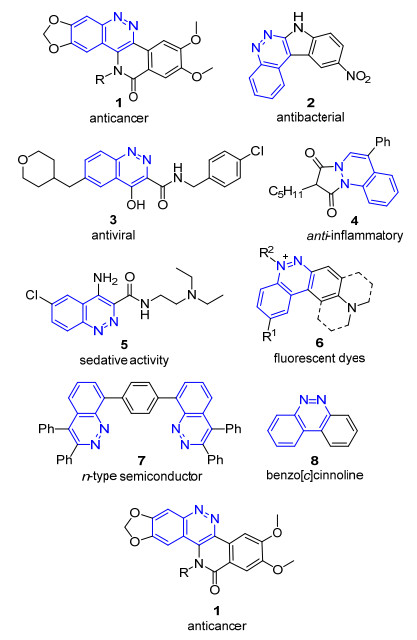
图 1.
代表性的噌啉类功能性分子
Figure 1.
Representative functional molecules of cinnolines

苯并[c]哒嗪(benzo[c]pyridazine), 俗称噌啉(cinnoline), 是一类重要的含氮有机杂环化合物(以下统称噌啉).噌啉本身具有细胞毒性, 对Escherichia coli菌种的生长有抑制作用[1], 引入取代基或并环结构的噌啉衍生物展现出更为广泛的生物活性[2], 例如抗癌(1)[3]、抗菌(2)[4]、抗病毒(3)[5]、抗炎(4)[6]和镇静作用(5)[7]等(图 1), 因此, 它已经成为药物研发中的“优势骨架”.此外, 由于噌啉环是一类良好的电子受体, 具有优良的电子传导能力, 因此噌啉分子及其配合物在光电材料的开发方面也有较为广阔的应用空间.例如化合物6系列是我国科学家研发的一类新型荧光染料[8], 化合物7被用于n型半导体的开发[9], 而苯并[c]噌啉类化合物8则被广泛应用于有机发光二极管的研究[10].
自然界中没有天然的噌啉类化合物存在, 所有文献中报道的噌啉类化合物都是通过有机合成得到的.过去几十年, 由于合成方法有限, 远远不能满足高效合成结构多样性的噌啉衍生物的需求, 因此, 对于噌啉类化合物的合成综述报道十分有限[11], 并且主要集中于对传统方法的总结.近十年来, 随着金属催化的偶联反应日渐成熟以及C—H活化策略的迅猛发展, 化学家们在传统方法的基础上应用这些新兴的策略建立了许多合成噌啉类化合物的新方法, 取得了较好的效果.对噌啉类化合物合成的传统方法和近年来发展的新方法进行了归纳, 并按照不同的合成策略和反应底物进行分类, 综述了近年来噌啉类化合物合成研究的最新进展(截至2018年7月).希望通过这种新的分类归纳思路, 从中获得一些启发, 从而设计出更好的合成方法.
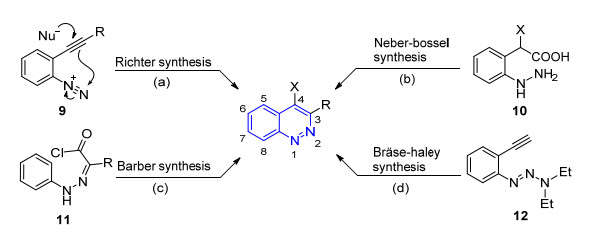
目前已有五篇论文对噌啉类化合物的经典合成方法进行了综述, 包括2017年两篇较新的综述论文.为避免赘述的同时又能够保持本文的完整性, 我们按照合成策略对传统合成方法进行归纳总结.如Scheme 1所示, 噌啉类化合物的经典合成方法按照合成策略主要分为四大类.
Richter合成法——邻炔基苯基重氮盐的分子内环化反应(Scheme 1a).这种方法是噌啉合成的最早方法, 由Richter于1883年完成[12].其中邻位炔基作为给电子体进攻分子内的重氮基团, 在体系中添加亲核试剂可以得到不同类型的3, 4-二取代噌啉类化合物[13].随后, 化学家们又将分子内的给电子基团拓展至烯基[14]和酮类(烯醇化)[15].目前, 这种合成策略已经成为3, 4-二取代噌啉类化合物最重要的合成方法之一, 在药物合成中有广泛的应用[16], 化合物1的合成即应用了这种方法[3, 17].近年来, 化学家们采用亚硝酸叔丁酯(TBN)合成重氮盐中间体, 可以避免强酸的使用, 使反应条件变得比较温和, 在室温下即可顺利进行[14e, 18].
Neber-Bossel合成法——邻肼基苯乙酸的分子内缩合反应(Scheme 1b).这种方法需要在酸性条件下缩合, 因此限制了其广泛使用[19].随后, 化学家们将亲电基团更换为炔基[20]、羰基[21]等官能团, 实现了类似转化. 2003年, Gomaa小组[22]将羧基替换为氰基, 无需酸性条件, 可以得到3-氨基取代的噌啉化合物.
Barber合成法——苯腙化合物的分子内傅克反应(Scheme 1c).这种策略由Barber小组[23]首先发现, 他们采用TiCl4催化酰氯的分子内傅克反应, 实现了3-羟基噌啉类化合物的合成.在此基础上, 化学家们将酰氯基团替换为醛基[24]、氰基[25]和酮类[26]等亲电官能团, 也实现了类似转化.这种方法在药物合成中得到了广泛的应用[27], 化合物3和5的合成即应用了该种方法.
Bräse-Haley合成法——邻炔基苯基三氮烯的分子内环化反应(Scheme 1d).这种方法本质上是Richter合成法的衍生[28], 1999年, Bräse小组[29]首次将其应用于固相合成.该方法的特点在于用氨基保护的重氮盐以稳定的三氮烯12的形式存在, Haley小组[30]通过控制反应条件可以将其分别转化为噌啉和吲唑化合物, 由于噌啉是热力学稳定产物, 需要在200 ℃高温下才能得到.
苯并[c]噌啉是一类重要的噌啉类化合物, 被广泛应用于有机光电材料的研究[10].由于结构的特殊性, 它们的合成方法也区别于普通的噌啉类化合物, 因此我们按照合成策略对其合成方法进行归纳总结.如Scheme 2所示, 苯并[c]噌啉类化合物的合成方法主要分为以下四类.
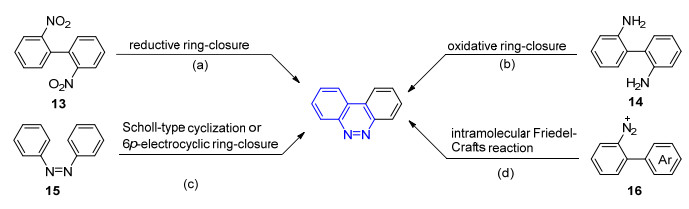
2, 2'-二硝基联苯的还原环化反应(Scheme 2a).采用Na2S[31a]、LiAlH4[31b]、Sm[32]或Pd/C[33]等不同的还原剂使13系列底物发生还原环化反应, 是合成苯并[c]噌啉类化合物最早的方法之一.尽管这种策略的合成应用较为广泛[34], 但在此类反应中难以避免N-氧化物副产物的生成[35].
2, 2'-二氨基联苯的氧化环化反应(Scheme 2b). Corbett和Holt[36]在早期报道了以NaBO3为氧化剂的此类反应, 但反应收率较低;在此基础上, Caronna小组[37]以m-CPBA氧化底物后再发生还原反应, 能以较好的收率得到苯并[c]噌啉类衍生物; 2015年, Minakata小组[38]使用t-BuOCl作为氧化剂, 能够在不产生N-氧化物副产物的条件下一步合成目标产物.最近, Cho小组[39]采用TBN生成重氮盐的策略, 在室温条件下以中等至较好的收率得到目标化合物, 并且对于酯基、氰基、硝基、羰基和卤素等基团耐受良好.
偶氮苯的Schroll反应或[6π]环化反应(Scheme 2c). 1963年, Went小组[40a]采用AlCl3作为催化剂(Schroll反应), 在加热条件下得到了一系列噌啉聚环类化合物, 但由于E式构型的偶氮不发生反应, 因此收率普遍较低.随后, Müller小组[41]采用光照条件发生[6π]环化反应, 使得收率有了明显改善(光照可以促使E式构型的偶氮转化为Z式). 2013年, Sharma小组[42]同样采用光照条件, 以77%~86%的收率得到10个噌啉并环类化合物, 这些化合物表现出与对照药吲哚美辛相当的抗炎活性.
邻芳基苯基重氮盐的分子内傅克反应(Scheme 2d).这种方法本质上也是Richter合成法的衍生, 1970年, Williams小组[43]采用该方法合成了2-甲氧基苯并[c]噌啉; 2003年, LaVoie小组[44]利用这种策略合成了一系列苯并[c]噌啉类衍生物, 并以拓扑异构酶I为靶标对这些化合物的细胞毒性进行了评价; 2015年, 杨有军小组[8]应用这一方法合成了一系列具有较大斯托克斯位移的新型荧光团分子6.这种合成方法具有条件温和、反应收率高、官能团耐受性好等优点, 具有一定的应用空间[45].
以上介绍的经典合成方法在噌啉骨架分子的合成中占有重要地位, 但同时也存在原料制备复杂、需强酸强碱性反应条件和反应温度高等缺点.因此, 这些方法的改进[14e, 18]以及发展原料廉价易得、条件温和、避免强酸强碱使用的新方法是噌啉类化合物合成研究的发展方向.以下我们将近年来开发的新方法按照合成策略分为四大类: (1)金属催化的偶联反应、(2)炔烃/烯烃的分子内芳氢化反应、(3) C—H活化策略和(4)其他策略, 并逐一进行介绍.
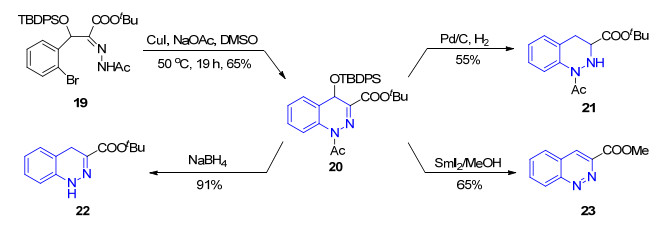
2008年, Nishida小组[46]采用腙17作为底物, 利用CuI催化的分子内Ullman反应以定量的收率实现了3-酯基噌啉18的合成(Eq. 1).当采用乙酰基取代腙19时, 可以中等收率得到对应的二氢噌啉类化合物20, 并且通过控制反应条件分别转化为四氢噌啉21, 二氢噌啉22和3-酯基噌啉23 (Scheme 3).这种方法条件温和, 避免了强酸强碱的使用, 但是原料的制备较为复杂.
2011年, Yamane小组[47]采用邻碘苯基三氮烯(24)作为底物, 在零价钯催化下与双取代炔25发生串联环化反应, 以中等至较好的收率得到多取代的噌啉类化合物26 (Eq. 2).该反应对酯基、氰基、硝基、羰基等基团耐受性良好, 但当采用不对称炔作为原料时, 区域选择性的问题仍有待解决.
|
|
(1) |
|
|
(2) |
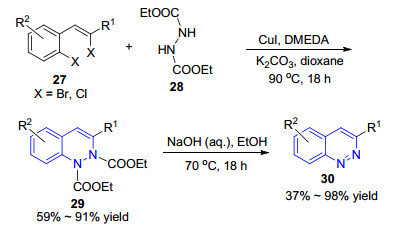
2012年, Willis小组[48]利用Cu(Ⅰ)催化的串联反应, 采用二卤代物27和1, 2-肼二羧酸二乙酯(28)作为原料, 通过两次C—N键的形成实现了3-取代二氢噌啉类化合物29的合成, 取得了59%~91%的收率.通过后续在碱性乙醇溶液中的脱保护-芳香化步骤, 进一步得到了多种3位官能团化的噌啉类化合物30 (Scheme 4).这种分子间C—N偶联法与传统方法相比, 原料制备简单, 操作简便, 为噌啉类化合物的多样性合成提供了新的思路.
2018年, Likhar小组[49]以碘代的三氮唑31为底物, 经Sonogashira偶联和分子内环化的串联反应, 获得了一系列全新结构的1, 2, 3-三唑并噌啉类化合物32, 收率60%~85% (Eq. 3).该反应具有优异的区域选择性[50], 选择性地发生2位氮原子对炔基的进攻.
|
|
(3) |
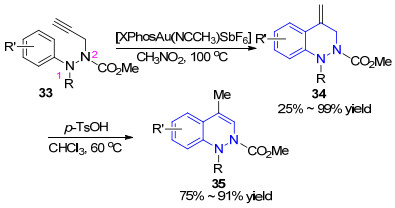
2011年, Gagosz小组[51]报道了一例Au(Ⅰ)催化的炔烃分子内芳氢化反应合成噌啉骨架的新方法.虽然底物33中2位的N原子容易与金催化剂发生配位作用, 但实际上并未对反应发生干扰.该工作首先以25%~99%的收率完成了对噌啉骨架的构建, 随后在对甲苯磺酸的作用下发生烯烃移位反应得到产物35 (Scheme 5).该方法采用的底物较易合成, 但当R基团为芳基或R'基团的取代位置不在芳环4位时, 反应存在的区域选择性问题尚待解决.
2014年, Gogoi小组[52]采用类似策略, 以烯醛苯腙36为底物, 在AlCl3催化下发生分子内的烯烃芳氢化反应(Eq. 4)得到系列4-苄基噌啉类化合物37.
2012年, Ge小组[53]以苯基腙类化合物38作为反应底物, 发展了一类铜催化的分子内氧化去氢偶联反应, 经sp3-C—H键氧化-环化-芳构化的级联反应过程, 实现了3位芳基取代的噌啉类化合物39的直接构建.这也是首例通过腙类化合物中sp3-C—H键的官能团化实现的铜催化的偶联反应, 产率为47%~96% (Eq. 5).该反应通过自由基机理实现, 对卤素、烷基、强吸电子基(CN、CF3)以及给电子基等都具有很好的适应性, 能够一步构建噌啉环骨架.当R2基团处于间位时, 得到具有一定比例的5位和7位取代的噌啉混合物.为避免在C—H键的氧化过程中产生选择性问题, 设计的底物中R1基团均为芳基, 是一种合成3位芳基取代的噌啉化合物的有效方法.
|
|
(4) |
|
|
(5) |
2017年, 许斌小组[54]采用醋酸铜水合物作为催化剂, 以空气作为氧化剂, 通过分子内的氧化去氢偶联-芳构化反应实现了3-取代噌啉类化合物41的合成.这一方法的原料易于制备, 反应操作简单, 具有潜在的应用价值, 反应收率为35%~97% (Eq. 6).值得一提的是, 当C-3位为芳基或烷基取代时均能取得较好的收率, 但C-3和C-4位同时引入取代时收率大幅下降.有趣的是, 当R1基团所在的芳环邻位有F、OMe、NMe2等官能团取代时, 即使另一个邻位的C—H键存在, 反应也能选择性地发生相对惰性的C—F、C—O、C—N键的断裂.
|
|
(6) |
2014年, 张翱小组[55]以上市药物依达拉奉(42)和碘苯类化合物43作为反应底物, 在钯催化剂和醋酸银的作用下发生第一次C—H活化反应, 完成了C—C键的构建; 在此基础上, 进一步发生钯催化的C—H/N—H氧化去氢偶联反应, 完成了C—N键的构建.他们采用这种方法合成了一系列噌啉并环类化合物44, 收率为32%~87% (Eq. 7).值得一提的是, 噌啉并环类化合物因其结构复杂往往不易合成, 由于1位和2位的芳香氮原子不能直接进行官能团化, 在此之前尚无有效构建1, 2-噌啉并环类化合物的合成方法.该工作巧妙利用底物42的固有氮原子同时作为导向基团和反应基团, 免去了定位基的引入和脱除引起的步骤浪费.此反应过程通过两步一锅法完成, 操作较为操作简便, 是一种合成此类杂环的有效手段.
|
|
(7) |
2015年, Reddy小组[56]采用类似的策略报道了一例苯并[c]噌啉类化合物的合成方法.他们采用双酯基保护的苯肼45作为底物, 首先在钯催化剂和当量AgOAc的作用下与碘化物46发生C—H活化-芳基化反应形成C—C键;随后在过硫酸氢钾和碘苯的作用下发生C—H/N—H氧化脱氢偶联反应形成C—N键, 同时脱除酯基形成48 (Scheme 6).这一工作的底物适应范围较广, 底物46中卤素、羰基、烷基、醚等官能团均可耐受, 可以用于合成多取代苯并[c]噌啉类化合物.但该方法需要采用当量的银盐作为氧化剂, 并且需要两步才能实现, 降低了反应效率.
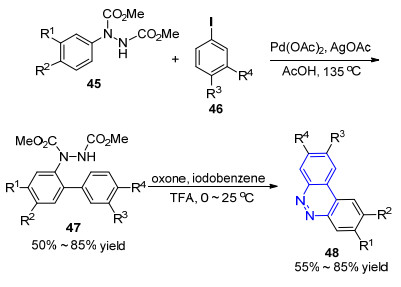
2013年, 游劲松小组[57]采用偶氮化合物49为底物, 以炔烃50为偶联试剂, 三价铑为催化剂, 通过串联反应成功实现了3, 4-二取代噌啉类化合物的合成.这一工作利用与异喹啉合成类似的策略[58], 将来源广泛、廉价易得的炔烃应用于噌啉类化合物的构建.通过控制R1基团的种类和反应条件, 可以分别得到噌啉-2-四氟硼酸盐和噌啉两类产物(Scheme 7):当R1基团不为叔丁基时, 反应主要得到2位噌啉季铵盐51, 收率为75%~ 94%, 在吡啶中进一步加热反应8 h可以得到噌啉类化合物52 (Scheme 7a); 当R1基团为叔丁基时, 对于烷基取代的炔烃, 通过调整反应条件, 可以直接得到电中性的噌啉化合物53 (Scheme 7b).该反应对酯基、氰基、卤素和酮等官能团均可耐受, 对于不对称的炔烃具有较好的区域选择性, 同时易发生副反应的端炔也能顺利发生反应, 是一种构建3, 4-二取代噌啉的有效方法.
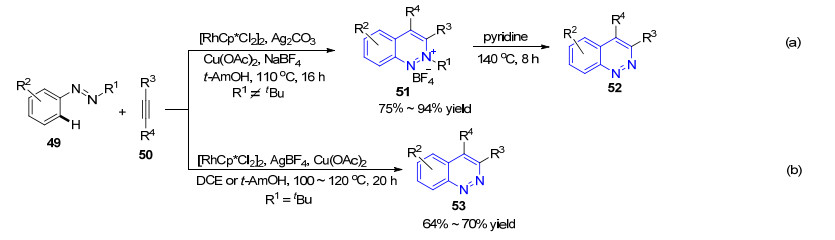
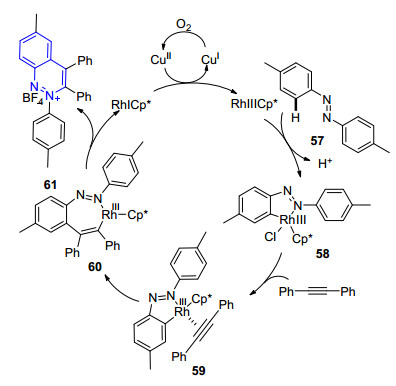
无独有偶, Cheng小组[59a]几乎在同时发表了类似的工作.他们同样以偶氮化合物作为底物, 以炔烃作为偶联试剂, 三价铑作为催化剂, 不同的是体系中采用六水合四氟硼酸铜作为氧化剂, 以叔丁醇为反应溶剂, 在70 ℃条件下发生反应, 得到3, 4-二取代的2位噌啉季铵盐56 (Eq. 8), 收率水平与游劲松小组相当, 产物可以进一步转化为吲哚和吲哚并吲哚两类化合物.他们在机理研究过程中成功合成并分离得到了环化铑中间体58, 将其用于催化反应, 以94%的收率顺利得到了目标产物, 进而验证了反应可能经历的碳氢活化-环化过程.作者据此提出了可能的反应机理(Scheme 8), 底物57首先与金属铑物种反应生成环化铑中间体58, 58与炔烃络合形成中间体59, 发生顺式插炔反应后进一步生成七元环中间体60, 随后发生还原消除反应得到噌啉季铵盐61.
在此基础上, 2016年, Cheng小组[59b]利用相同的策略, 采用Co(Ⅲ)催化剂, 以78%~90%的收率得到了3, 4-二取代的噌啉季铵盐64 (Eq. 9).值得一提的是, 催化剂的种类在这个反应中十分重要.在筛选过程中, 除了[CoCp*(CO)I2]之外, 其它的二价钴和三价钴催化剂均无效.作者选取反应得到的代表性噌啉类化合物用于光学性质的研究, 发现部分化合物在490 nm波长位置展现出强的蓝色荧光发射, 说明这些化合物在蓝色荧光有机发光二极管方面具有潜在的应用价值.
2014年, 张翱小组[60]利用类似的策略以廉价易得的依达拉奉(65)和炔烃66作为原料, 三价铑作为催化剂, 六氟异丙醇为溶剂, 在70 ℃条件下反应, 以中等到优秀(41%~96%)的收率得到了系列吡唑啉酮并噌啉类化合物67 (Eq. 10), 硝基、酯基等敏感基团均能良好耐受.另外, 由于依达拉奉是一种已知的上市药物, 这一方法也为依达拉奉的修饰提供了新的思路.
|
|
(8) |
|
|
(9) |
|
|
(10) |
2016年, Gandhi小组[61]以相对廉价的二价钌作为金属催化剂, 报道了另一例噌啉并环类化合物的合成方法.该方法采用哒嗪酮和酞嗪酮68作为底物, 与苯基炔丙醇69发生区域选择性的氧化环化反应, 同样以分子内固有官能团作为定位基, 醋酸作为仅有的副产物脱除, 展示出较好的原子经济性.该反应底物适应性广泛, 对酮羰基、硝基等官能团耐受性良好, 产率为65%~94% (Eq. 11).值得一提的是, 羟基通常被视为一种离去能力较差的基团, 然而在这个反应中得到的却是炔丙醇底物去氧化的产物, 其反应机理值得进一步深入研究.
|
|
(11) |
2016年, Perumal小组[62]采用与Gandhi小组报道的工作中类似的底物71, 在三价铑的催化作用下合成了哒嗪并噌啉类化合物72 (Eq. 12), 收率与上述工作维持在同一水平.除此之外, 他们还以吲唑衍生物73作为反应底物与不同的炔发生反应, 合成了吲唑酮并噌啉类化合物74 (Eq. 13).这些杂环化合物展现了良好的聚集诱导发光性质, 同时, 良好的生物相容性使得它们具有用于细胞成像研究的潜在价值.
2015年, 易伟小组[63a]报道了以重氮化的麦氏酸76作为偶联试剂, 与依达拉奉衍生物75发生三价铑催化的C—H活化/环化反应, 合成了9个噌啉酮并环类衍生物77, 产率为43%~90% (Eq. 14).该反应底物便于合成, 不需添加其他配体, 体系简单. 2016年, 他们[63b]采用三价铱为催化剂, 甲醇作为溶剂实现了类似转化, 反应时间缩短至0.5 h (Eq. 15).
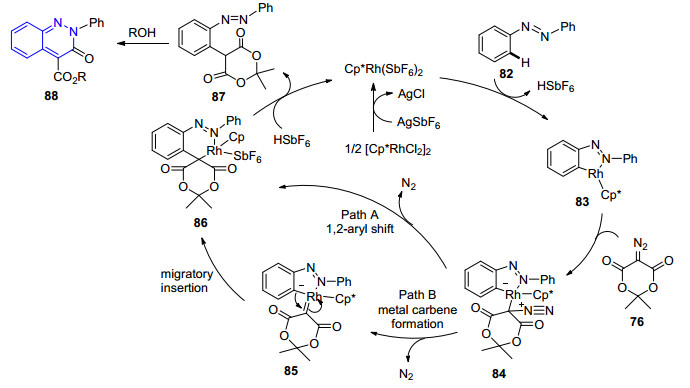
同年, Lee课题组[64]同样以76作为偶联试剂, 与芳基偶氮底物80在三价铑的催化作用下发生C—H活化/环化反应, 实现了2-芳基取代噌啉酮81的构建.在[RhCp*Cl2]2/AgSbF6的催化体系下, 以乙醇为溶剂, 在80 ℃条件下发生反应, 取得了45%~99%的收率(Eq. 16).反应产物中C-4位的酯基为进一步官能团转化提供了有利条件.在这个反应中, 当底物为非对称的偶氮化合物时, 不能得到选择性的产物, 而是以一定比例得到两种混合产物.反应机理如Scheme 9所示, 通过亲电去质子化得到环化铑中间体83后, 底物76与其发生络合作用进而形成中间体84; 84可能在形成铑卡宾中间体85后发生迁移插入反应得到中间体86, 也可能直接发生1, 2-芳基迁移反应得到中间体86, 随后发生质子化作用形成87, 进一步得到产物88.
|
|
(12) |
|
|
(13) |
|
|
(14) |
|
|
(15) |
几乎同时, Kim小组[65]报道了类似的反应.他们采用相同的催化体系, 以甲醇作为溶剂, 得到相应的甲酯化产物(Eq. 17), 但是收率较低, 并且底物大都局限在对称的芳基偶氮化合物.有趣的是, 在与重氮化的丙二酸二甲酯(91)反应时, 增加重氮的用量至3 equiv., 可以得到噌啉酮化合物C-8位进一步C—H烷基化的产物92 (Eq. 18).
|
|
(16) |
2016年, 林爱俊和姚和权小组[66]报道了在Rh(Ⅲ)的催化作用下, 偶氮底物93和重氮化底物94发生C—H烷基化/环化反应, 一步合成了3, 4-二取代的噌啉类化合物95 (Eq. 19).该反应在50 ℃下即可发生, 条件温和, 反应体系简单, 底物适用范围广, 除了硝基取代的底物收率较低外, 其他常见的取代基都能获得很好的产率.另外, 由于底物中的Boc基团在反应完成后自动脱除, 避免了不对称二芳基偶氮底物引起的区域选择性问题, 这些优点使得该方法成为一种有效地合成3, 4-二取代噌啉化合物的方法.
同年, 朱进小组[67]采用1-取代苯肼96作为底物, 重氮化合物97作为偶联试剂, 三价铑为催化剂, 通过串联反应实现了1, 3, 4-三取代噌啉骨架化合物98的合成.该反应以甲醇为溶剂, 以醋酸或碱金属醋酸盐作为唯一添加剂, 反应体系简单(Eq. 20).反应在室温下即可进行, 对氰基、酯基等敏感基团均能良好耐受, 能够以良好至优秀的收率(60%~98%)获得目标产物, 具有较好的应用价值.
|
|
(17) |
|
|
(18) |
|
|
(19) |
|
|
(20) |
2018年, 林爱俊和姚和权小组[68]采用菲尼酮99为底物, 以α-羰基重氮类化合物100为偶联试剂, 发展了一类三价铑催化的C—H活化/环化反应, 合成了一系列二氢吡唑酮并[1, 2-a]噌啉类化合物101.该反应在40 ℃下发生, 条件温和, 对硝基、氰基、酮羰基和端位烯基等敏感基团均能良好耐受, 收率为44%~87% (Eq. 21).在这个工作中, 反应仅生成无毒无害的N2和H2O副产物, 具有较好的原子经济性, 符合现代绿色化学的发展方向.
2018年, 李兴伟小组[69]采用偶氮102为底物, 在三价铑催化下, 与α-羰基重氮类化合物103发生C—H活化/环化反应, 建立了合成噌啉三氟甲磺酸盐104的新方法(Eq. 22).该反应在室温进行, 反应体系简单, 但当采用不对称偶氮底物时区域选择性的问题仍待解决.
2017年, 黄湧小组[70]以菲尼酮105作为底物, α-甲磺酰氧基酮106作为偶联试剂, 报道了一例Rh(Ⅲ)催化的C—H活化/环化反应合成噌啉并环骨架.在这个工作中, 碱的种类可以调节后续的脱水步骤是否发生, 从而控制反应的产物类型.当采用NaOCN作为碱时, 反应停留在醇的阶段(Eq. 23);当采用NaOPiv作为碱时, 反应进一步脱水(Eq. 24).该反应条件温和, 底物适应性好, 卤素、硝基、三氟甲基、三氟甲氧基等取代的底物都能获得很好的收率, 且反应能够放大至克级进行.
|
|
(21) |
|
|
(22) |
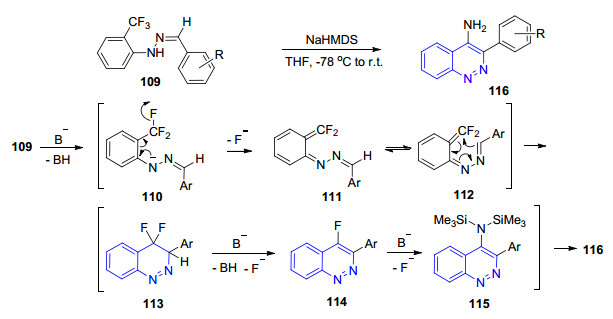
1995年, Kiselyov小组[71a]发展了一类4-氨基噌啉类化合物的新型合成方法. 1999年, 他们[71b]利用该方法以邻三氟甲基苯肼与芳香醛缩合成苯腙类底物109后, 在强碱NaHMDS的作用下得到一系列3-芳基-4-氨基噌啉类化合物116.三氟甲基是该反应的必需基团, 反应机理如Scheme 10所示.首先在碱的作用下拔氢形成氮负离子110, 为稳定负电荷发生分子内电荷转移同时离去氟离子得到中间体111, 进一步发生[6π]环化反应得到中间体113, 随后在碱的作用下芳构化得到中间体114, 再与碱发生芳香亲核取代得到中间体115, 酸处理后即得到目标化合物116.
|
|
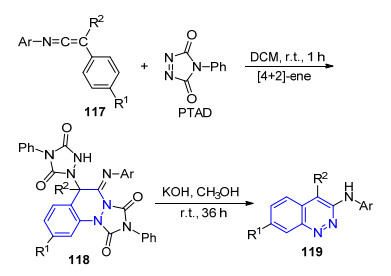
4-苯基-3H-1, 2, 4-三唑啉-3, 5-二酮(PTAD)是一种高活性的亲双烯体, 在有机合成中常常用作氮气的合成等价物参与反应[72]. 2009年, Alajarin小组[73]采用烯酮亚胺117作为底物, 与PTAD发生D-A/ene串联反应得到中间体118, 最后在KOH的作用下发生消除反应得到3-氨基取代噌啉化合物119 (Scheme 11).
2009年, 杨春皓小组[74]采用邻硝基苯乙腈120作为底物, 利用苯基格氏试剂对氰基进攻引发的串联环化反应实现了系列二氢噌啉类化合物121的合成, 部分化合物对急性早幼粒白血病细胞HL-60表现出中等抑制活性(Eq. 25).
|
|
(25) |
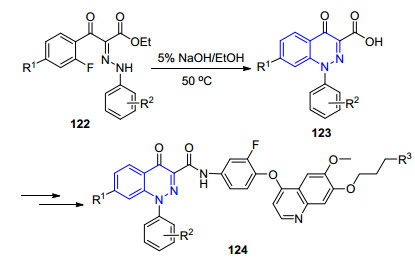
2013年, 宫平小组[75]以苯腙122作为原料, 在碱性条件下发生分子内的芳香亲核取代反应, 得到系列4-氧代噌啉-3-羧酸化合物123, 进一步合成得到了系列小分子c-Met抑制剂124, 优选化合物的IC50值为0.59 nmol/L, 展示出优异的抗肿瘤活性(Scheme 12).
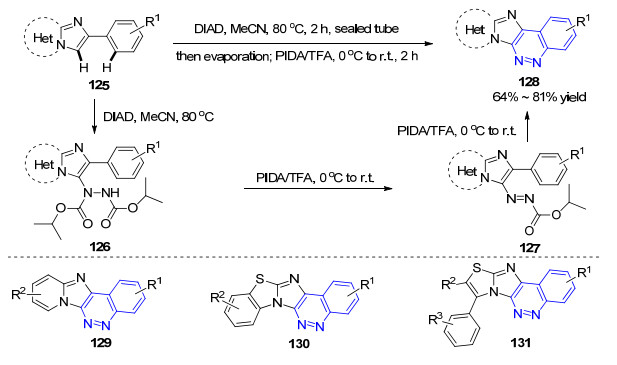
2016年, Sabitha课题组[76]采用咪唑并环类化合物125作为底物, 首先与偶氮二甲酸二异丙酯(DIAD)发生C-3区域选择性的联氨化反应, 再在PIDA的作用下发生C—H/N—H氧化去氢偶联反应, 完成了两个C—N键的构建, 合成了一系列噌啉并杂环类化合物128 (Scheme 13).通过控制反应条件, 可以分离得到联氨化中间体118, 随后通过控制PIDA的当量, 分离得到了偶氮中间体127, 127可以继续在PIDA的作用下发生环合得到目标产物128.该方法实际上是通过两步反应来实现目标噌啉环的合成, 但可以经一锅法完成.反应过程中无需过渡金属参与, 条件温和.基于该方法, 作者合成了三种杂环并噌啉类化合物129~131. 2017年, 汤日元小组[77]也用类似的策略合成了多种杂环化合物, 其中报道了三个构建噌啉并环类化合物的例子.
2015年, Chenoweth小组[78]以四嗪133为底物, 与原位生成的苯炔中间体发生Diels-Alder反应, 得到了系列噌啉类聚环化合物134 (Eq. 26). 134具有较好的荧光性质, 其光学性质可通过取代基调控, 具有应用于肝细胞影像检测的潜力.该反应采用一锅法在室温条件下进行, 对氧气不敏感, 反应完成时间不到5 min, 较为便捷, 但收率较低.
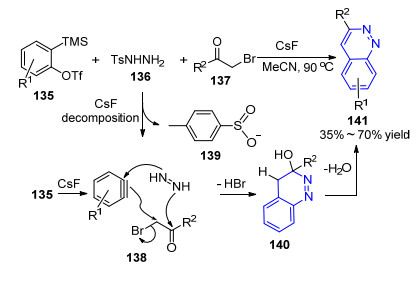
2016年, Wu小组[79]报道了一例通过苯炔中间体、偶氮中间体与α-溴代酮底物发生的三组分环化反应合成3-取代噌啉类化合物的方法, 产率35%~70%.他们提出的可能反应机理如Scheme 14所示:化合物135在氟化铯的作用下原位生成苯炔中间体138; 与此同时, 对甲苯磺酰肼(136)在氟化铯的作用下形成偶氮中间体139; 二者与α-溴代酮137发生[2+2+2]环化反应生成140, 再进一步脱水得到目标产物141.该反应的优点在于无需过渡金属催化剂参与, 原料简单易得, 反应体系简单, 一步完成两个C—N键和一个C—C键的构建; 不足之处在于反应收率不够理想, 且仅当R2基团为芳基时才能发生反应.
2017年, 范学森小组[80]报道了以邻溴苯乙酮和邻氨基苯甲酰肼为原料在I2的介导下发生的氧化-环化串联反应, 得到了一系列全新骨架的喹唑啉并噌啉酮骨架类杂环分子(Eq. 27).该方法具有原料简单易得, 无需金属催化剂参与, 操作简便等优点.
|
|
(26) |
|
|
(27) |
综上所述, 近年来噌啉类化合物的合成新方法呈现快速发展的趋势, 其中金属催化的偶联反应和C—H活化策略等现代合成技术的不断发展为噌啉类化合物的合成新方法研究注入了强大的活力.化学家们通过巧妙的反应设计, 利用定位基和偶联试剂的变化, 建立了许多合成新方法, 使得噌啉类化合物特别是噌啉并环类化合物的合成变得直接、高效.新方法的发展不但为噌啉类化合物的合成带来了便利, 也大大丰富了其分子多样性, 因此, 也将促进新型荧光团、有机发光材料和生物活性分子的发现.但是, 某些方法也存在催化剂价格昂贵、区域选择性差以及官能团耐受性差等不利因素, 这些客观存在的问题是化学家们前进的动力.相信未来随着化学家们的不断努力, 噌啉类化合物的合成新方法会进一步朝着简单、经济、高效、绿色、选择性和耐受性好的方向发展.另外, 虽然利用许多新方法合成了一系列全新结构的噌啉类化合物, 但是对其功能性的研究却往往滞后, 未来多学科的紧密合作将有助于加速噌啉类化合物的功能性研究发展.
Bekhit, A. A. Boll. Chim. Farm. 2001, 140, 243.
Lewgowd, W.; Stanczak, A. Arch. Pharm. Chem. Life Sci. 2007, 340, 65. doi: 10.1002/(ISSN)1521-4184
Ruchelman, A. L.; Singh, S. K.; Ray, A.; Wu, X.; Yang, J.-M.; Zhou, N.; Liu, A.; Liu, L.-F.; LaVoie, E. J. Bioorg. Med. Chem. 2004, 12, 795. doi: 10.1016/j.bmc.2003.10.061
Barraja, P.; Diana, P.; Lauria, A.; Passannanti, A.; Almerico, A. M.; Minnei, C.; Longu, S.; Cogiu, D.; Musiu, C.; La Colla, P. Bioorg. Med. Chem. 1999, 7, 1591. doi: 10.1016/S0968-0896(99)00096-6
Vaillancourt, V. A.; Larsen, S. D.; Nair, S. K. WO 0170706, 2001[Chem. Abstr. 2001, 135, 257250]
(a) Siegfried, A.-G. FR 1393596, 1965 [Chem. Abstr. 1965, 63, 2983].
(b) Schatz, F.; Wagner-Jauregg, T. Helv. Chim. Acta 1968, 51, 1919.
Stanczak, A.; Lewgowd, W.; Ochocki, Z.; Pakulska, W.; Szadowska, A. Pharmazie 1997, 52, 91. https://www.onacademic.com/detail/journal_1000033797117110_33ab.html
Shen, Y.; Shang, Z.; Yang, Y.; Zhu, S.; Qian, X.; Shi, P.; Zheng, J.; Yang, Y. J. Org. Chem. 2015, 80, 5906. doi: 10.1021/acs.joc.5b00242
Tsuji, H.; Yokoi, Y.; Sato, Y.; Tanaka, H.; Nakamura, E. Chem. Asian J. 2011, 6, 2005. doi: 10.1002/asia.201100234
Chen, J.-C.; Wu, H.-C.; Chiang, C.-J.; Peng, L.-C.; Chen, T.; Xing, L.; Liu, S.-W. Polymer 2011, 52, 6011. doi: 10.1016/j.polymer.2011.10.059
(a) Leonard, N. J. Chem. Rev. 1945, 37, 269.
(b) Vinogradova, O. V.; Balova, I. A. Chem. Heterocycl. Compd. 2008, 44, 501.
(c) Han, Y.-T.; Jung, J.-W.; Kim, N.-J. Curr. Org. Chem. 2017, 21, 1265.
(d) Mathew, T.; Papp, A. A.; Paknia, F.; Fustero, S.; Prakash, G. K. S. Chem. Soc. Rev. 2017, 46, 3060.
Richter, V. V. Berichte. 1883, 16, 677.
Senadi, G. C.; Gore, B.; Hu, W.-P.; Wang, J.-J. Org. Lett. 2016, 18, 2890. doi: 10.1021/acs.orglett.6b01207
(a) Widman, O. Berichte 1884, 17, 722.
(b) Widman, O. Berichte 1909, 42, 4216.
(c) Stoermer, R.; Fincke, H. Berichte 1909, 42, 3115.
(d) Stoermer, R.; Gaus, O. Berichte 1912, 45, 3104.
(e) Pang, X.-B.; Zhao, L.-B.; Zhou, D.-G.; He, P.-Y.; An, Z.; Ni, J.-X.; Yan, R.-L. Org. Biomol. Chem. 2017, 15, 6318.
(a) Keneford, J. R.; Simpson, J. С. Е. J. Chem. Soc. 1947, 917.
(b) Keneford, J. R.; Simpson, J. С. Е. J. Chem. Soc. 1948, 354.
(c) Schofield, K.; Theobald, R. S. J. Chem. Soc. 1949, 2404.
(a) Hu, E.; Kunz, R. K.; Rumfelt, S.; Andrews, K. L.; Li, C.; Hitchcock, S. A.; Lindstrom, M.; Treanor, J. Bioorg. Med. Chem. Lett. 2012, 22, 6938.
(b) Yang, H.; Murigi, F. N.; Wang, Z.-J.; Li, J.-F.; Jin, H.-J.; Tu, Z.-D. Bioorg. Med. Chem. Lett. 2015, 25, 919.
(c) Hu, E.; Ma, J.; Biorn, C.; Lester-Zeiner, D.; Cho, R.; Rumfelt, S.; Kunz, R. K.; Nixey, T.; Michelsen, K.; Miller, S. J. Med. Chem. 2012, 55, 4776.
Hennequin, L. F.; Thomas, A. P.; Johnstone, C.; Stokes, E. S. E.; Plé, P. A.; Lohmann, J. J. M.; Ogilvie, D. J.; Dukes, M.; Wedge, S. R.; Curwen, J. O.; Kendrew, J.; Brempt, C. L. J. Med. Chem. 1999, 42, 5369. doi: 10.1021/jm990345w
(a) Senadi, G. C.; Gore, B. S.; Hu, W. P.; Wang, J. J. Org. Lett. 2016, 18, 2890.
(b) Khaligh, N. G.; Mihankhah, T.; Johan, M. R.; Ching, J. J. Monatsh. Chem. 2018, 149, 1083.
(a) Neber, P. W.; Knoller, G.; Herrst, K.; Trissler, A. Liebigs Ann. Chem. 1929, 471, 113.
(b) Alford, E. J.; Schofield, K. J. Chem. Soc. 1952, 2102.
Shvartsberg, M. S.; Ivanchikova, I. D. Tetrahedron Lett. 2000, 41, 771. doi: 10.1016/S0040-4039(99)02151-6
Stefańska, B.; Arciemiuk, M.; Bontemps-Gracz, M. M.; Dzieduszycka, M.; Kupiec, A.; Martellib, S.; Borowskia, E. Bioorg. Med. Chem. 2003, 11, 561. doi: 10.1016/S0968-0896(02)00425-X
(a) Gomaa, M. A. M. Tetrahedron Lett. 2003, 44, 3493.
(b) Ryu, C. K.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2006, 16, 1850.
(c) Lambert, D. J.; Parikh, N.; Messham, S. J.; Edwards, G.; Truong, H. V.; Dempster, N. M.; Drew, M. G. B.; Nahar, L.; Sarker, S. D.; Ismail, F. M. D. Tetrahedron Lett. 2015, 56, 6980.
(a) Barber, H. J.; Washbourn, K.; Wragg, W. R.; Lunt, E. J. Chem. Soc. 1961, 2828.
(b) Barber, H. J.; Lunt, E. J. Chem. Soc., Perkin Trans. 1 1968, 9, 1156.
Al-Awadi, N. A.; Elnagdi, M. H.; Ibrahim, Y. A.; Kaul, K.; Kumar, A. Tetrahedron 2001, 57, 1609. doi: 10.1016/S0040-4020(00)01141-8
Liu, X.-F.; Chang, H.-F.; Schmiesing, R. J.; Wesolowski, S. S.; Knappenberger, K. S.; Arriza, J. L.; Chapdelaine, M. J. Bioorg. Med. Chem. 2010, 18, 8374. doi: 10.1016/j.bmc.2010.09.058
Awad, E. D.; El-Abadelah, M. M.; Matar, S.; Zihlif, M. A.; Naffa, R. G.; AlMomani, E. Q. Molecules 2012, 17, 227. https://www.mdpi.com/1420-3049/17/1/227
Alhambra, C.; Becker, C.; Blake, T.; Chang, A. H.; Damewood, J. R.; Daniels, T.; Dembofsky, B. T.; Gurley, D. A.; Hall, J. E.; Herzog, K. J.; Horchler, C. L.; Ohnmacht, C. J.; Schmiesing, R. J.; Dudley, A.; Ribadeneira, M. D.; Knappenberger, K. S.; Maciag, C.; Stein, M. M.; Chopra, M.; Liu, X.-F.; Christian, E. P.; Arriza, J. L.; Chapdelaine, M. J. Bioorg. Med. Chem. 2011, 19, 2927. doi: 10.1016/j.bmc.2011.03.035
Vinogradova, O. V.; Balova, I. A.; Popik, V. V. J. Org. Chem. 2011, 76, 6937. doi: 10.1021/jo201148h
Bräse, S.; Dahmen, S.; Heuts, J. Tetrahedron Lett. 1999, 40, 6201. doi: 10.1016/S0040-4039(99)01166-1
(a) Kimball, D. В.; Hayes, A. G.; Haley, M. M. Org. Lett. 2000, 2, 3825.
(b) Kimball, D. В.; Weakly, T. J. R.; Herges, R.; Haley, M. M. J. Am. Chem. Soc. 2002, 124, 1572.
(c) Kimball, D. В.; Weakly, T. J. R.; Herges, R.; Hayes, A. G.; Haley, M. M. J. Am. Chem. Soc. 2002, 124, 13463.
(d) Kimball, D. В.; Weakly, T. J. R.; Haley, M. M. J. Org. Chem. 2002, 67, 6395.
(e) Kimball, D. В.; Haley, M. M. Angew. Chem., Int. Ed. 2002, 41, 3338.
(f) Young, B. S.; Marshall, J. L.; MacDonald, E.; Vonnegut, C. L.; Haley, M. M. Chem. Commun. 2012, 48, 5166.
(g) Young, B. S.; Köhler, F.; Herges, R.; Haley, M. M. J. Org. Chem. 2011, 76, 8483.
(a) Ross, S. D.; Kuntz, I. J. Am. Chem. Soc. 1952, 74, 1297.
(b) Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1960, 3646.
Hou, Z.; Fujiwara, Y.; Taniguchi, H. J. Org. Chem. 1988, 53, 311.
Dehghanpour, S.; Afshariazar, F.; Assoud, J. Polyhedron 2012, 35, 69. doi: 10.1016/j.poly.2011.12.041
(a) Braithwaite, R. S. W.; Holt, P. F.; Hughes, A. N. J. Chem. Soc. 1958, 4073.
(b) Barton, J. W.; Rowe, D. J. Tetrahedron Lett. 1983, 24, 299.
(c) Barton, J. W.; Sheperd, M. K. Tetrahedron Lett. 1984, 25, 4967.
(d) Benin, V.; Kaszynski, P. J. Org. Chem. 2000, 65, 6388.
(a) Bjorsvik, H.-R.; Gonzalez, R. R.; Liguori, L. J. Org. Chem. 2004, 69, 7720.
(b) Elumalai, V.; Bjorsvik, H.-R. ChemistrySelect 2018, 3, 2092.
Corbett, J. F.; Holt, P. F. J. Chem. Soc. 1961, 3695.
Caronna, T.; Fontana, F.; Mele, A.; Sora, I. N.; Panzeri, W.; Viganò, L. Synthesis 2008, 413.
Takeda, Y.; Okazaki, M.; Maruoka, Y.; Minakata, S. Beilstein J. Org. Chem. 2015, 11, 9. doi: 10.3762/bjoc.11.2
Lee, D. S.; Chatterjee, T; Ban, J.; Rhee, H.; Cho, E. J. ChemistrySelect 2018, 3, 2092. doi: 10.1002/slct.201800278
(a) Holt, P. F.; Went, C. W. J. Chem. Soc. 1963, 4099.
(b) Al-Mousawi, S. M.; El-Apasery, M. A. Molecules 2012, 17, 6547.
(c) Al-Awadi, N. A.; Ibrahim, Y. A.; Elnagdi, M. H.; Adam, A. Y.; John, E. J. Anal. Appl. Pyrol. 2017, 124, 602.
Wimmer, R.; Müller, N. Monatsh. Chem. 1998, 129, 1161.
Sharma, M.; Maheshwari, A.; Bindal, N. J. Heterocycl. Chem. 2013, 50, 116. doi: 10.1002/jhet.1086
Swenton, J. S.; Ikeler, T, J.; Williams, B. H. J. Am. Chem. Soc. 1970, 92, 3103. doi: 10.1021/ja00713a031
Yu, Y.; Singh, S. K.; Liu, A.; Li, T.-K.; Liu, L. F.; LaVoie, E. J. Bioorg. Med. Chem. 2003, 11, 1475. doi: 10.1016/S0968-0896(02)00604-1
(a) Parrino, B.; Carbone, A.; Muscarella, M.; Spanò, V.; Montalbano, A.; Barraja, P.; Salvador, A.; Vedaldi, D.; Cirrincione, G.; Diana, P. J. Med. Chem. 2014, 57, 9495.
(b) Shen, Y.; Zhang, Q.; Qian, X.; Yang, Y. Anal. Chem. 2015, 87, 1274.
Hasegawa, K.; Kimura, N.; Arai, S.; Nishida, A. J. Org. Chem. 2008, 73, 6363. doi: 10.1021/jo8010864
Zhu, C.; Yamane, M. Tetrahedron 2011, 67, 4933. doi: 10.1016/j.tet.2011.04.079
Ball, C. J.; Gilmore, J.; Willis, M. C. Angew. Chem., Int. Ed. 2012, 51, 5718. doi: 10.1002/anie.201201529
Kumar, A.; Tiwari, D. K.; Sridhard, B.; Likhar, P. R. Org. Biomol. Chem. 2018, 16, 4840. doi: 10.1039/C8OB01152D
Zhao, S.; Yu, R.; Chen, W.; Liu, M.; Wu, H. Org. Lett. 2015, 17, 2828. doi: 10.1021/acs.orglett.5b01247
Jurberg, I. D.; Gagosz, F. J. Organomet. Chem. 2011, 696, 37. doi: 10.1016/j.jorganchem.2010.06.017
Gogoi, P.; Gogoi, S. R.; Devi, N.; Barman, P. Synth. Commun. 2014, 44, 1142. doi: 10.1080/00397911.2013.853799
Zhang, G.; Miao, J.; Zhao, Y.; Ge, H. Angew. Chem., Int. Ed. 2012, 51, 8318. doi: 10.1002/anie.v51.33
Lan, C.; Tian, Z.; Liang, X.; Gao, M.; Liu, W.; An, Y.; Fu, W.; Jiao, G.; Xiao, J.; Xu, B. Adv. Synth. Catal. 2017, 359, 3735. doi: 10.1002/adsc.v359.21
Fan, Z.; Wu, K.; Xing, L.; Yao, Q.; Zhang, A. Chem. Commun. 2014, 50, 1682. doi: 10.1039/C3CC47989G
Reddy, B. V. S.; Reddy, C. R.; Reddy, M. R.; Yarlagadda, S.; Sridhar, B. Org. Lett. 2015, 17, 3730. doi: 10.1021/acs.orglett.5b01717
Zhao, D.; Wu, Q.; Huang, X.; Song, F.; Lv, T.; You, J. Chem.-Eur. J. 2013, 19, 6239. doi: 10.1002/chem.201300155
Guimond, N.; Gouliaras, C.; Fagnou, K. J. Am. Chem. Soc. 2010, 132, 6908. doi: 10.1021/ja102571b
(a) Muralirajan, K. Cheng, C.-H. Chem.-Eur. J. 2013, 19, 6198.
(b) Prakash, S.; Muralirajan, K.; Cheng, C.-H. Angew. Chem., Int. Ed. 2016, 55, 1844.
Xing, L.; Fan, Z.; Hou, C.; Yong, G.; Zhang, A. Adv. Synth. Catal. 2014, 356, 972. doi: 10.1002/adsc.v356.5
Rajkumar, S.; Savarimuthu, S. A.; Kumaran, R. S.; Nagarajad, C. M.; Gandhi, T. Chem. Commun. 2016, 52, 2509. doi: 10.1039/C5CC09347C
Mayakrishnan, S.; Arun, Y.; Balachandran, C.; Emi, N.; Muralidharana, D.; Perumal, P. T. Org. Biomol. Chem. 2016, 14, 1958. doi: 10.1039/C5OB02045J
(a) Shi, J.; Zhou, J.; Yan, Y.; Jia, J.; Liu, X.; Song, H.; Xu, H. E.; Yi, W. Chem. Commun. 2015, 51, 668.
(b) Lv, H.; Xu, W. L.; Lin, K.; Shi, J.; Yi, W. Eur. J. Org. Chem. 2016, 5637.
Son, J.-Y.; Kim, S.; Jeon, W. H.; Lee, P. H. Org. Lett. 2015, 46, 2518.
Sharma, S.; Han, S. H.; Han, S.; Ji, W.; Oh, J.; Lee, S.-Y.; Oh, J. S.; Jung, Y. H.; Kim, I. S. Org. Lett. 2015, 46, 2852.
Sun, P.; Wu, Y.; Huang, Y.; Wu, X.; Xu, J.; Yao, H.; Lin, A. Org. Chem. Front. 2016, 3, 91. doi: 10.1039/C5QO00331H
Song, C.; Yang, C.; Zhang, F.; Wang, J.; Zhu, J. Org. Lett. 2016, 18, 4510. doi: 10.1021/acs.orglett.6b02103
Li, P.; Xu, X.; Chen, J.; Yao, H.; Lin, A. Org. Chem. Front. 2018, 5, 1777. doi: 10.1039/C8QO00209F
Chen, X.; Zheng, G.; Song, G.; Li, X. Adv. Synth. Catal. 2018, 360, 2836. doi: 10.1002/adsc.v360.15
Yang, C.; Song, F.; Chen, J.; Huang. Y. Adv. Synth. Catal. 2017, 359, 3496. doi: 10.1002/adsc.v359.20
(a) Kiselyov, A. S. Tetrahedron Lett. 1995, 36, 1383.
(b) Kiselyov, A. S.; Dominguez, C. Tetrahedron Lett. 1999, 40, 5111.
(a) Kobayashi, S.; Furuya, T.; Otani, T.; Saito, T. Tetrahedron 2008, 64, 9705.
(b) Süennemann, H. W.; Banwell, M. G.; de Meijere, A. Chem.-Eur. J. 2008, 14, 7236.
(c) Kobayashi, S.; Furuya, T.; Otani, T.; Saito, T. Tetrahedron Lett. 2008, 49, 4513.
(d) Alajarín, M.; Cabrera, J.; Pastor, A.; Sánchez-Andrada, P.; Bautista, D. J. Org. Chem. 2008, 73, 963.
Alajarin, M.; Bonillo, B.; Marin-Luna, M.; Vidal, A.; Orenes, R.-A. J. Org. Chem. 2009, 74, 3558. doi: 10.1021/jo900304a
Chen, D.; Yang, C.; Xie, Y.; Ding, J. Heterocycles 2009, 77, 273. doi: 10.3987/COM-08-S(F)7
Li, S.; Zhao, Y.; Wang, K.; Gao, Y.; Han, J.; Cui, B.; Gong, P. Bioorg. Med. Chem. 2013, 21, 2843. doi: 10.1016/j.bmc.2013.04.013
Kandimalla, S. R.; Sabitha, G. RSC Adv. 2016, 6, 67086. doi: 10.1039/C6RA15418B
Jiao, J.; Xu, L.; Zheng, W.; Xiong, P.; Hu, M.-L.; Tang, R.-Y. Synthesis 2017, 49, 1839.
Suh, S.-E.; Barros, S. A.; Chenoweth, D. M. Chem. Sci. 2015, 6, 5128. doi: 10.1039/C5SC01726B
Shu, W.-M.; Ma, J.-R.; Zheng, K.-L.; Wu, A.-X. Org. Lett. 2016, 18, 196. doi: 10.1021/acs.orglett.5b03236
Guo, S.; Zhai, J.; Fan, X. Org. Biomol. Chem. 2017, 15, 1521. doi: 10.1039/C6OB02699K

图式 2 苯并噌啉类化合物的经典合成方法
Scheme 2 Classical methods for the synthesis of benzo[c]cinnolines

图式 4 Cu(Ⅰ)催化的C—N偶联法合成噌啉化合物
Scheme 4 Synthesis of cinnolines by Cu(Ⅰ) catalyzed tandem C—N bond formation

图式 5 Au(Ⅰ)催化的分子内芳氢化反应
Scheme 5 Synthesis of cinnolines by Au(Ⅰ) catalyzed intramolecular hydroarylation

图式 6 通过连续形成C—C键和C—N键合成苯并[c]噌啉类化合物
Scheme 6 Synthesis of benzo[c]cinnolines by sequential C—C and C—N bonds formation

图式 9 与重氮化合物的C—H活化/环化过程机理
Scheme 9 Proposed mechanism of C—H activation/annulation with diazo compounds

图式 10 邻三氟甲基苯腙的[6π]环化反应机理
Scheme 10 [6π] annulation of o-trifluoromethyl phenylhydrazones and proposed mechanism

图式 13 C-3选择性氢胺化/氧化脱氢偶联一锅法合成噌啉化合物
Scheme 13 One-pot synthesis of cinnolines through C-3 regioselective hydrazination and oxidative N-arylation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



























 下载:
下载:







 下载:
下载: