m, 4H), 7.37 (dd, J=8.4, 2.5 Hz, 1H), 7.03~7.00 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 164.79, 160.19, 147.11, 142.87, 131.86, 129.00, 127.70, 126.85, 112.60, 110.82, 106.30.
2-Phenylbenzthiazole (3df): White powder, m.p. 113~115 ℃ (lit.[25] m.p. 113~115 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.02 (dd, J=7.0, 4.5 Hz, 3H), 7.84 (d, J=8.0 Hz, 1H), 7.46~7.39 (m, 4H), 7.35~7.28 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 168.18, 154.24, 135.16, 133.71, 131.08, 129.13, 127.66, 126.43, 125.30, 123.34, 121.73.
1.2 mmol of benzoxazole (143 mg), 1.0 mmol of LiOtBu (80 mg), 0.1 mol% PdCl2 (0.2 mg) and 0.1 mol% dppp (0.5 mg) were added into a Schlenk tube under N2 atmoshere, and then 1.0 mmol of bromobenzene (105 μL) and 4.0 mL of dry DME were added to the reaction tube with syringe. The sealed reaction tube was stirred at 363 K for 6 h. At the end of reaction, the mixture was cooled to room temperature and added with 1.0 mL of ether and 3.0 mL of H2O. Then palladium particles in the mixture were separated by high-speed centrifuging (10000 r/min), the supernate was removed and Pd particles were washed by acetone and used for analysis.
Supporting Information The detail of KIE experimental, Hot filtration test, Hg(0) poisoning test, HAADF-STEM image of the catalyst after reaction, TEM image of Pd nanoparticles without dppp, the experiments of supports for the ring-opening pathway, the possible structures of transition states in the reaction, and NMR spectra for target products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Seth, K.; Garg, S. K.; Kumar, R.; Purohit, P.; Meena, V. S.; Goyal, R.; Banerjee, U. C.; Chakraborti, A. K. ACS Med. Chem. Lett. 2014, 5, 512.
(b) Noel, S.; Cadet, S.; Gras, E.; Hureau, C. Chem. Soc. Rev. 2013, 42, 7747.
He, J.; Lin, F.; Yang, X. F.; Wang, D.; Tan, X. H.; Zhang, S. J. Org. Process Res. Dev. 2016, 20, 1093. doi: 10.1021/acs.oprd.6b00168
Zhang, Z.; Zhao, D.; Dai, Y.; Cheng, M. S.; Geng, M. Y.; Shen, J. K.; Ma, Y. C.; Ai, J.; Xiong, B. Molecules 2016, 21, 1407. doi: 10.3390/molecules21101407
Abdellaoui, F.; Youssef, C.; Ammar, H. B.; Roisnel, T.; Soulé, J. F.; Doucet, H. ACS Catal. 2016, 6, 4248. doi: 10.1021/acscatal.6b00678
Kosugi, M.; Koshiba, M.; Atoh, A.; Sano, H.; Migita, T. B. Chem. Soc. Jpn. 1986, 59, 677. doi: 10.1246/bcsj.59.677
Derridja, F.; Djebbar, S.; Benali-Baitich, O.; Doucet, H. J. Organomet. Chem. 2008, 693, 135. doi: 10.1016/j.jorganchem.2007.10.028
Gao, F.; Kim, B. S.; Walsh, P. J. Chem. Commun. 2014, 50, 10661. doi: 10.1039/C4CC05307A
(a) Yan, X. M.; Mao, X. R.; Huang, Z. Z. Heterocycles 2011, 83, 1371.
(b) Alagille, D.; Baldwin, R. M.; Tamagnan, G. D. Tetrahedron 2005, 46, 1349.
(c) Wang, M.; Li, D.; Zhou, W.; Wang, L. Tetrahedron 2012, 68, 1926.
Huang, J. k.; Chan, J.; Chen, Y.; Borths, C. J.; Baucom, K. D.; Larsen, R. D.; Faul, M. M. J. Am. Chem. Soc. 2010, 132, 3674. doi: 10.1021/ja100354j
Li, S.; Wan, P. H.; Ai, J.; Sheng, R.; Hu, Y. Z.; Hu, Y. H. Adv. Synth. Catal. 2017, 359, 772. doi: 10.1002/adsc.v359.5
Seth, K.; Purohit, P.; Chakraborti, A. K. Org. Lett. 2014, 16, 2334. doi: 10.1021/ol500587m
(a) Arslan, H.; Özdemİr, İ.; Vanderveer, D.; Demİr, S.; Çetİnkaya, B. J. Coord. Chem. 2009, 62, 2591.
(b) Yang, L. G.; Yuan, J. W.; Mao, P.; Guo, Q. RSC Adv. 2015, 5, 107601.
(a) Lewis, J. C.; Wu, J. Y.; Bergman, R. G.; Ellman, J. A. Angew. Chem., Int. Ed. 2006, 45, 1589.
(b) Strotman, N. A.; Chobanian, H. R.; Guo, Y.; He, J. F.; Wilson, J. E. Org. Lett. 2010, 12, 3578.
(a) Zhao, D. B.; Wang, W. D.; Lian, S.; Yang, F.; Lan, J. B.; You, J. S. Chem.-Eur. J. 2009, 15, 1337.
(b) Wang, X.; Gribkov, D. V.; Sames, D. J. Org. Chem. 2007, 72, 1476.
(c) Le, H. T. N.; Nguyen, T. T.; Vu, P. H. L.; Truong, T.; Phan, N. T. S. J. Mol. Catal. A: Chem. 2014, 391, 74.
(a) Jiang, Z. J.; Wang, W.; Zhou, R.; Zhang, L.; Fu, H. Y.; Zheng, X. L.; Chen, H.; Li, R. X. Catal. Commun. 2014, 57, 14.
(b) Zhou, R.; Wang, W.; Jiang, Z. J.; Fu, H. Y.; Zheng, X. L.; Zhang, C. C.; Chen, H.; Li, R. X. Catal. Sci. Technol. 2014, 4, 746.
(c) Zhou, R.; Wang, W.; Jiang, Z. J.; Wang, K.; Fu, H. Y.; Zheng, X. L.; Chen, H.; Li, R. X. Chem. Commun. 2014, 50, 6023.
Yu, P.; Zhang, G. Y.; Chen, F.; Cheng, J. Tetrahedron Lett. 2012, 53, 4588. doi: 10.1016/j.tetlet.2012.06.076
Huang, J. K.; Chan, J.; Chen, Y.; Borths, C. J.; Baucom, K. D.; Larsen, R. D.; Faul, M. M. J. Am. Chem. Soc. 2010, 132, 3674. doi: 10.1021/ja100354j
Simmons, E. M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. doi: 10.1002/anie.v51.13
(a) Kim, Y.; Kim, H.; Kim, J. D. RSC Adv. 2018, 8, 2441.
(b) Ho, C. E.; Hsieh, W. Z.; Lee, P. T.; Huang; Y. H.; Kuo, T. T. Appl. Surf. Sci. 2018, 434, 1353.
Zhang, Y.; Zhao, Y.; Luo, Y.; Xiao, L.Q.; Huang, Y. X.; Li, X. R.; Peng, Q. T.; Liu, Y. Z.; Yang, B.; Zhu, C. Z.; Zhou, X. C.; Zhang, J. M. Org. Lett. 2017, 19, 6470. doi: 10.1021/acs.orglett.7b02967
Sánchez, R. S.; Zhuravlev, F. A. J. Am. Chem. Soc. 2007, 129, 5824. doi: 10.1021/ja0679580
(a) Jutzi, P.; Gilge, U. J. Organomet. Chem. 1983, 246, 159.
(b) Kernbach, U.; Lugger, T.; Hahn, F. E.; Fehlhammer, W. P. J. Organomet. Chem. 1997, 541, 51.
Patra, A.; James, A.; Das, T. K.; Biju, A. T. J. Org. Chem. 2018, 83, 14820. doi: 10.1021/acs.joc.8b02598
Yin, Y. Z.; Yue, X. Y.; Zhong, Q.; Jiang, H.; Bai, R. P.; Lan, Y.; Zhang, H. Adv. Synth. Catal. 2018, 360, 1639. doi: 10.1002/adsc.v360.8
Ueda, S.; Nagasawa, H. Angew. Chem., Int. Ed. 2008, 47, 6411. doi: 10.1002/anie.v47:34
Chang, W. C.; Sun, Y. K.; Huang, Y. Heteroat. Chem. 2017, 28, e21360. doi: 10.1002/hc.2017.28.issue-2
Yuan, X. l.; Liu, Y. F.; Qin, M. D.; Yang, X. Y., Chen, B. H. ChemistrySelect 2018, 3, 5541. doi: 10.1002/slct.201800874
Ackermann, L.; Althammer, A.; Fenner, S. Angew. Chem., Int. Ed. 2009, 48, 201. doi: 10.1002/anie.200804517
Naeimi, H.; Rahmatinejad, S. Synth. React. Inorg. Met.-Org. Chem. 2016, 46, 471. doi: 10.1080/15533174.2014.988794
Ghodbane, A.; D'Altério, S.; Saffon, N.; McClenaghan, N. D.; Scarpantonio, L.; Jolinat, P.; Fery-Forgues; S. Langmuir 2012, 28, 855. doi: 10.1021/la2036554
Hachiya, H.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2009, 11, 1737. doi: 10.1021/ol900159a
Shibahara, F.; Yamaguchi, E.; Murai, T. Chem. Commun. 2010, 46, 2471. doi: 10.1039/b920794e
Li, Y. Q.; Yu, X. J.; Wang, Y. D. D.; Fu, H. Y.; Zheng, X. L.; Chen, H.; Li, R. X. Organometallics 2018, 37, 979. doi: 10.1021/acs.organomet.8b00005
Eastman, K. C. US 3458506, 1967[Chem. Abstr. 1969, 71, 125998d].
Singh, V.; Singh, A.; Singh, G.; Verma, R. K.; Mall, R. Med. Chem. Res. 2018, 27, 735. doi: 10.1007/s00044-017-2097-1
Jung, M. R.; Choi, S. W.; Cho, K. W. J. Heterocycl. Chem. 2000, 37, 969. doi: 10.1002/jhet.v37:4
Evindar, G.; Batey, R. A. J. Org. Chem. 2006, 71, 1802. doi: 10.1021/jo051927q
Dai, W. C.; Wang, Z. X. Org. Chem. Front. 2017, 4, 1281. doi: 10.1039/C7QO00174F

Figure 2 (a) Relationship between time and conversion in the presence of dppp, (b) relationship between time and conversion in the absence of dppp, (c) TEM image of catalyst with dppp after 6 h, (d) TEM image of catalyst with dppp after 14 h, (e) XPS spectrum of palladium particles, and (f) high-resolution XPS spectra of palladium particles
Table 1. Optimization of the reaction conditions for the direct arylation of benzoxazole with bromobenzene
|
|||||
| Entry | Catalyst | Ligand | Base | Solvent | Yieldd/% |
| 1 | PdCl2 | dppe | LiOtBu | DME | 10 |
| 2 | PdCl2 | dppp | LiOtBu | DME | 98 |
| 3 | PdCl2 | dppb | LiOtBu | DME | 94 |
| 4 | PdCl2 | xantphos | LiOtBu | DME | 93 |
| 5 | PdCl2 | dppf | LiOtBu | DME | 92 |
| 6 | PdCl2(CNMe)2 | dppp | LiOtBu | DME | 21 |
| 7 | [PdCl(C3H5)]2 | dppp | LiOtBu | DME | 50 |
| 8 | Pd2(dba)3 | dppp | LiOtBu | DME | 32 |
| 9 | PdCl2 | dppp | NaOH | DME | 69 |
| 10 | PdCl2 | dppp | KOH | DME | N.R. |
| 11 | PdCl2 | dppp | Cs2CO3 | DME | 10 |
| 12 | PdCl2 | dppp | KOtBu | DME | N.R |
| 13 | PdCl2 | dppp | NaOtBu | DME | 72 |
| 14 | PdCl2 | dppp | LiOtBu | DME | 98 |
| 15 | PdCl2 | dppp | LiOtBu | DMF | 78 |
| 16 | PdCl2 | dppp | LiOtBu | Toluene | 85 |
| 17 | PdCl2 | dppp | LiOtBu | DMA | 53 |
| 18 | PdCl2 | dppp | LiOtBu | Dioxane | 80 |
| 19b | PdCl2 | dppp | LiOtBu | DME | 87 |
| 20c | PdCl2 | dppp | LiOtBu | DME | N.R. |
| a PhBr (1.0 mmol), benzoxazole (1.2 mmol), base (1.0 equiv.), Pd (0.10 mol%), ligand (0.10 mol%), solvent (4.0 mL), temperature 90 ℃, time 4 h. b PhBr (1.0 mmol), benzoxazole (1.2 mmol), LiOtBu (1.0 equiv, ) PdCl2 (0.10 mol%), dppp (0.050 mol%), DME (4.0 mL), temperature 90 ℃, time 4 h. c PdCl2 (0.10 mol%), dppp (0.20 mol%), DME (4.0 mL), temperature 90 ℃, time 4 h. d GC yield. |
|||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Substrate scope in this reaction
|
|
|
| a ArBr (1.0 equiv.), benzoxazole (1.2 equiv.), LiOtBu (1.0 equiv.), PdCl2 (0.10 mol%), dppp (0.10 mol%), DME (4.0 mL), temperature 90 ℃, time 6 h. Isolated yields.b PdCl2 (0.01 mol%), time 24 h. Isolated yields in parenthesis. c PdCl2 (0.01 mol%), time 48 h. Isolated yields in parenthesis. d PdCl2 (0.50 mol%), time 24 h. Isolated yields. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: