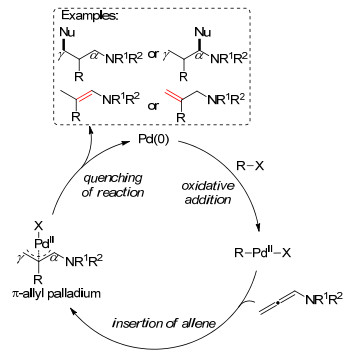
图式 1.
钯催化联烯胺反应的主要步骤
Scheme 1.
Main steps for Pd-catalyzed allenamides reaction

现代合成化学和精细化学品工业不仅为人类生活提供了赖以生存的物质基础, 而且对于人工智能和航天等高科技领域的飞速发展也具有重要的推动作用, 在医药、农药、保健品、环保等行业表现的尤为突出.近几年, 随着世界医药产业的飞速发展, 合成新药物分子本体的难度不断增大, 有机合成化学也面临着更大的机遇和挑战, 发展新颖的合成方法学显得尤为重要.
化学合成的核心为化学键的构建, 而如何高效地构建C—X键(X=C, N, O, S, 卤素等)一直是有机化学界关注的热点.在过去的几十年中, 过渡金属催化的交叉偶联反应已经发展成为化学键构建必不可少的工具[1], 同时, 提高合成效率、降低能耗、减少废物排放及发展环境友好型化学更加符合当今的时代背景.
近四十年来, 有关联烯胺的研究逐渐增多, 日趋完善, 联烯胺类化合物逐渐成为有机合成中的重要前驱体和结构单元, 能在多种过渡金属的催化作用下反应, 尤其是π酸性的金属元素, 如Au、Pd、Ru、Rh等, 这些过渡金属催化剂能够与中心碳原子配位, 产生活性中间体, 发生偶联、环化等, 生成二烯、杂环类化合物.这类反应大多呈现出反应条件温和、适用底物范围广等特点, 因此, 可以通过引入联烯胺的方式, 生成新的可作为亲电、亲核试剂连续进攻的中间体, 进一步转化实现C—X键的高效构建, 发展新的方法学.
自1968年第一例报道制备联烯胺以来, 联烯胺的研究吸引了众多科研工作者的兴趣.其中, 过渡金属催化联烯胺环化, 合成特定的分子结构成为了联烯胺研究中的主要内容, 包括钯催化、金催化、铑催化、钌催化以及镍催化等.相较于传统的催化反应体系, 过渡金属催化反应具有效率更高、热稳定性以及化学稳定性更好, 催化循环寿命长等优点.同时, 由于联烯胺其独特的反应性, 选择性、易得性和稳定性, 已被广泛用于联烯胺[2+2], [3+2], [4+2], [2+2+2]等加成环化和串联环化反应中, 以构建四至九元碳环或杂环骨架.在新型有机反应的探索中, 过渡金属催化联烯胺环化反应丰富了含氮、氧杂环合成方法学, 同时为有机化学发展应用奠定了良好的基础.本文根据金属催化剂进行分类, 对联烯胺环化反应进行了归纳和总结, 并对相应的反应机理进行了讨论.
过渡金属钯催化剂具有性质稳定、活性表面积大、原子经济性相对较高等特点, 被广泛地应用于天然产物全合成等催化体系, 偶联反应高效构建目标分子骨架, 包括C—C键、C—X键等.钯催化联烯胺环化过程的反应机理大体可以概括为Scheme 1所示的反应途径, 主要包括碳卤键对Pd(0)的氧化加成, 联烯胺插入并形成π-烯丙基钯中间体, 最后以β-H消除、亲核取代等淬灭方式, 选择性地在α位或γ位形成新的化学键.
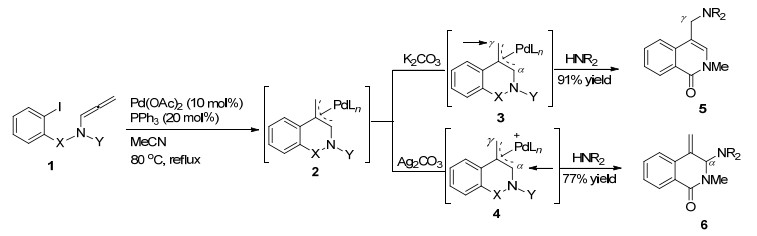
1995年, Grigg课题组[2]报道了第一例Pd催化的分子内碳钯化/环化-阴离子捕获串联反应实现联烯胺分子内的环化反应(Scheme 2). Pd(0)启动该反应循环, 最后在碳酸钾存在的条件下, 经还原消除得到γ位取代的环化产物.若加入碳酸银, 中间体可能脱掉一分子AgI, 形成钯阳离子中间体, 更倾向于进攻α位, 两种不同的环化方式产率分别为91%和77%.
Grigg课题组[3]随后对Pd催化联烯胺, 阴离子捕获环化反应进行了进一步的探究, 并在2001年报道了一例联烯胺环化反应(Scheme 3).该转化经历相似的氧化加成、环化、形成π-烯丙基钯中间体8过程.与之前工作不同的是, 本次报道是将亲核试剂引入联烯胺分子内, 完成分子内α位的亲核进攻, 得到一系列多环异喹啉酮的化合物.
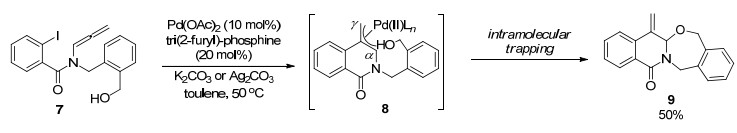
2006年, Grigg课题组[4]又报道了一例钯/铟双金属催化的联烯胺环化反应.机理如Scheme 4所示, 在该转化中首先形成π-烯丙基钯中间体, 随后完成钯与铟的转金属交换过程, 再与手性叔丁基亚磺酰亚胺配位形成六元椅式过渡态, 最后得到包含连续两个手性中心的化合物13.
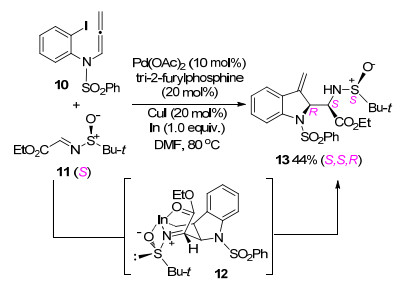
2005年, Hiroya课题组[5]报道了一例钯催化联烯胺与芳基碘化物的分子间环化反应(Scheme 5).在反应过程中, 尽管存在取代基空间位阻的影响, O或N等亲核基团仍然优先对α位进攻, 以5-或6-exo-trig环化的方式得到产物.作者认为, 由于氮原子对邻位α碳的电性影响, 使得α碳原子周围的电子云偏向于N原子, 相比于γ位碳原子更加具有亲电性, 因此亲核基团更倾向于进攻烯丙基α位, 得到环化产物16, 并没有得到γ位碳原子进攻环化产物17.
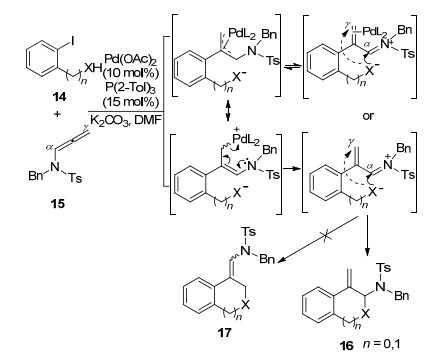
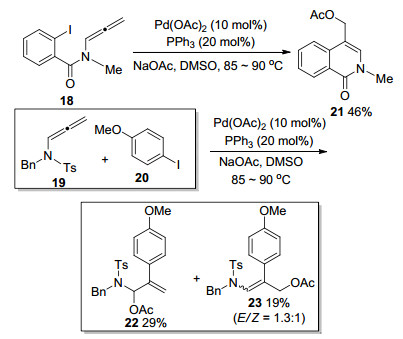
Savic课题组[6]在2012年也对该类型反应进行了相关的探索, 并进行了报道(Scheme 6).作者以醋酸根阴离子作为捕获剂, 较好完成了分子间亲核取代的过程, 得到进攻γ位的环化产物.值得注意的是, 在作者试图进行分子间一锅法实现联烯胺环化的探索中, 发现联烯胺和4-碘苯甲醚在相同的条件下反应并没有得到环化的产物, 而是以29%和19%的低产率得到联烯胺选择性异构化的乙酸酯22和23.
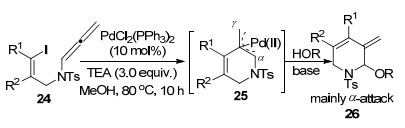
2014年, 童晓峰课题组[7]继续完成了钯催化联烯胺分子内环化的工作(Scheme 7), 作者以PdCl2(PPh3)2为催化剂, 得到π-烯丙基钯中间体25, 与Grigg等工作不同之处在于, 作者构建的是烯基联烯胺, 而非芳基取代的联烯胺, 随后以甲醇作为亲核试剂进攻π-烯丙基钯中间体25, 得到进攻α位的环化产物26.
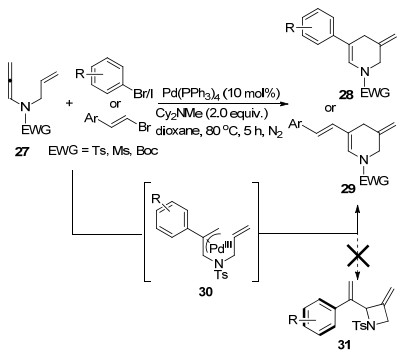
2016年, 刘会课题组[8]实现了一例联烯胺与芳基碘之间的环化-Heck反应(Scheme 8).该反应起始于Pd(0)对芳基碘的氧化加成, 得到π-烯丙基钯中间体, 在实验的探索和条件筛选阶段, 作者并没有检测到联烯胺α位碳选择性四元环化产物31, 而是经过6-exo环化过程, 联烯胺γ位碳原子选择性环化.通过该转化可以得到一系列3-亚甲基-5-苯基-四氢吡啶类化合物28, 该骨架结构在治疗糖尿病、帕金森以及肥胖症等活性药物分子中广泛存在.同时, 烯基溴化物也能在该转化中表现出较好的兼容性, 得到分子内包含共轭二烯结构的产物29, 具有更广泛的潜在应用价值.
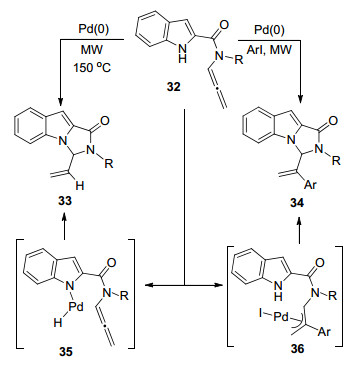
2010年, Broggini课题组[9]报道了一例钯催化联烯胺分子内的环加成反应(Scheme 9).在微波条件下, Pd与吲哚N原子配位生成H-Pd(Ⅱ)物种35, 随后插入联烯基团, 形成π-烯丙基钯中间体, 最后还原消除得到环化产物33.若加入芳基碘, 则首先钯与碳碘键氧化加成, 再对联烯胺插入形成π-烯丙基把中间体36, 随后碘原子完成对吲哚N原子的亲核取代过程, 最后还原消除, 得到进攻α位产物34.
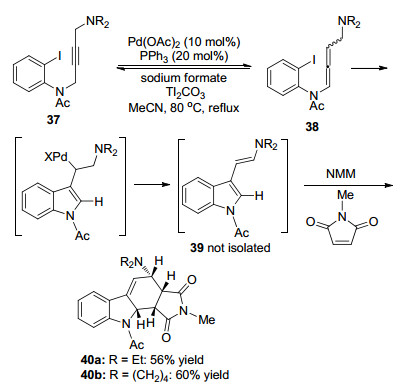
1996年, Grigg课题组[10]报道了一例多组分间的高区域和立体选择性5-或6-exo-dig环化反应(Scheme 10).首先, 在Tl2CO3存在的条件下, 丙炔胺化合物37异构得到联烯胺化合物38.随后在Pd的作用下完成碳卤键的氧化加成、分子内环化等过程得到不稳定的化合物39, 该化合物虽未能分离得到, 但能够被N-马来酰亚胺(NMM)捕获, 发生Diels-Alder偶联反应得到手性环化产物40.
当捕获剂为NaN3时, 得到环化产物42, 在丁炔二酸二甲酯(DMAD)存在下, 可以得到1, 3-偶极环加成产物43.而当降冰片二烯参与反应时, 则会经历中间体44, 发生Diels-Alder环加成反应, 获得三唑化合物45[11](Scheme 11).
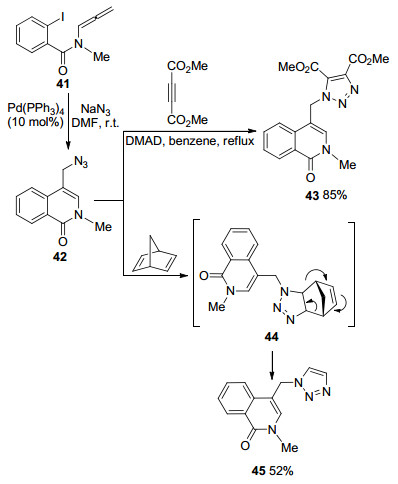
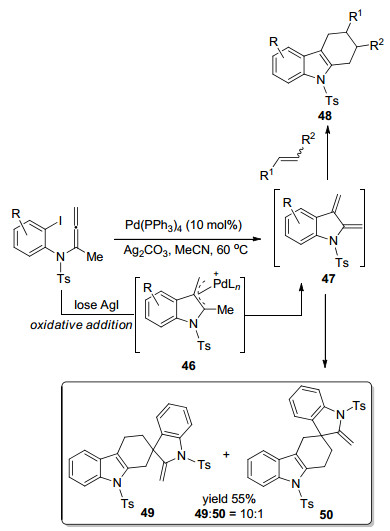
基于同样的设计思路, Sasaki课题组[12]在2007年实现了分子内联烯胺Diels-Alder加成环化得到2, 3位取代的吲哚衍生物(Scheme 12).首先, Pd对碳碘键氧化加成, 失去一分子AgI, 得到Pd阳离子中间体46, 还原消除得到吲哚-2, 3-醌二甲烷47.在外部亲双烯体存在的情况下, 分子间发生Diels-Alder环加成反应, 以较好的产率得到四氢咔唑类化合物48.在没有外部亲双烯体存在的情况下, 两分子的47可以发生分子间Diels-Alder环加成反应, 分离出同型二聚体49和50.
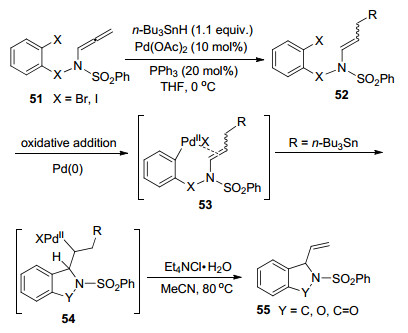
在Pd催化联烯胺环化的研究进展中, Grigg课题组[13]在1996年报道了一例锡烷基化-碳钯化串联反应构建含氮、氧杂环的反应(Scheme 13).转化经历联烯胺α位碳原子区域选择性环化, 得到中环(5~7)和大环(7~11)类型的杂环化合物.反应由Pd(0)启动, 首先对碳卤键氧化加成, 经碳钯化、环化、还原消除等过程, 最终再生Pd(0)催化剂完成催化循化.在中间体54环化的过程中, n-Bu3SnPdX的消除速度高于β-H的消除速度, 以较理想的产率得到最终环化产物55.
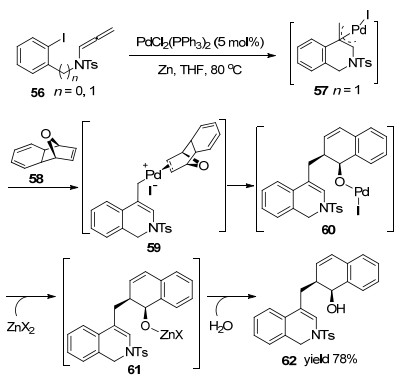
2006年, Cheng课题组[14]报道了一例新型Pd催化联烯胺环化反应(Scheme 14).反应起始于Pd(0)对烯基碘的氧化加成, 随后得到π-烯丙基钯中间体57, 与58的双键配位得到中间体59, 随后经过迁移插入和β-氧同步消除产生钯-氧络合物60.在锌的作用下发生Pd转移过程, 再生Pd(0)催化剂, 中间体61经水解处理后, 最终得到1, 2-二氢异喹啉甲基-1, 2-二氢-1-萘酚衍生物.
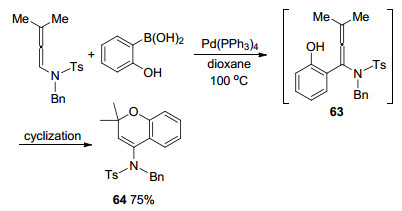
2012年, 当Lai课题组[15]试图通过Suzuki-Miyaura偶联构建四取代的联烯胺化合物时, 发现并没有获得偶联产物.作者认为, 联烯胺63可能以中间体的形式存在, 通过分子内环化, 得到六元含氧化合物64 (Scheme 15).
Grigg等[16]在1997年报道了一例Pd催化Suzuki- Miyaura偶联环化反应(Scheme 16).与之前描述的机理非常相似, 该反应首先也是经历氧化加成、环化形成烯丙基中间体66.接着中间体66与芳基硼酸完成转金属交换过程, 选择性环化得到环化产物67和68.虽然获得少量的进攻α位环化产物, 但是Suzuki-Miyaura型交叉偶联总体上是具有高度区域选择性的, 在碱性条件下, 反应主要还是发生在空间位阻较小的γ位碳.
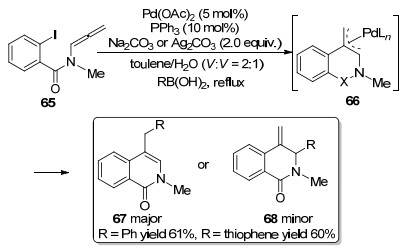
2007年, Sasaki课题组[17]报道了一种合成2, 3位取代吲哚的合成方法(Scheme 17).利用π-烯丙基钯中间体69与一系列芳基硼酸进行Suzuki-Miyaura偶联, 可以高效地合成二取代吲哚及其衍生物.
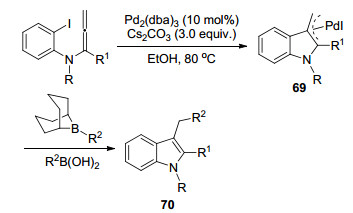
2017年, 刘会研究组[18]报道了一篇Pd催化联烯胺Metallo-Ene/Suzuki串联环化反应(Scheme 18).该反应在PdCl2(PPh3)2为催化剂, K3PO4为碱, 二氧六环为溶剂, 50 ℃的条件下进行, 一锅法实现C(sp3)—C(sp2)键的高效构建.
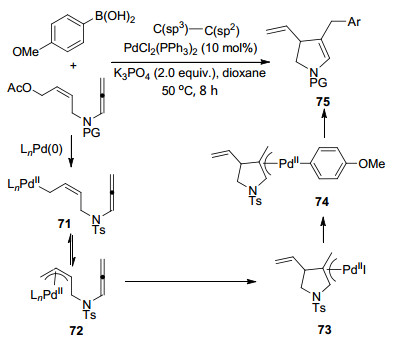
结合Bäckvall等课题组相关工作, 作者提出了可能的反应机理(Scheme 18).首先, Pd(0)完成对醋酸乙烯酯的氧化加成得到中间体71, 同时可能形成π-烯丙基钯中间体72, 并且两个中间体之间可能存在相互转化.接下来, Pd对联烯插入, 分子内环化形成第一个C(sp3)—C(sp3)键, 与此同时, Metallo-Ene反应产生另一个π-烯丙基钯中间体73.随后, 中间体73与甲氧基苯硼酸发生转金属交换, 得到中间体74.最后经还原消除构建第二个C(sp3)—C(sp3)键, 并得到最终环化产物75.在该循环过程中, 并没有发生γ位的还原消除过程, 而是高选择性地完成α位还原消除反应, 得到最终产物.
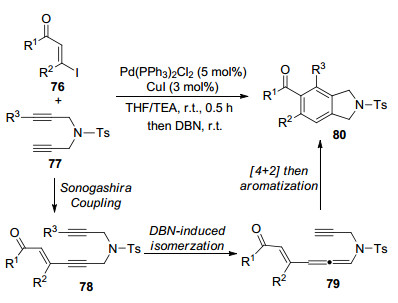
2012年, Huang课题组[19]报道了一例一锅法合成取代的异吲哚啉衍生物的反应(Scheme 19).在该转化中, 首先是双炔丙基酰胺与β-碘烯酮Sonogashira发生偶联反应, 生成化合物78, 并在1, 5-二氮杂二环[4.3.0]壬烯-5 (DBN)的诱导下产生异构化反应, 得到联烯胺79.随后经历分子内的[4+2]环加成反应, 完成芳构化过程, 以较好的产率获得异吲哚啉衍生物80.
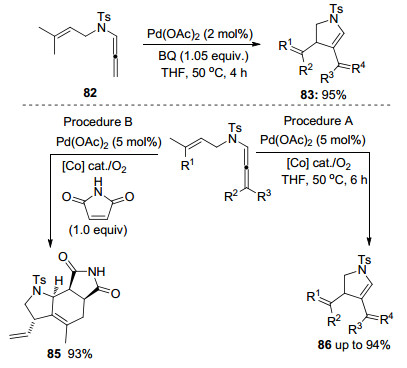
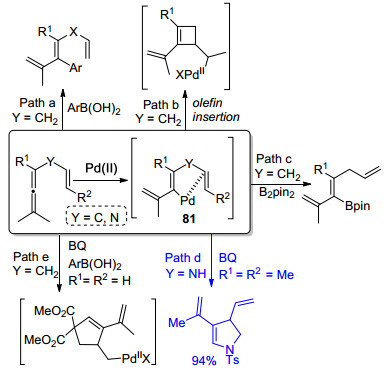
在联烯和联烯胺的研究领域中, Bäckvall课题组[20]做了大量的工作.他们主要研究联烯化合物在Pd(Ⅱ)的催化下形成中间体81之后, 进行Suzuki偶联、环化、亲核取代等过程(Scheme 20).如Path d所示, 该团队[21]报道了一种新颖的钯催化联烯胺氧化环化反应, 通过该转化可以得到一系列共扼二烯化合物.
作者研究发现, 烷烃、芳烃以及环状烯烃取代的联烯胺都能够较好地适配于该转化.在适当的反应时间下, 联烯胺能够达到100%的转化率, 高产率得到产物83, 同时存在极少量的Diels-Alder加成环化产物(Scheme 21).但是当苯醌的量增加到2.1 equiv.时, 主要发生的反应是联烯胺和苯醌之间的Diels-Alder环加成. Bäckvall小组进一步研究了该转化, 开发并使用了一种新型醌衍生物混合催化剂, 如Scheme 21所示, 以氧气为末端催化剂, 在路径A的条件下, 联烯胺的分子内环化产率达到94%, 并且没有形成任何可检测的Diels-Alder加合物.在相同的反应条件下, 联烯胺和1.0 equiv.的马来酰亚胺发生氧化-Diels-Alder反应, 得到环加成产物85的产率达到93%.反应生成的杂环产物是合成复杂分子的重要中间体.该氧化策略能够和Diels- Alder环加成反应相结合, 一锅法实现分子间串联环化反应, 成为具有潜在应用价值的合成方法学.
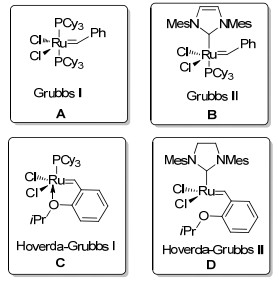
以钌卡宾催化剂催化α-烯烃复分解反应已经成为构建碳碳双键的常用的策略之一, 被广泛用于制备复杂的有机化合物, 钌亚烷基催化剂如A~D (Scheme 22).随着对钌催化剂的研究不断深入, 在烯烃异构化[22]、氢化[23]、自由基反应[24]、硅烷活化[25]、环丙烷化[26]、环丙烷差向异构化[27]、氧化[28]、氢化乙烯化[29]、[4+2]环加成[30]、[3+2]环加成[31]、[2+2+2]环加成[32]反应中有着广泛的应用.
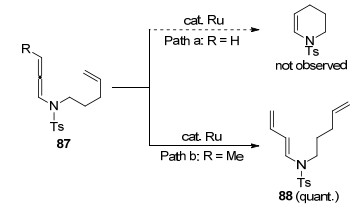
关于钌催化烯烃或者烯胺的关环反应的报道较多, 但是, 钌催化联烯胺加成环化却鲜有报道.在2011年, Hiemstra课题组[33]对联烯胺闭环复分解(RCM)反应进行了探索(Scheme 23).以联烯胺87作为反应底物, 在钌催化剂Grubbs Ⅰ和Grubbs Ⅱ的作用下, 最终没有得到关环的产物, 只得到了联烯胺异构化的产物88.作者认为这可能是由于钌催化剂对分子的丙二烯结构具有更大的亲和力, 一旦最初的复分解发生在这一端, 将不会与双键进一步反应.
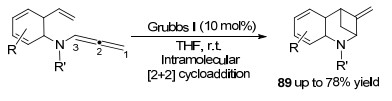
2016年, Arisawa课题组[34]实现了钌催化联烯胺分子内的[2+2]环加成反应(Scheme 24), 构筑双环[3.1.1]杂环庚烷化合物89.这是第一例钌卡宾催化剂催化的[2+2]环加成反应报道, 转化在室温下进行, 没有发生自由基和经典的烯烃复分解反应.该方法的实现, 将有助于钌卡宾催化剂更加广泛地应用于功能性分子合成领域.
2006年, Barluenga课题组[35]首次实现了铑催化联烯胺与铬烯基-(甲氧基)卡宾复合物的[3+2]环加成反应(Scheme 25), 此过程具有化学和区域选择性, 生成一系列的酰胺基环戊烯及其衍生物90.作者提出了可能的反应机理:首先, 铑催化剂与烯基铬卡宾发生转金属交换, 生成活化的烯基Rh(Ⅰ)卡宾配合物和Cr(CO)6.然后与联烯胺加成环化, 经还原消除过程得到环戊烯衍生物, 并再生催化剂.在这里, 作者推测CO在该反应中有两种作用: (1)通过改变反应途径, 提高催化反应效率; (2)可以当量回收Cr(CO)6.作者发现, 当联烯带有富电子基团时, 在铑卡宾的作用下, 联烯胺α碳选择性环化得到[3+2]环加成产物, 若联烯带有的取代基为拉电子基时, 则是联烯胺γ位碳参与反应.
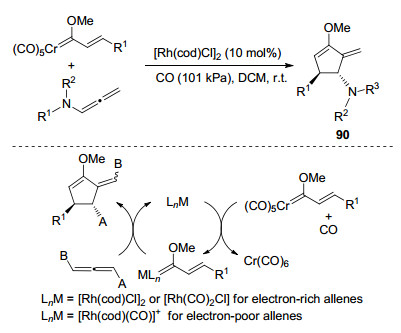
作者探究机理时认为, 无论CO的存在与否, 铑卡宾的获得是氧化铬配合物与[Rh(cod)(naphthalene)]+相互作用的结果.同时, 在反应过程中, 卡宾配合物自身不会发生配体COD-CO的相互交换.该环加成过程具有的区域选择性和非对映选择性, 以及所得环加成物具有易水解的特性, 使得该方法成为制备官能化的3, 4-二取代的2-亚烷基环戊酮的简单而有效的方式.
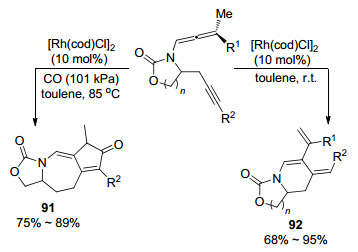
2008年, Brummond课题组[36]首次报道了Rh(Ⅰ)催化炔基联烯胺分子内的环化反应(Scheme 26).与Barluenga等的工作不同点在于:无论取代基的电性如何, 最终只能得到α碳的环加成产物, 且化合物分子内同时含有三个不饱和烯烃, 可以进行更多转化, 因此具有更大的潜在应用价值.在该转化中, 将气氛从氩气转变为一氧化碳时, 反应以Pauson-Khand方式生成具有非对映选择性的复杂三并环结构化合物91.
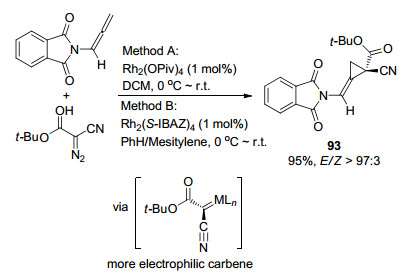
2013年, Charette课题组[37]报道了一例Rh(Ⅱ)催化的联烯胺和α-氰基重氮酯分子间环丙烷化反应(Scheme 27).该反应的区域选择性发生在联烯胺空间位阻更小的端位碳上, 与Barluenga工作类似, 铑催化剂与α-氰基重氮酯生成氰基取代的Rh(Ⅱ)金属卡宾, 相继的环加成和还原消除后得到环丙烷环产物93, 具有较高的E/Z比和ee值.
在过去的几十年中, 虽然联烯分子的[2+2]环加成反应的报道较多, 但头对头、尾对尾、头对尾的区域选择性的控制仍然是一个较大的挑战.最近, 康强课题组和张俊良课题组分别报道了Rh催化的联烯胺[2+2]和[3+2]环加成反应.
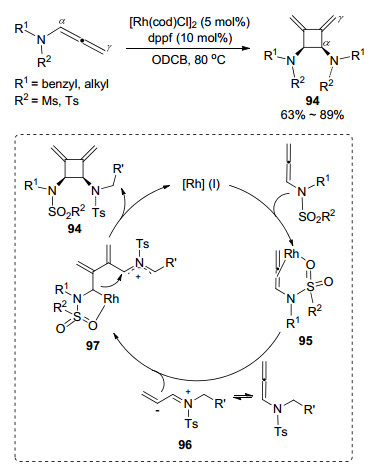
2016年, 康强课题组[38]报道的联烯胺分子自身环加成方式为头对头, 反应生成具有高度区域和立体选择性的反式-二亚甲基环丁烷-二胺及其衍生物(Scheme 28).该反应在高沸点的邻二氯苯为溶剂的条件下实现的, 首先是Rh(Ⅰ)催化剂与磺酰基氧进行配位, 同时与联烯胺反应得到中间体95, 另一分子的联烯胺异构化得到亚胺中间体96.随后, 亚胺中间体作为亲核试剂进攻中间体95上的联烯中心碳原子, 得到偶联中间体97.最后, 消除Rh配合物关环产生[2+2]环加成产物94.值得注意的是, 当联烯胺的α位碳原子上带有取代基时, 反应则不能够顺利进行, 作者认为α位碳原子上的取代基阻止了稳定亚胺中间体的生成, 终止了反应循环.
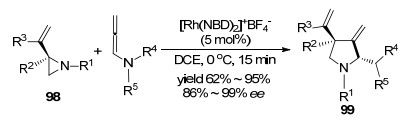
相比于Rh卡宾催化, 张俊良课题组[39]报道的是铑催化联烯胺与烯基氮杂环丙烷化合物间的[3+2]环加成反应(Scheme 29).首先是Rh(Ⅰ)催化剂对环丙烷化合物氧化加成, 开环得到Rh(Ⅲ)中间体, 然后选择性地对联烯胺N邻位C=C双键插入, 分子内环化, 最后经还原消除得到环加成产物99, 并再生Rh(Ⅰ)催化剂参与反应循环, 制备了一系列的手性四氢吡咯类化合物.
近几十年来, 金催化联烯胺的反应的报道较多, 催化联烯胺的反应类型主要有环化反应、环加成反应、不对称催化以及亲核取代等.金催化联烯胺是钯催化之后又一重要的反应类型, 由于金催化的研究报道众多, 在本节中, 我们从环化、[2+2]、[3+2]、[4+2]、[2+2+2]环加成分别进行介绍.
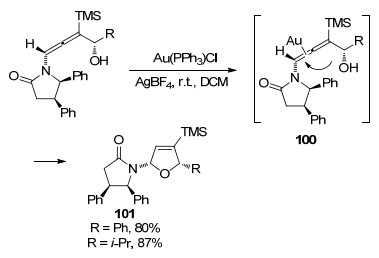
2006年, Hegedus[40]实现了Au催化手性γ-取代的联烯胺化合物环化过程(Scheme 30), 合成一系列高度官能化的顺式二氢呋喃.首先, 金完成对联烯胺的插入, 分子内的氧亲核试剂容易进攻氮原子邻位的碳, 发生分子内亲核取代反应, 环化得到最终的含氧五元产物101.
2007年, Fujii和Ohno研究团队[41]首次实现了Au(Ⅰ)催化联烯胺分子内环化制备二氢喹啉类化合的方法(Scheme 31).作者认为, Au(Ⅰ)催化剂在配体的作用下, 完成对联烯胺的活化, 分子内环化得到烯基金中间体, 由于正离子的不稳定性, 发生质子化转移, 最终进行芳烃异构化, 完成苯并哌啶环的构建.
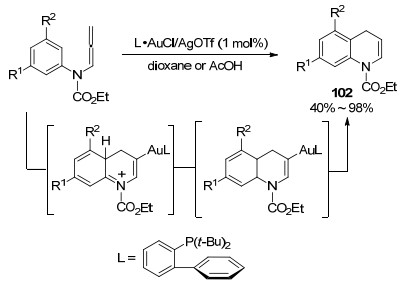
2009年, Pérez-Castells等[42]报道了Au催化的联烯胺和亲核试剂分子间的环化反应(Scheme 32).作者认为金作为催化剂启动反应, 优先完成对丙二烯结构的插入, 而不是像在烯炔环化中的首先对叁键的插入, 在亲核试剂的作用下发生偶联.随后, Au催化剂完成对叁键插入活化, 环化得到最终产物104, 值得注意的是, 如果氮原子上取代基的拉电子效应弱于磺酰基, 该环化过程则不能顺利的进行.
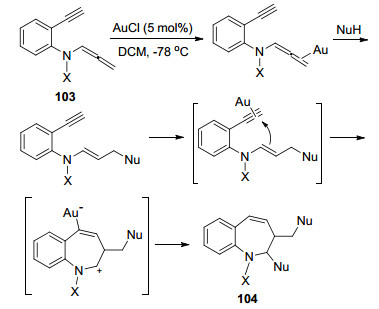
2012年, Hsung课题组[43]报道了一例α取代的联烯胺环化反应(Scheme 33).在作者提出的反应机理中, Au催化剂首先完成对丙二烯的插入, 得到亚胺中间体, 然后发生Imino-Nazarov环化反应, 得到区域选择性产物108.该转化为构建稠合的环戊烯酰胺提供了一种有效的合成方法, 并且产率高达97%.
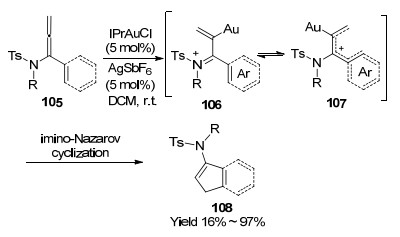
Au催化联烯胺环加成反应, 同样还是有效构建四元、五元、以及七元、八元大环有用的方法之一. 2011年, 陈自立课题组[44]报道了第一例金催化联烯胺[2+2]环加成反应(Scheme 34).该反应是在金催化联烯胺和富电子烯烃完成环加成后实现的, 如果联烯胺的α或者γ位上存在取代基, 则不能够分离到环化产物.作者认为, 由于四元环的张力较大, 如果存在取代基, 较大的空间位阻会阻碍环加成反应的顺利进行.同时, 在不存在富电子烯烃的情况下, 金能够催化联烯胺自身之间以头-尾的方式发生聚合反应, 得到二烯的环丁烷化合物109.为了避免在富电子烯烃的环加成反应中得到二聚化合物, 作者筛选了合适的配体, 在配体调控下, 以更高的区域和立体选择性得到目标产物110.
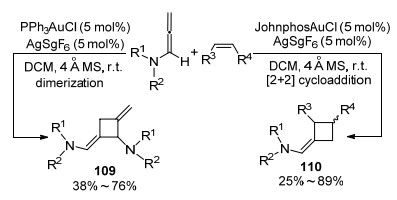
2012年, Mascareñas课题组[45]报道了相似的工作, 即联烯胺和烯烃在金的催化下, 高区域选择性地完成[2+2]环加成反应(Scheme 35).与陈自立课题组工作不同的是, 在Mascareñas教授实现的转化中, 烯烃的选择范围更加广泛, 并且当作者将此环加成方案应用于不同构型的烯烃时, 具有非常好的选择性, 可以得到单一的反式-立体异构体.作者认为该串联反应的机理如下:金催化剂完成对丙二烯的插入, 得到阳离子金中间体111, 接受烯烃的进攻, 再次得到阳离子中间体112.当使用环状或非环状烯酰胺时, 形成更稳定的苄基或N-酰基亚胺鎓阳离子会增加该反应的区域选择性.但是, 由于δ碳-碳键的易旋转性, 得到更小的空间位阻, 使得烯烃立体化学性降低.最后, 在氮原子的辅助下, 完成闭环, 并经过还原消除过程, 再生Au催化剂.
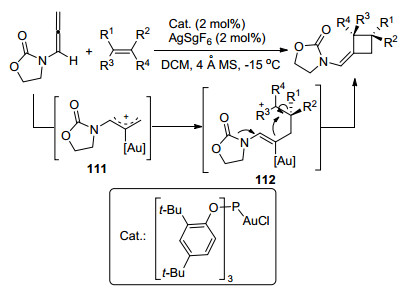
同年, González课题组[46]完成了联烯胺和烯醇醚的金催化[2+2]环加成反应(Scheme 36).与Mascareñas课题组报道的催化剂相同, 但是用量却少很多, 经济节能效果更好.同时得注意的是, 作者还突破了陈自立课题组实现环加成反应的局限性, 即在该策略下, γ位取代的联烯胺同样能够便显出较好的反应性, 得到4-取代环丁烷结构.
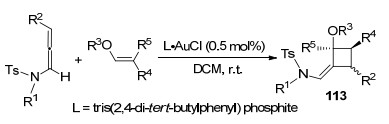
2015年, Bandini课题组[47]实现了金催化联烯胺和吲哚及其衍生物之间的环加成反应(Scheme 37).该转化中所使用的[JohnPhosAu(NCMe)]SbF6催化剂为商业化产品, 容易获取, 并且底物的适应性更好, 条件更温和.同时, 该[2+2]环加成反应的区域和非对映选择性更强.但是, 作者研究发现, 吲哚取代基只能是拉电子基如Boc等, 才能够表现出较好的适应性, 给电子保护基则没有反应性.该方法的实现有助于(-)-tubifoline等天然产物的全合成.
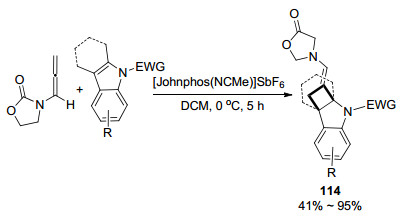
随着对金催化联烯胺研究的不断深入, 环加成反应也被陆续报道. 2013年, 陈自立课题组[48]探索出了联烯胺和硝酮之间[3+2]环加成反应制备手性异噁唑烷115的实用新型方案(Scheme 38).该反应是在改性的BINOL衍生的手性氨基磷酸酯Au(Ⅰ)催化剂催化下实现的, 表现出良好的区域选择性.其中加入过量(5 mol%)的银盐以减少额外的金-硝酮配位数, 并提高金阳离子的催化活性.在联烯胺取代基为Ts等酰基情况下, 配体L表现出较好的适应性, 但是, 当氮原子上的取代基为呋喃酮等环状结构时, 配体L'能展现出更良好的对应选择性.
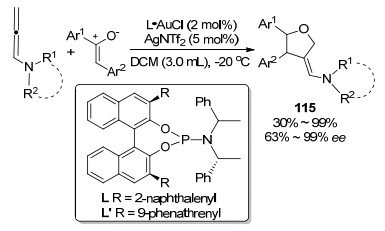
与之前大量报道的联烯胺α碳参与环加成的方式不同, 2015年, 张俊良课题组[49]报道了一例Au(Ⅰ)催化的联烯胺邻位碳碳双键和烯炔基酮化合物116之间的[3+2]环加成反应(Scheme 39).在配体的调控下, 反应所得产物均具有较好的区域选择性和对映选择性.并且在温和的条件下产率优良, 同时, 该转化原料的易得性, 使得该方法在合成有机分子骨架时具有较高的实用性和高效性.
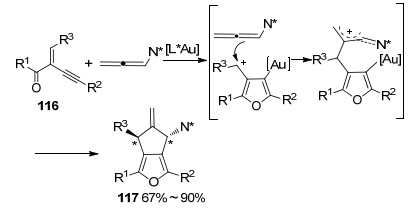
2016年, 刘瑞雄课题组[50]报道了金催化联烯胺[3+2]环加成反应(Scheme 40), α-芳基重氮腈与联烯胺在二氯甲烷中反应产生1-氨基-1H-茚.在底物的适配性探索时, 作者发现, 只有当联烯胺γ位的取代基都为甲基时, 才能使联烯胺氮邻位的C=C双键参与反应, 得到产物119, 这些杂环结构分子的成功获得依赖于α-氰基芳基卡宾碳烯的高亲电子性.当X取代基为卤素时, 会对该转化造成一定影响.
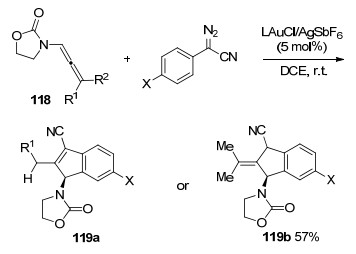
2011年, López和Mascareñas等[51]报道了首例金催化联烯胺和非环状共轭二烯烃之间的[4+2]环加成反应(Scheme 41), 转化表现出良好的原子利用率和较高的Z选择性.联烯胺相比于其他的底物如联烯或者联烯基醚, 具有更大的优越性, 更高的适配性.该方法成功的关键是使用新型轴向手性N-杂环卡宾-金催化剂.该反应为合成各种环己烯化合物提供了一种通用的方法. 2012年, 该小组对[4+2]环加成反应做了进一步的探索[52], 非对映选择性的实现了第一例联烯胺[4+2]环加成(Scheme 41), 在含有轴向手性三唑并异喹啉-3-亚基金配体的调控下, 实现联烯胺和共轭双烯之间的反应, 获得一系列环己烯化合物121.
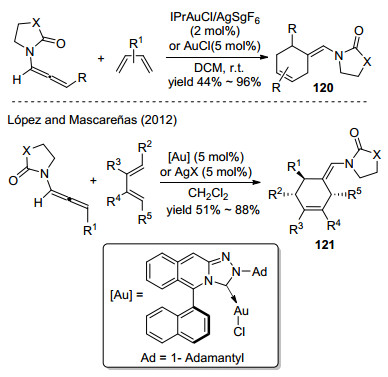
2013年, Vicente课题组[53]实现了JohnPhos-Au催化联烯胺和2-烯基吲哚间的[4+2]加成环化(Scheme 42), 反应得到取代的四氢咔唑类化合物122, 丰富了[4+2]环加成反应方法学.
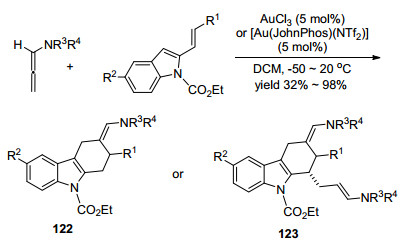
López和Mascareñas等[54]在2013年报道了一例联烯胺与烯基酮之间的新型的环加成反应(Scheme 43).作者通过内部亲核试剂截取阳离子中间体I, 以endo型或者exo型串联环化反应得到稠合或桥接双环骨架.通过该反应途径, 可以实现合成六元-九元的氧桥分子结构.转化经历相似的反应机理:金首先完成对联烯的活化, 得到碳正离子中间体, 随后, 该中间体被底物124中的羰基氧捕获形成桥联中间体, 最后, 烯胺进攻活泼的碳正离子, 环化得到化合物125并再生催化剂, 该反应的配体结构如Scheme 43所示.
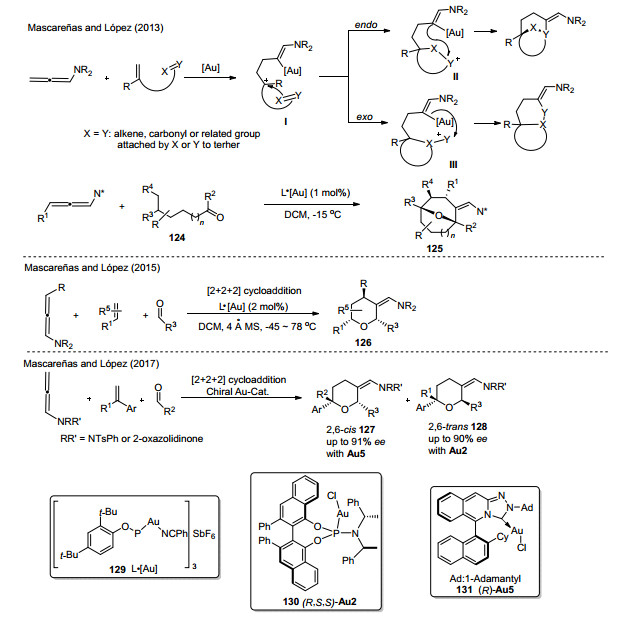
2015年, 该课题组对联烯胺环加成反应进行持续的探索[55], 利用相同的催化剂129 (Scheme 43), 实现了联烯胺和烯烃以及不饱和醛之间三组分的[2+2+2]加成环化, 得到了多取代的四氢吡喃化合物.该分子骨架中的酰胺结构为进一步活化制备生物活性产物Aspergi- llide A和Decytospolide B提供了更多的潜在可能性.同时, 这一策略的实现, 为多组分环加成反应制备含氮、氧杂环开辟了新的途径.
2017年, Mascareñas和López等[56]报道了一种简便直接, 更加具有原子经济性地制备多取代四氢吡喃衍生物的方法(Scheme 43).三组分联烯胺和烯烃以及不饱和醛在手性N-杂环卡宾-金催化剂130或131的催化下, 完成具有非对映选择性和对映选择性的[2+2+2]环加成过程, 并为具有光学活性的2, 6-二取代的cis和trans THPs的合成提供了更加合理有效方法.
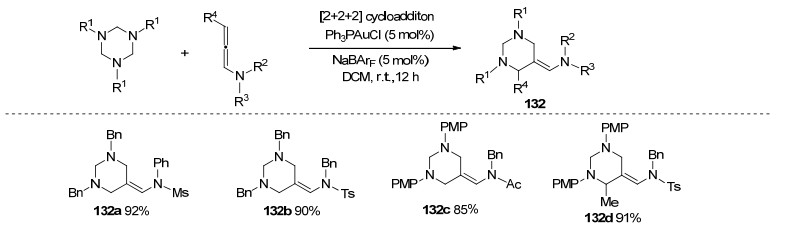
2017年, 孙江涛课题组[57]进一步完善了金催化[2+2+2]加成环化策略(Scheme 44).该反应以PPh3AuCl为催化剂, 1, 3, 5-三嗪与联烯胺在温和条件下转化得到含氮六元杂环化合物132, 产率较高, 同时具有良好的区域选择性.当联烯胺γ位存在取代基时, 联烯胺也能表现出较好的反应性.该工作与张俊良课题组[39]2016年发表工作相似之处在于, 不同的官能化的联烯呈现完全不同的环加成途径.经过控制实验与机理验证表明, Au催化循环经历碳正离子形成、亲核加成、亚胺中间体形成、环化异构化等过程, 逐步完成[2+2+2]加成环化.
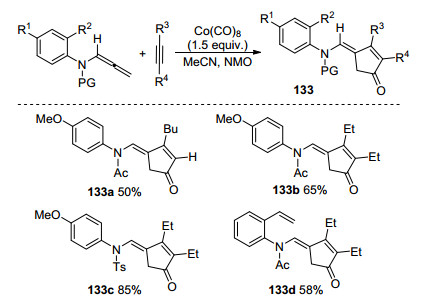
2004年, Pérex-Castells课题组[58]首次实现了钴催化联烯胺与炔烃分子间Pauson-Khand环化反应(Scheme 45), 得到带有E酰胺的官能化环戊烯酮类化合物, 具有良好的产率以及区域选择性.
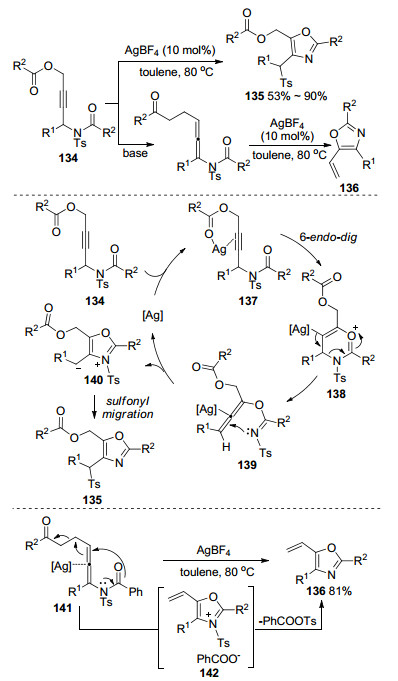
2013年, Wan课题组[59]以N-炔丙基脂134作为底物, 报道了一例Ag催化的联烯胺环化反应(Scheme 46).作者将易脱去的羧基引入底物结构, 通过脱羧反应, 分子内环化得到乙烯基噁唑类五元环化产物.他们对该转化提出了可能的反应机理(Scheme 46):首先, 银催化剂和炔丙基酯相互作用得到π-配合物137, 同时, 酰氧基还与Ag(Ⅰ)配位以促进随后的转化, 经过分子内6-endo-dig对酰胺上的羰基氧进行亲核进攻形成中间体138, 进一步形成联烯中间体139.随后, 亚胺上的氮接受联烯中心碳原子的亲核攻击, 接着产生两性离子中间体140, 通过磺酰基[3, 3]重排的方式得到噁唑化合物135.作者设想在Ag的催化下, 经历和炔丙基酯重排的相似过程, 但结果发现, 并没有得到相应的环化产物.作者认为, 丙二烯结构被Ag(Ⅰ)阳离子活化, 随后在酰胺部分氧原子分子内亲核攻击后环化, 磺酰基和酰氧基两者的消除产生最终的乙烯基噁唑产物136, 由于这一独有的特征, 该反应为合成乙烯基噁唑提供了一种可行的替代方案.
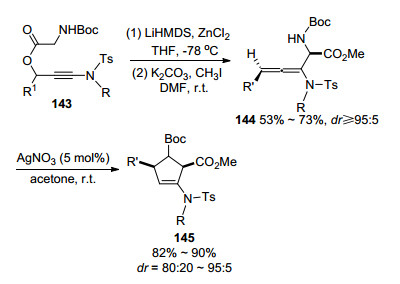
同年, Crossy课题组[60]实现了银催化的联烯胺环化合成3-吡咯啉(Scheme 47).首先, N-Boc保护的炔丙基酯143在碳酸钾以及碘甲烷的存在下, 完成Enolate- Claisen重排, 得到γ取代的联烯胺化合物144.接着在Ag的催化下分子内环化, 高区域选择性地得到环戊烯化合物145.作者认为, 该方法实现了立体选择性的功能化联烯胺的合成, 并且在Ag的催化下可以成为合成吡咯烷行之有效的方法.
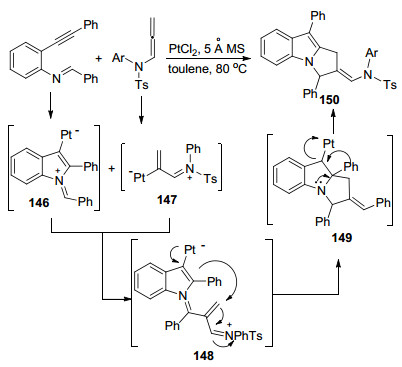
2017年, Patil课题组[61]首次报道了Pt催化联烯胺与亚胺炔基化合物间的[3+2]环化反应(Scheme 48), 实现了联烯胺γ位碳选择性环化, 通过该方法可以制备一系列吡咯[1, 2-a]吲哚化合物.作者同样对反应机理进行了探讨, 首先完成的是Pt对炔烃的活化, 亚胺捕获产生甲亚胺叶立德, 接受联烯胺的进攻, 接下来, Pt(Ⅱ)以1, 4-方式将电子密度贡献给缺电子亚胺离子以产生148, 一旦产生中间体148, 将发生1, 2-芳基迁移以形成中间体149, 最后还原消除完成Pt催化剂的再生, 得到吡咯[1, 2-a]吲哚化合物150.
2017年, 贾义霞课题组[62]首次实现了Ni催化联烯胺与亚胺的[2+2]环加成(Scheme 49), 高对应选择性地生成含季碳中心的四元含氮杂环化合物.该转化是在手性噁唑啉配体的调控下完成的, 作者为该反应拟定了可能的机理.首先, 底物151在手性Ni催化剂的作用下以1, 4-结合的方式活化得到中间体154, 随后对联烯胺选择性亲核加成生成N正离子亚胺中间体155, 然后亚胺结构中的碳接收亲核试剂的进攻, 通过区域选择性的分子内环化, 得到[2+2]环加成产物152 (Path a). Path b中丙烯醛153并不是环加成的产物, 可能是由于使用N-烯丙基噁唑烷酮作为反应底物时, 中间体155发生水解.该[2+2]环加成催化循环的实现, 完善了Ni催化联烯胺环化方法学, 能够高效合成带有四元立体构型中心的多取代手性氮杂环丁烷骨架分子, 同时具有优异的对映选择性.
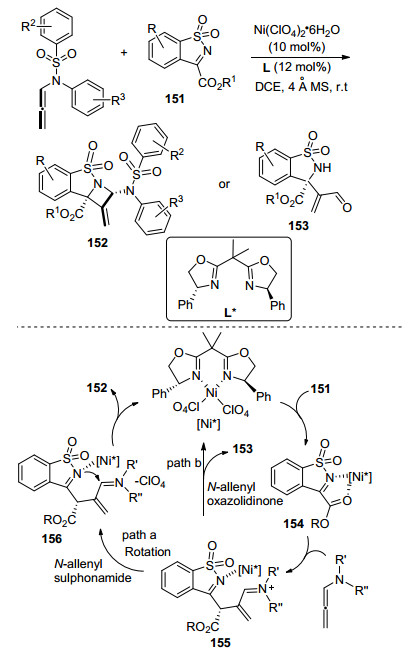
联烯胺类化合物独特的反应特点, 使其在钯、钌、铑、金、钴、银、铂、镍金属催化剂作用下都能高效地选择性地进行环化反应.在不同的碱性环境、配体和溶剂等的影响下, 能够构建不同种类的含有氮原子的杂环化合物.因此, 该联烯胺环化反应在现代药物和天然产物全合成, 以及精细化工合成中都具有非常大的潜在应用价值.联烯胺类化合物具有独特的化学反应性和选择性, 通过改变胺的保护基团可以调控氮原子的电子云密度, 在合适的催化剂和配体作用下, 对不同选择性的反应位点进行环化, 从而构建不同原子数的环化产物.目前, 在联烯胺类化合物环化反应中, 对反应区域选择性的探索并不完善, 还有待化学工作者继续探索, 此外, 在联烯胺环化反应中实现可控的手性碳原子构建也是亟待解决的研究课题.在此, 希望该综述能为化学工作者提供帮助, 促进联烯胺环化领域的发展, 研究出更多新颖优异的方法.
(a) Colacot, T. New Trends in Cross Coupling: Theory and Applications, RSC, Cambridge, UK, 2015.
(b) Meijere, A.; Oestreich, M. Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, Germany, 2014.
(c) Hao, E.; Jiang, X.; Fu, D.; Wang, D.; Xie, M.; Qu, G.; Guo, H. Chin. J. Org. Chem. 2016, 36, 2746 (in Chinese).
(郝二军, 蒋小涵, 付丹丹, 王东超, 谢明胜, 渠桂荣, 郭海明, 有机化学, 2016, 36, 2746.)
(d) Liu, C.; Liu, G.; Zhao, H. Chin. J. Chem. 2016, 34, 1048.
(e) Dong, X.; Hou, Y.; Meng, F.; Liu, H.; Liu, H. Chin. J. Org. Chem. 2017, 37, 1088 (in Chinese).
(董旭, 侯永正, 孟凡威, 刘洪波, 刘会, 有机化学, 2017, 37, 1088.)
(f) Tang, H. M.; Huo, X. H.; Meng, Q. H.; Zhang, W. B. Acta Chim. Sinica 2016, 74, 219 (in Chinese).
(汤淏溟, 霍小红, 孟庆华, 张万斌, 化学学报, 2016, 74, 219.
Grigg, R. Sridharan, V. Xu, L.-H. J. Chem. Soc., Chem. Commun. 1995, 1903.
Grigg, R.; Köppen, I.; Rasparini, M.; Sridharan, V. Chem. Commun. 2001, 96.
Grigg, R.; McCaffrey, S.; Sridharan, V.; Fishwick, C. W. G.; Kilner, C.; Korn, S.; Bailey, K.; Blacker, J. Tetrahedron 2006, 62, 12159. doi: 10.1016/j.tet.2006.09.098
Inamoto, K.; Yamamoto, A.; Ohsawa, K.; Hiroya, K.; Sakamoto, T. Chem. Pharm. Bull. 2005, 53, 1502. doi: 10.1248/cpb.53.1502
Husinec, S.; Petkovic, M.; Savic, V.; Simic, M. Synthesis 2012, 44, 399. doi: 10.1055/s-0031-1289658
Xie, Z.; Wu, P.; Cai, L.; Tong, X. Tetrahedron Lett. 2014, 55, 2160. doi: 10.1016/j.tetlet.2014.02.087
Yan, F.; Liang, H, ; Song, J.; Cui, J.; Liu, Q.; Liu, S.; Wang, P.; Dong, Y.; Liu, H. Org. Lett. 2017, 19, 86. doi: 10.1021/acs.orglett.6b03364
Beccalli, E. M.; Bernasconi, A.; Borsini, E.; Broggini, G.; Rigamonti, M.; Zecchi, G. J. Org. Chem. 2010, 75, 6923 doi: 10.1021/jo101501u
Grigg, R.; Loganathan, V.; Sridharan, V.; Stevenson, P.; Sukirthalingam, S.; Worakun, T. Tetrahedron 1996, 52, 11479. doi: 10.1016/0040-4020(96)00638-2
Gardiner, M.; Grigg, R.; Sridharan, V.; Vicker, N. Tetrahedron Lett. 1998, 39, 435. doi: 10.1016/S0040-4039(97)10541-X
Fuwa, H.; Sasaki, M. Org. Biomol. Chem. 2007, 5, 2214. doi: 10.1039/b707338k
Grigg, R.; Sansano, J. M. Tetrahedron 1996, 52, 13441. doi: 10.1016/0040-4020(96)00801-0
Parthasarathy, K.; Jeganmohan, M.; Cheng, C. Org. Lett. 2006, 8, 621. doi: 10.1021/ol0527936
Cao, J.; Kong, Y.; Deng, Y.; Lai, G.; Cui, Y.; Hu, Z.; Wang, G. Org. Biomol. Chem. 2012, 10, 9556. doi: 10.1039/c2ob26727f
Grigg, R.; Sansano, J. M.; Santhakumar, V.; Sridharan, V.; Thangavelanthum, R.; Thornton-Pett, M.; Wilson, D. Tetrahedron 1997, 53, 11803. doi: 10.1016/S0040-4020(97)00754-0
Fuwa, H.; Sasaki, M. Org. Biomol. Chem. 2007, 5, 2214. doi: 10.1039/b707338k
Liang, H.; Yan, F.; Dong, X.; Liu, Q.; Wei, X.; Liu, S.; Dong, Y.; Liu, H. Chem. Commun. 2017, 53, 3138. doi: 10.1039/C7CC00191F
Zhu, S.; Cao, J.; Wu, L.; Huang, X. J. Org. Chem. 2012, 77, 1049.
(a) Zhu, C.; Yang, B.; Jiang, T.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2015, 54, 9066.
(b) Qiu, Y.; Yang, B.; Zhu, C.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2016, 128, 6630.
Persson, A. K. Å.; Bäckvall, J.-E. Angew. Chem., Int. Ed. 2010, 49, 4624. doi: 10.1002/anie.201000726
(a) Arisawa, M.; Terada, Y.; Takahashi, K.; Nakagawa, M.; Nishida, A. J. Org. Chem. 2006, 71, 4255.
(b) Donohoe, T. J.; O'Riordan, T. J. C.; Rosa, C. P. Angew. Chem., Int. Ed. 2009, 48, 1014.
Poeylaut-Palena, A. A.; Testero, S. A.; Mata, E. G. Chem. Commun. 2011, 47, 1565. doi: 10.1039/C0CC04115G
Edlin, C. D.; Faulkner, J.; Quayle, P. Tetrahedron Lett. 2006, 47, 1145. doi: 10.1016/j.tetlet.2005.12.018
Bokka, A.; Hua, Y.; Berlin, A. S.; Jeon, J. ACS Catal. 2015, 5, 3189. doi: 10.1021/acscatal.5b00431
(a) Kim, B. G.; Snapper, M. L. J. Am. Chem. Soc. 2006, 128, 52.
(b) Peppers, B. P.; Diver, S. T. J. Am. Chem. Soc. 2004, 126, 9524.
Zeng, X.; Wei, Z.; Farina, V.; Napolitano, E.; Xu, Y.; Zhang, L.; Haddad, N.; Yee, N. K.; Grinberg, N.; Shen, S.; Senanayake, C. H. J. Org. Chem. 2006, 71, 8864. doi: 10.1021/jo061587o
Dornan, P. K.; Wickens, Z. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2015, 54, 7134. doi: 10.1002/anie.201501505
Gavenonis, J.; Arroyo, R. V.; Snapper, M. L. Chem. Commun. 2010, 46, 5692. doi: 10.1039/c0cc00008f
(a) Rosillo, M.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. Tetrahedron Lett. 2001, 42, 7029.
(b) Rosillo, M.; Domínguez, G.; Casarrubios, L.; Amador, U.; Pérez-Castells, J. J. Org. Chem. 2004, 69, 2084.
(a) López, F.; Delgado, A.; Rodríguez, J. R.; Castedo, L.; Mascareñas, J. L. J. Am. Chem. Soc. 2004, 126, 10262.
(b) Arisawa, M.; Fujii, Y.; Kato, H.; Fukuda, H.; Matsumoto, T.; Ito, M.; Abe, H.; Ito, Y.; Shuto, S. Angew. Chem., Int. Ed. 2013, 52, 1003.
Desroy, N.; Robert-Peillard, F.; Toueg, J.; Hénaut, C.; Duboc, R.; Rager, M.-N.; Savignac, M.; Gênet, J.-P. Synthesis 2004, 2665.
Kinderman, S. S.; Van Maarseveen, J. H.; Schoemaker, H. E.; Hiemstra, H.; Rutjes, F. P. T. Org. Lett. 2001, 3, 2045. doi: 10.1021/ol016013e
Nada, T.; Yoneshige, Y.; Ii, Y.; Matsumoto, T.; Fujioka, H.; Shuto, S.; Arisawa, M. ACS Catal. 2016, 6, 3168. doi: 10.1021/acscatal.6b00628
Barluenga, J.; Vicente, R.; López, L. A.; Tomás. M. J. Am. Chem. Soc. 2006, 128, 7050.
Brummond, K. M.; Yan, B. Synlett 2008, 2303.
Lindsay, V. N. G.; Fiset, D.; Gritsch, P. J.; Azzi, S.; Charette, A. B. J. Am. Chem. Soc. 2013, 135, 1463. doi: 10.1021/ja3099728
Zheng, W.; Bora, P. P.; Sun, G.; Kang. Q. Org. Lett. 2016, 18, 3694. doi: 10.1021/acs.orglett.6b01731
Lin, T.; Zhu, C.; Zhang, P.; Wang, Y.; Wu, H.; Feng, J.; Zhang, J. Angew. Chem., Int. Ed. 2016, 55, 10844. doi: 10.1002/anie.201605530
Hyland, C. J. T.; Hegedus, L. S. J. Org. Chem. 2006, 71, 8658. doi: 10.1021/jo061340r
Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Org. Lett. 2007, 9, 4821. doi: 10.1021/ol702179n
González-Gómez, A.; Domínguez, G.; Pérez-Castells, J. Eur. J. Org. Chem. 2009, 5057.
Ma, Z.; He, S.; Song, W.; Hsung, R. P. Org. Lett. 2012, 14, 5736. doi: 10.1021/ol302743k
Li, X.; Zhu, L.; Zhou, W.; Chen. Z. Org. Lett. 2012, 14, 436. doi: 10.1021/ol202703a
Faustino, H.; Bernal, P.; Castedo, L.; López, F.; Mascareñas, J. L. Adv. Synth. Catal. 2012, 354, 1658. doi: 10.1002/adsc.201200047
Suarez-Pantiga, S.; Hernández-Díaz, C.; Piedrafita, M.; Rubio, E.; Gonzáleza, J. M. Adv Synth. Catal. 2012, 354, 1651. doi: 10.1002/adsc.201200043
Ocello, R.; Nisi, A. D.; Jia, M.; Yang, Q.; Monari, M.; Giacinto, P.; Bottoni, A.; Miscione, G. P.; Bandini. M. Chem. Eur. J. 2015, 21, 18445. doi: 10.1002/chem.201503598
Li, G.; Zhou, E.; Li, X.; Bi, Q.; Wang, Z.; Zhao, Z.; Hu, W.; Chen, Z. Chem. Commun. 2013, 49, 4770. doi: 10.1039/c3cc41769g
Wang, Y.; Zhang, P.; Qian, D.; Zhang, J. Angew. Chem., Int. Ed. 2015, 54, 14849. doi: 10.1002/anie.201507165
Singh, R. R.; Pawar, S. K.; Huang, M.; Liu, R. Chem. Commun. 2016, 52, 11434. doi: 10.1039/C6CC04308A
Faustino, H.; López, F.; Castedo, L.; Mascareñas, J. L. Chem. Sci. 2011, 2, 633. doi: 10.1039/c0sc00630k
Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigüenza, J.; Díez, E.; Alonso, I.; Fernández, R.; Lassaletta, J. M.; López, F.; Mascareñas, J. L. J. Am. Chem. Soc. 2012, 134, 14322. doi: 10.1021/ja3065446
Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594. doi: 10.1039/c3cc41514g
Faustino, H.; Alonso, I.; Mascareñas, J. L.; López, F. Angew. Chem., Int. Ed. 2013, 52, 6526. doi: 10.1002/anie.201302713
Faustino, H.; Varela, I.; Mascareñas, J. L.; López, F. Chem. Sci. 2015, 6, 2903. doi: 10.1039/C5SC00295H
Varela, I.; Faustino, H.; Díez, E.; Iglesias-Sigüenza, J.; Grande- Carmona, F.; Fernandez, R.; Lassaletta, J. M.; Mascareñas, J. L.; López. F. ACS Catal. 2017, 7, 2397. doi: 10.1021/acscatal.6b03651
Peng, S.; Cao, S.; Sun, J. Org. Lett. 2017, 19, 524. doi: 10.1021/acs.orglett.6b03691
Anorbe, L.; Poblador, A.; Domínguez, G.; Pérez-Castells, J. Tetrahedron Lett. 2004, 45, 4441. doi: 10.1016/j.tetlet.2004.04.061
Hu, Y.; Yi, R.; Wu, F.; Wan, B. J. Org. Chem. 2013, 78, 7714. doi: 10.1021/jo401330t
Brioche, J.; Meyer, C.; Cossy, J. Org. Lett. 2013, 15, 1626. doi: 10.1021/ol400402n
Chakrabarty, I.; Inamdar, S. M.; Akram, M. O.; Gade, A. B.; Banerjee, S.; Berac, S.; Patil, N. T. Chem. Commun. 2017, 53, 196. doi: 10.1039/C6CC07874E
Liu, R.; Hu, J.; Hong, J.; Lu, C.; Gao, J.; Jia, Y. Chem. Sci. 2017, 8, 2811. doi: 10.1039/C6SC05450A

图式 3 钯催化联烯胺分子内亲核进攻环化
Scheme 3 Pd catalyzed the intramolecular nucleophilic offensive cyclization of allenamides

图式 4 钯-铟双金属催化的联烯胺环化反应
Scheme 4 Palladium/indium bimetallic catalyzed allenamides cyclization

图式 5 钯催化联烯胺与芳基碘化物的分子间环化反应
Scheme 5 Pd-catalyzed intermolecular cyclization of allenamides with aryl iodides

图式 6 钯催化联烯胺与芳基碘化物的选择性异构化反应
Scheme 6 Pd-catalyzed selective isomerization of allenamides with aryl iodides

图式 7 钯催化联烯胺与醇之间的亲核进攻反应
Scheme 7 Pd-catalyzed cyclization of vinyl iodide-tethered allensulfonamides

图式 8 钯催化联烯胺与芳基碘间环化Heck反应
Scheme 8 Pd-catalyzed cyclization-Heck of allenamides and aryl iodides

图式 10 钯催化联烯胺分子间Diels-Alder环加成反应
Scheme 10 Pd-catalyzed Diels-Alder cycloaddition of allenamides

图式 11 钯催化联烯胺与降冰片二烯Diels-Alder反应
Scheme 11 Pd-catalyzed Diels-Alder cycloaddition of allenamides and 2, 5-norbornadiene

图式 12 钯催化联烯胺分子间Diels-Alder环加成反应构建吲哚衍生物
Scheme 12 Synthesis of indole derivatives by Diels-Alder cycloaddition of Pd-catalyzed allenamides

图式 13 锡烷基化-碳钯化串联反应构建含氮、氧杂环的反应
Scheme 13 Tin alkylation-carbon palladium tandem reaction for the reaction of nitrogen and oxygen heterocycles

图式 15 钯催化通过Suzuki-Miyaura偶联的水解/环化反应
Scheme 15 Pd-catalyzed carbopalladation/cyclization via Suzuki-Miyaura coupling

图式 16 钯催化Suzuki-Miyaura偶联环化反应
Scheme 16 Pd-catalyzed cyclization of Suzuki-Miyaura coupling

图式 17 钯催化联烯胺环化构建2, 3取代吲哚反应
Scheme 17 Pd-catalyzed allenamides cyclization to build 2, 3 substituted indole reactions

图式 18 钯催化联烯胺Metallo-Ene/Suzuki串联反应机理
Scheme 18 Cascade reaction mechanism of Pd-catalyzed allenamides Metallo-Ene/Suzuki

图式 19 一锅法合成取代的异吲哚啉衍生物的反应
Scheme 19 One-pot synthesis of substituted isoindoline derivatives

图式 21 Bäckvall课题组新型氧化联烯胺环化
Scheme 21 Novel oxidation of allene condensates by Bäckvall's group

图式 23 Hiemstra对联烯胺的RCM反应探索
Scheme 23 Hiemstra's exploration for RCM reaction of allenamides

图式 24 Arisaw钌催化联烯胺分子内的[2+2]环加成反应与机理
Scheme 24 [2+2] cycloaddition in Arisaw's Ru catalyzed allenamides

图式 25 铑卡宾催化剂催化联烯胺环加成反应
Scheme 25 Rhodium carbene catalyst catalyzed cycloaddition reaction of allenamides

图式 27 铑催化的联烯胺和α-氰基重氮酯分子间环丙烷化反应
Scheme 27 Rh(Ⅱ) catalyzed cyclopropanation of allenamides and α-cyanodiazonium esters

图式 29 Zhang课题组铑催化的联烯胺环加成反应
Scheme 29 Rh-catalyzed ring cycloaddition reaction of allenamides from Zhang group

图式 30 金催化手性γ-取代的联烯胺化合物环化过程
Scheme 30 Au catalyzed cyclization of chiral γ-substituted allenamide compounds

图式 31 联烯胺分子内环化制备二氢喹啉类化合的方法
Scheme 31 Method for preparing dihydroquinoline compound by intramolecular cyclization of allenamides

图式 32 金催化的联烯胺和亲核试剂分子间的环化反应
Scheme 32 Au-catalyzed cyclization between allenamides and nucleophile molecules

图式 33 金催化α位芳基取代联烯胺的Imino-Nazarov环化反应
Scheme 33 Au-catalyzed Imino-Nazarov cyclization of a-aryl substituted allenamide

图式 34 金催化联烯胺与烯烃的[2+2]环加成反应
Scheme 34 Au-catalyzed allenamides [2+2] cycloaddition with olefins

图式 35 金催化联烯胺与烯烃间[2+2]环加成反应
Scheme 35 Au-catalyzed [2+2] cycloaddition reaction between allenamides and olefins

图式 36 金催化联烯胺与烯醇醚[2+2]环加成反应
Scheme 36 Au-catalyzed [2+2] cycloaddition of allenamides with enol ethers

图式 37 金催化联烯胺和吲哚及其衍生物之间的[2+2]环加成反应
Scheme 37 Au-catalyzed [2+2] cycloaddition between allenamides and hydrazines derivatives

图式 38 金催化联烯胺和硝酮之间[3+2]环加成反应
Scheme 38 Au-catalyzed [3+2] cycloaddition reaction of allenamides with nitrones

图式 39 金催化的联烯胺和烯炔基酮化合物之间的[3+2]环加成反应
Scheme 39 Au(Ⅰ) catalyzed [3+2] cycloaddition reaction between allenamides and alkynyl ketone compounds

图式 40 金催化联烯胺与α-芳基重氮腈间[3+2]环加成反应
Scheme 40 Au-catalyzed allenamines with α-aryl diazonitriles [3+2] cycloaddition reaction

图式 41 金催化联烯胺和非环状共轭二烯烃之间的[4+2]环加成反应
Scheme 41 Au-catalyzed [4+2] cycloaddition between allenamides and acyclic conjugated

图式 42 金催化联烯胺和2-烯基吲哚间的[4+2]环加成反应
Scheme 42 Au-catalyzed [4+2] cycloaddition reaction between allenamides and 2-vinyl indoles

图式 43 金催化新型多组分环加成反应
Scheme 43 Au-catalyzed novel multicomponent cycloaddition reaction

图式 46 银催化联烯胺活化脱羧环化反应
Scheme 46 Ag-catalyzed allenamides activation decarboxylation cyclization

图式 47 Enolate-Claisen重排制备联烯胺及其环化反应
Scheme 47 Enolate-Claisen rearrangement for allenamides cyclization

图式 48 铂催化联烯胺与亚胺炔基化合物间的[3+2]环化反应
Scheme 48 Pt-catalyzed [3+2] cyclization reaction between allenamides and iminoalkynyl compounds

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:










 下载:
下载: