 图 图式5
室温水相下纳米Cu (0)-Fe3O4@SiO2/NH2催化三唑类化合物的合成
Figure 图式5.
Synthesis of triazole derivatives catalyzed by Cu (0)-Fe3O4@SiO2/NH2 NPs in water at room temperature
图 图式5
室温水相下纳米Cu (0)-Fe3O4@SiO2/NH2催化三唑类化合物的合成
Figure 图式5.
Synthesis of triazole derivatives catalyzed by Cu (0)-Fe3O4@SiO2/NH2 NPs in water at room temperature
引用本文:
张金, 刘佳, 马养民, 程佩. 纳米金属氧化物催化含氮杂环类化合物的合成研究进展[J]. 有机化学,
2017, 37(3): 555-565.
doi:
10.6023/cjoc201609006

Citation: Zhang Jin, Liu Jia, Ma Yangmin, Cheng Pei. Progress in the Synthesis of N-Fused Heterocycles Catalyzed by Nanocrystalline Metal Oxides[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 555-565. doi: 10.6023/cjoc201609006
Citation: Zhang Jin, Liu Jia, Ma Yangmin, Cheng Pei. Progress in the Synthesis of N-Fused Heterocycles Catalyzed by Nanocrystalline Metal Oxides[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 555-565. doi: 10.6023/cjoc201609006
纳米金属氧化物催化含氮杂环类化合物的合成研究进展
English
Progress in the Synthesis of N-Fused Heterocycles Catalyzed by Nanocrystalline Metal Oxides
Abstract:
N-Fused heterocycles are an important class of organic compounds. As a basic scaffold of a wide variety of biologically active alkaloids and synthetic pharmaceuticals and agrochemicals, the construction methods of N-fused heterocycles have attracted much attention. Significant efforts have been devoted to the application of the nanocrystalline metal oxides as heterogeneous, efficient and environmentally friendly catalyst in organic reaction. This review provides a summary of the synthesis of the different kinds of N-fused heterocycles in recent 5 years catalyzed by the nanocrystalline metal oxides.
-
Key words:
- nanocrystalline metal oxides
- / N-fused heterocycles
- / catalyst
-
含氮的杂环化合物是有机化合物的重要组成部分, 多数药物、维生素、天然产物、生物大分子等杂环均具有含氮杂环的结构[1], 如喹啉、吡啶、吡咯、咔啉是天然产物和药物分子的重要骨架, 生物DNA中碱基对嘌呤、嘧啶氮杂环等.含氮的杂环化合物广泛应用于合成工业的医药活性结构和中间体, 合成过程的保护基团, 手性助剂, 有机催化剂, 有机金属配体中.在生物医学和农业科学有大量的文献报道[2], 如在抑菌[3]、抗炎[4]、抗肿瘤[5]、抗抑郁[6]、抗疟疾[7]等有广泛的应用, 同时在农业科学中亦可用作农药, 如除草剂[8]、杀虫剂等, 还可以作为染料[9]、荧光剂[10]、分析检测试剂的主要骨架在材料科学方面有重要应用, 在信息存储、塑料工业也有巨大的应用前景.因此含氮杂环的构建成为研究的热点[11~13], 本文旨在从催化剂的角度对含氮杂环的合成进行综述.
从廉价易得的原料出发, 探索一种绿色、温和、高效的催化方法是合成含氮杂环化合物方法中的重点研究方向, 每年有大量的关于这类化合物的合成新方法报道.其中, 非均相纳米金属氧化物催化剂在催化有机反应中的应用被大大拓展, 2014年, 吴磊、范青华等[14]的综述中提到了纳米金属氧化物在不对称氢化和氢转移反应中的研究进展, 这种纳米粒子本身可以作为催化活性的位点, 还可以作为催化剂的载体, 进一步手性修饰后可以应用于不对称氢化的合成反应. 2010年以来, 纳米粒子因具有易分离、可回收、催化效率高等优点而得到快速发展, 不断有新型纳米金属氧化物被应用在温和条件下高效催化合成杂环化合物[15].这类催化剂具有特殊的纳米尺寸, 能在反应物、溶剂中更好地分散; 具有大的比表面积, 从而大大地增加了催化活性和反应速率; 反应条件更绿色环保, 高产率得到目标化合物; 反应结束时, 催化剂及产物后处理简单, 催化剂稳定性高, 回收重复使用催化活性没有明显改变.纳米金属氧化物粒子在催化杂环形成过程中不但可以作为催化剂催化反应, 同时还可以作为其他贵重金属催化剂的载体, 使成本高的催化剂更易分离且回收使用.
按照合成的含氮杂环目标化合物的类别, 本文对近年来报道的纳米金属氧化物作为催化剂催化的含氮杂环的合成方法及机理做了综述.
1 纳米金属氧化物催化单环含氮杂环类化合物合成
1.1 纳米金属氧化物催化含1个氮原子杂环化合物合成
1.2 纳米金属氧化物催化含2个氮原子杂环化合物合成
1.3 纳米金属氧化物催化含3个氮原子杂环化合物合成
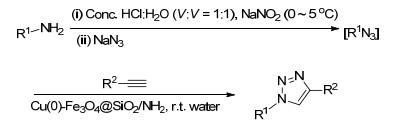
2013年, Kamal等[33]设计了用纳米Fe2O3催化合成2, 4, 5-三取代-1, 2, 3-三唑类化合物的新方法.通过查尔酮类化合物、叠氮化钠和卤代芳基类化合物三组分串联反应, 可以立体选择性地合成芳基取代的三唑类化合物 (Eq. 8).通过反应条件的控制实验, 发现反应中的氧化剂来自于空气中的氧气.该合成方法底物适用范围广, 产物产率高, 并且催化剂可以回收利用没有明显的催化活性的变化.但使用DMSO为溶剂对环境污染大, 叠氮化钠剧毒, 易爆炸, 后处理相对复杂.
端炔和叠氮类化合物催化合成三氮唑类化合物的反应也有广泛的报道, 2013年, Jana和Islam等[34]引入一种石墨烯为载体的γ-Fe2O3纳米粒子 (G-γ-Fe2O3) 催化剂在温和的条件下合成三氮唑类化合物, 使用水作为溶剂, 在氩气保护下进行 (Eq. 9).
2016年, Paul等[35]采用Cu (0)-Fe3O4负载在二氧化硅上 (Cu (0)-Fe3O4@SiO2/NH2) 的新型催化剂一锅法高效合成1, 4-二取代-1, 2, 3-三唑类化合物, 该方法减少了有毒叠氮化合物的使用, 而且以水为溶剂, 反应在室温条件下进行, 产率高达97% (Scheme 5).该反应更加绿色环保, 适用性更广阔.
图 图式5
室温水相下纳米Cu (0)-Fe3O4@SiO2/NH2催化三唑类化合物的合成
Figure 图式5.
Synthesis of triazole derivatives catalyzed by Cu (0)-Fe3O4@SiO2/NH2 NPs in water at room temperature
1.1.1 吡咯类化合物的合成
纳米金属氧化物可以用于催化经典的反应制备吡咯类化合物. 2012年, Teimouri等[16]利用硫酸化的纳米氧化锆为催化剂, 催化2, 5-二羰基化合物与芳香胺在乙醇中回流一锅法进行Paal-Knorr反应, 取得较高的产率. 2013年, Zhang课题组[17]报道了一种简洁的方法, 制备出具有磁性的纳米粒子负载的锑盐离子液体 (Fe2O3@ SiO2-Sb-IL) 催化剂, 通过Clauson-Kaas反应利用2, 5-二烷氧基四氢呋喃和伯胺出发合成取代吡咯类化合物.这类催化剂呈现出良好的催化活性, 可以重复使用至少6次, 反应在以水为溶剂的条件下进行.第二年, 其课题组设计出两种不同的纳米氧化物负载的催化剂, 磁性纳米CoFe2O4负载的锑盐离子液体 ([CoFe2O4@SiO2-DABCO-Sb])[18]和CoFe2O4负载的钼催化剂 ([CoFe2O4@ SiO2-PrNH2-Mo (acac)2])[19], 分别催化三组分芳香胺、硝基烯类化合物及1, 3-二羰基化合物和四组分芳香醛、芳香胺、1, 3-二羰基化合物和硝基甲烷一锅法合成多取代吡咯化合物 (Scheme 1).这类磁性的非均相催化剂可以通过其他磁性物质吸出回收利用而不损失其催化活性, 在工业上有很大的应用前景.
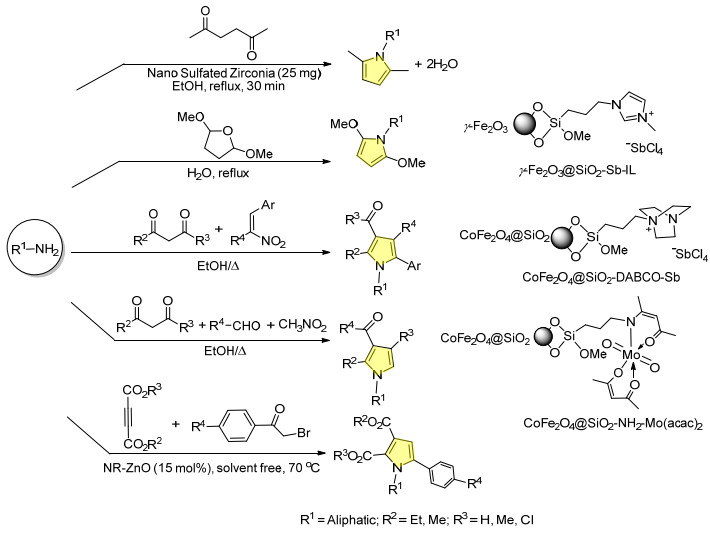 图 图式1
N-取代吡咯类化合物合成
Figure 图式1.
Synthesis of N-substituted pyrroles
图 图式1
N-取代吡咯类化合物合成
Figure 图式1.
Synthesis of N-substituted pyrroles
2014年, Sabbaghan等[20]报道了用纳米ZnO为催化剂, 催化脂肪胺、不同取代的溴代苯乙酮与二乙炔酯三组分在温和条件下高效合成多取代的吡咯类化合物, 并且反应不需要溶剂参与, 更加绿色环保 (Scheme 1).同时还研究了不同的纳米结构的催化剂对催化活性的影响, 结果发现当催化剂纳米ZnO为棒状结构时, 产物产率最高.催化的机理研究表明, 纳米ZnO粒子催化反应可以视为路易斯酸 (Zn2+) 及路易斯碱 (O2-) 共同作用, Zn2+与丁炔二甲酯的酯基作用, O2-与苯甲酰基相互作用, 从而与氨基成环形成吡咯骨架, 机理如Scheme 2所示.
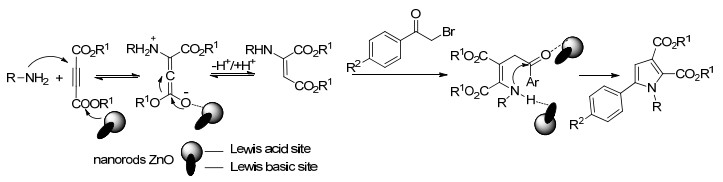 图 图式2
纳米氧化锌催化生成吡咯类化合物机理图
Figure 图式2.
Possible mechanism for the formation of substituted pyrroles catalyzed by ZnO nanorods
图 图式2
纳米氧化锌催化生成吡咯类化合物机理图
Figure 图式2.
Possible mechanism for the formation of substituted pyrroles catalyzed by ZnO nanorods
1.1.3 吡啶类化合物的合成
2013年, Sobhani等[23]将离子液体乙二胺磺酸盐固定在纳米γ-Fe2O3粒子上 (γ-Fe2O3-2-HEAS) 作为一种非均相催化剂用于一锅法合成多取代吡啶的衍生物中.反应原料为芳香醛、丙二腈、硫酚类化合物 (Eq. 2), 在催化剂的作用下反应不需要溶剂参与, 反应时间短, 只需要10 min.并且催化剂可以重复使用多次, 环境污染小.
2015年, Eshghi等[24]设计了一种磁性的纳米材料, 用于三组分合成四氢吡啶类化合物 (Eq. 3), 催化反应在温和的条件下进行并取得较高产率.反应完成后, 催化剂易分离, 大大减小了后处理的步骤, 使反应更加简洁.
1.1.2 噻唑类化合物的合成
噻唑类化合物是一类重要的含氮和硫的五元杂环化合物, 在农业生产中有广泛的应用, 例如已经商品化的农药土菌灵、噻唑菌胺等, 这类化合物的合成被学者广泛关注. Hajinasiri等[21]以脂肪胺、异硫氰酸酯、溴代羰基化合物为原料, 在室温无溶剂条件下, 利用实验室制备的纳米ZnO为催化剂催化多组分合成1, 3-噻唑类衍生物 (Eq. 1), 取得较高产率, 并且催化剂形貌用扫描电子显微镜 (SEM) 表征也呈现棒状结构, 并对反应机理进行了探索.推测纳米ZnO棒状结构上具有路易斯酸和路易斯碱作用位点催化反应的进行.
Mokhtary等[22]利用离子液体和纳米Fe3O4负载的SiO2催化剂 (MNPs@SiO2-IL) 在无溶剂条件下三组分合成了噻唑酮类化合物, 反应原料为芳香醛、芳香胺及巯基乙酸 (Scheme 3).反应推测离子液体甲基咪唑阳离子在催化过程中与芳香醛羰基结合, 增加了羰基的亲电性, 加快了亚胺中间体的形成.
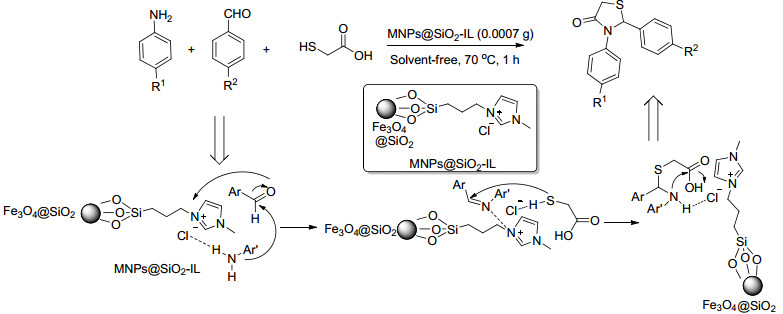 图 图式3
MNPs@SiO2-IL催化合成噻唑类化合物机理图
Figure 图式3.
Possible mechanism for the formation of thiazole derivatives catalyzed by MNPs@SiO2-IL
图 图式3
MNPs@SiO2-IL催化合成噻唑类化合物机理图
Figure 图式3.
Possible mechanism for the formation of thiazole derivatives catalyzed by MNPs@SiO2-IL
1.2.3 噁二唑类杂环化合物的合成
2013年, Sangshetti等[31]发明了一种合成4, 5, 6, 7-四氢-6-[(5-取代-1, 3, 4-噁二唑) 甲基]噻吩并[2, 3-c]吡啶类化合物方法.以芳香醛和取代的酰肼类化合物为原料, 乙醇为溶剂, 在微波辅助下纳米 (ZnO-TiO2) 催化取得87%以上的产率 (Eq. 6).试验还研究了这类化合物的抑菌效果, 可以作为一种潜在的抑菌药物.
2014年, Murty等[32]利用纳米CuO为催化剂, 催化N-芳基-N-酰肼衍生物中C—H键断裂形成C—O键, 得到1, 3, 4-噁二唑类化合物, 产率较高, 并且催化剂可以回收利用不影响其催化活性, 但该方法使用DMSO为溶剂, 产物后处理比较复杂 (Eq. 7).
1.2.2 吡唑类杂环化合物的合成
2015年, Zakerinasab等[30]设计了一种核壳结构的Fe3O4负载的SiO2纳米颗粒Fe3O4@SiO2@PDETSA用于吡唑类衍生物的合成中, 以α, β-二羰基化合物和肼或酰肼衍生物为原料, 在室温条件下以水为溶剂反应, 条件更加绿色温和, 高产率地得到目标化合物.并且催化剂用量少, 可回收重复利用 (Eq. 5).
1.2.1 咪唑类杂环化合物的合成
咪唑类化合物因其重要的药用价值受到广泛关注.有许多文献报道纳米粒子催化下2, 4, 5-三取代咪唑类化合物的合成.其中以二苯基乙二酮、脂肪醛或芳香醛、乙酸铵为原料的合成路线研究比较广泛.报道过的纳米粒子催化剂有:硫酸化的纳米氧化锆 (A) 催化剂[25], 这类催化剂催化条件温和, 以乙醇为溶剂, 环境污染小, 原料中吸电子基团和供电子基团对产率没有影响.氨基磺酸改性的磁性纳米Fe3O4粒子 (Fe3O4-MNPs, B) 催化剂[26], 反应过程不需要溶剂参与, 微波辅助下大大减少了常规加热时所需要的时间, 产率均在93%以上.反应结束时, 催化剂可以回收使用, 催化活性没有明显变化.纳米Fe3O4负载壳聚糖 (C) 催化剂[27], 其合成中利用共沉淀法将Fe2+和Fe3+负载在壳聚糖上, 使反应更加绿色环保 (Eq. 4).在纳米催化剂催化下, 以二苯基乙二酮、有机胺类、乙酸铵和一系列芳香醛四组分一锅法合成1, 2, 4, 5-四取代咪唑的方法也有广泛报道.所用的纳米催化剂有:前文提到的硫酸化的纳米氧化锆 (D) 催化剂[25], 同样可以应用到1, 2, 4, 5-四取代咪唑的合成, 反应条件温和.纳米Al2O3粒子 (E) 催化剂[28], 在紫外光照下, 乙醇中回流反应, 反应时间只有25 min (Eq. 4).
2013年, Majee等[29]采用纳米In2O3催化剂, 高产率地合成了不对称取代的1H-咪唑.反应原料为脒类化合物和不同取代基的硝基烯烃, 推测其机理为In3+催化脒类化合物和硝基烯进行Michael加成反应 (Scheme 4).
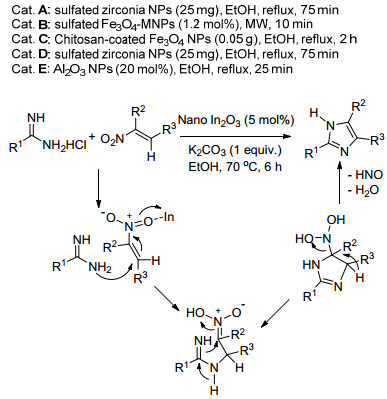 图 图式4
纳米In2O3催化咪唑类化合物合成机理
Figure 图式4.
The possible mechanism for the formation of substituted imidazoles catalyzed by In2O3NPs
图 图式4
纳米In2O3催化咪唑类化合物合成机理
Figure 图式4.
The possible mechanism for the formation of substituted imidazoles catalyzed by In2O3NPs
2 纳米金属氧化物催化多环含氮杂环化合物的合成
2.1 含有两个环的含氮杂环类化合物的合成
2.2 含有多个环的含氮杂环类化合物的合成
2013年, Majee等[64]设计一种纳米CuO催化异吲哚喹唑啉酮类化合物合成方法, 反应水溶液中进行.水通过氢键与反应物和催化剂结合, 在加速反应中起到重要的电子桥梁作用.这种方法反应适用性广, 催化靛红酸酐, 2-甲酰基苯甲酸和胺类化合物反应, 其中脂肪胺和芳香胺为原料均取得较高的产率, 反应中催化剂可以重复使用多次, 这种异吲哚并喹唑啉酮类化合物的绿色的合成方法在工业上有很大的应用前景 (Eq. 23).
2015年, Yavari等[65]设计了一种新型的非均相核壳结构的纳米粒子催化剂Fe3O4/SiO2/(CH2)3N+Me3Br3-, 在无溶剂的条件下催化合成苯并噻唑及咪唑和嘧啶等复杂稠环含氮化合物.由于催化剂的引入, 大大缩短了反应时间, 提高了产物产率.该方法底物适用性好, 反应操作及后处理简单, 成为绿色合成复杂稠环化合物的一种新方法 (Eq. 24).
2015年, Maleki等[66]利用磁性Fe3O4粒子负载在壳聚糖上作为催化剂, 在温和的反应条件下 (40 ℃, 乙醇为溶剂) 催化2-氨基苯并噻唑或2-氨基苯并咪唑、二甲基环已烷二酮与系列芳香醛反应, 一锅法高产率地得到四环的稠杂环苯并咪唑并喹唑啉酮类化合物.这类催化剂对环境无污染且可回收多次使用, 并且合成方法具有很多优点, 如反应过程中避免了有毒溶剂的使用, 过程简单易操作, 产率高 (Eq. 25).
本课题组在纳米金属氧化物催化合成喹唑啉酮衍生物的合成研究取得一定的进展, 喹唑啉酮类化合物因其特殊的含氮杂环骨架及特殊的生理活性而吸引了合成研究者的广泛关注.本课题组使用纳米CuO为催化剂, 靛红酸酐、芳香醛、芳香胺为原料, 一锅法合成了2, 3-2(H) 二取代的喹唑啉酮类化合物, 当使用无机盐氯化铵替代芳香胺作为氮源时, 得到了3位取代的喹唑啉酮类化合物[67].利用纳米CeO2为催化剂合成了螺环吲哚喹唑啉酮类化合物[68]及3-酰胺基喹唑啉酮类化合物[69].这两类化合物分别具有一定的抗细菌及抗真菌的作用, 利用超声辅助的方法, 在纳米氧化铜催化条件下得到硫代的喹唑啉酮类化合物, 该类化合物对植物病原菌有着良好的抑制活性[70].在纳米TiO2的催化下, 靛红酸酐、邻甲酰基苯甲酸分别与肼和酰肼类化合物反应, 可得到喹唑啉酮并酞嗪酮及3-酰胺基喹唑啉酮并异吲哚酮类化合物[71].这几种合成方法简洁高效, 反应条件温和, 同时取得了较高的产率 (Scheme 9).
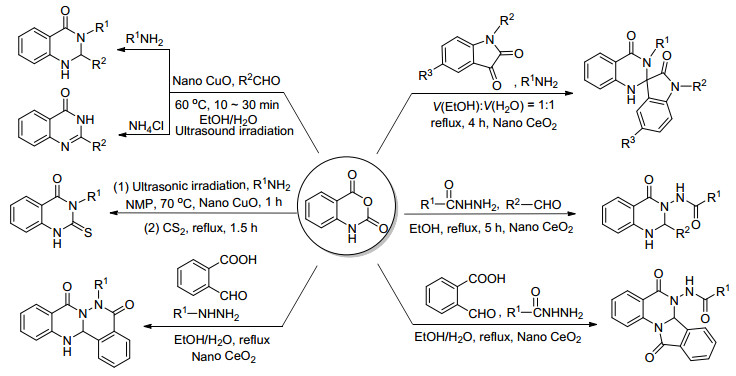 图 图式9
本课题组用纳米金属氧化物在合成喹唑啉酮类含氮杂环类化合物
Figure 图式9.
Synthesis ofN-fused quinazolinones catalyzed by nanoparticles in our group
图 图式9
本课题组用纳米金属氧化物在合成喹唑啉酮类含氮杂环类化合物
Figure 图式9.
Synthesis ofN-fused quinazolinones catalyzed by nanoparticles in our group
2.1.3 苯并噻唑的合成
2015年, Heydari等[50]采用一种新的途径合成2-氨基苯并噻唑衍生物, 利用邻碘苯胺和异硫氰酸酯为原料, 用改性后的纳米Fe3O4[Fe3O4@EDTA-Cu (Ⅱ)]为催化剂, 以水为溶剂, 高效地得到目标产物.该反应催化条件温和, 但催化过程中要加入碱性物质三乙烯二胺 (DABCO) 和添加剂四丁基溴化铵 (TABA), 使反应复杂.该反应是合成2-氨基苯并噻唑的一种重要方法 (Eq. 14).
2015年, Amoozadeh等[51]在无溶剂条件下用磺酸负载的纳米TiO2催化剂成功催化2-氨基苯硫酚和芳香醛类化合物合成苯并噻唑类衍生物, 带有不同电子基团的芳香醛为原料参与反应均得到良好的产率. 2016年, Bahrami等[52]同样引入纳米TiO2作为催化剂, 与H2O2共同催化苯并咪唑和苯并噻唑两类化合物的合成 (Eq. 15).原料分别为取代的邻苯二胺或2-氨基苯硫酚与系列的芳香醛类化合物.反应简单易操作, 用光照50 ℃下反应, 反应过程污染小, 反应时间短, 产物产率高.不同取代的原料均取得较高产率, 但是进一步实验证明, 该方法不适用于催化邻氨基苯酚和芳香醛合成苯并噁唑类化合物.
2016年, Paul等[35]同样采用Cu (0)-Fe3O4负载在二氧化硅上[Cu (0)-Fe3O4@SiO2/NH2] (Scheme 15) 中的催化剂结构) 的新型催化剂一锅法高效合成2-取代的苯并噻唑类化合物.催化剂重复使用6次催化活性没有明显变化, 反应在水溶剂中进行, 这种合成方法将在绿色有机合成中有更广泛的应用 (Eq. 16).
2.1.5 吡唑并酞嗪酮类化合物的合成
2012年, Azarifar等[60]利用纳米ZnO为催化剂合成了一系列吡咯并酞嗪酮类化合物.该方法具有反应时间短, 产物后处理简单且产率高的优点 (Eq. 19).
2.1.2 苯并咪唑的合成
纳米氧化物催化剂催化由不同原料合成苯并咪唑类化合物的研究有广泛报道, Shiraishi等[39]报道了在光和Pt负载在TiO2纳米粒子为催化剂同时催化下, 邻苯二胺和伯醇反应在N2保护室温条件下合成2位取代的苯并咪唑类化合物, 产率高达99%. Karami等[40]以可回收的磁性Fe3O4为催化剂, 无溶剂条件下以邻苯二胺和原甲酸三甲酯或原乙酸三甲酯为原料合成2位取代的苯并咪唑类化合物. Alinezhad等[41]利用纳米ZnO催化邻苯二胺和甲酸在无溶剂条件下合成苯并咪唑类衍生物, 原料简单易得, 反应条件也比较温和. Majee等[42]发明了以纳米In2O3为催化剂合成1, 2-二取代的苯并咪唑类化合物, 用乙醇和水的体积比2:1为溶剂, 绿色环保, 反应条件温和, 产率在84%~92%之间 (Scheme 6).
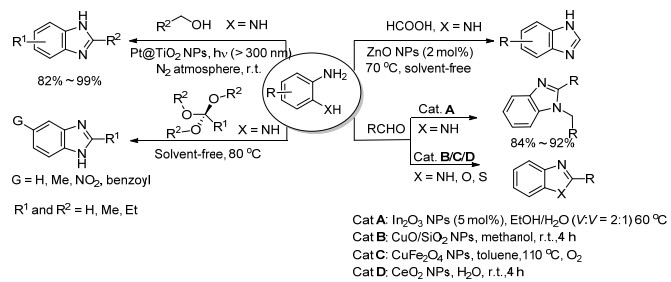 图 图式6
不同纳米金属氧化物催化苯并咪唑类化合物的合成
Figure 图式6.
One-pot synthesis of benzimidazole derivatives using different nanoparticles
图 图式6
不同纳米金属氧化物催化苯并咪唑类化合物的合成
Figure 图式6.
One-pot synthesis of benzimidazole derivatives using different nanoparticles
在由邻二苯胺合成苯并咪唑的同时, 同一种催化剂对不同的反应原料也有很好的适用性, 由邻氨基苯酚和邻氨基苯硫酚为原料分别可以得到苯并噁唑及苯并噻唑. 2013年, Mandal等[43]将纳米CuO颗粒负载在二氧化硅上, 作为一种新的催化剂催化一系列的苯并杂环类化合物的反应, 如苯并噁唑、苯并咪唑、苯并噻唑等.该反应利用甲醇为溶剂, 在室温下反应4 h, 催化剂可多次回收利用, 反应应用范围广, 产物产率高. 2014年, Wang等[44]设计采用纳米CuFe2O4为催化剂合成这三类衍生物, 并证明空气中氧气的存在使反应中间体氧化, 最终得到杂环产物. 2013年, Nagarkar等[45]利用非均相可回收纳米CeO2为催化剂用同样的原料合成苯并咪唑、苯并噁唑及苯并噻唑类衍生物, 反应在室温条件下进行, 反应时间仅为30 min左右.采用水作为溶剂, 更加绿色环保, 并且底物适用范围广, 对不同种类的醛 (芳香醛、杂环芳香醛、不饱和醛、脂肪醛) 为原料均得到高产率的化合物. Hajipour等[46]通过对此类化合物与乳腺癌细胞雌性激素的受体之间的作用研究表明, 这类化合物是一类潜在的雌性激素抑制剂 (Scheme 6).
在苯并咪唑类化合物合成中, 采用不同的活泼羰基的化合物为原料时, 同样的催化剂可能得到完全不同的产物. 2014年, Pal等[47]使用磁性纳米Fe3O4在无溶剂条件下催化两种化合物的合成, 当使用邻苯二胺和醛类为原料时, 合成得到1, 2-苯并咪唑类衍生物; 当使用邻苯二胺和芳香酮类或脂肪酮类为原料时, 合成得到1, 5-吩噻嗪类衍生物.反应温度为80 ℃, 反应时间为10 min左右.同年, Maleki等[48]设计合成一种生物大分子 (壳聚糖) 负载的磁性纳米非均相催化剂, 使这两类化合物的合成向更加环境友好的方向发展, 而且反应均在室温下进行, 溶剂为乙醇, 产率高达97%.这种合成方法更加简单易行, 合成条件温和, 催化剂可重复利用, 原子经济性高 (Scheme 7).
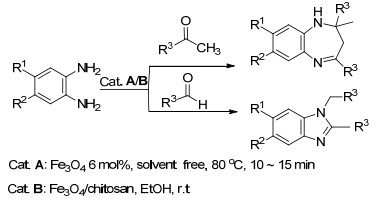 图 图式7
不同纳米金属氧化物催化苯并咪唑和1, 5-吩噻嗪类衍生物的合成
Figure 图式7.
Synthesis of benzimidazoles and 1, 5-benzodiaze-pines using different NPs
图 图式7
不同纳米金属氧化物催化苯并咪唑和1, 5-吩噻嗪类衍生物的合成
Figure 图式7.
Synthesis of benzimidazoles and 1, 5-benzodiaze-pines using different NPs
2015年, Singh等[49]通过离子液体负载的纳米ZnO颗粒, 使用球磨法在室温下选择性地催化合成1, 2-双取代的苯并咪唑类衍生物.在协同催化的作用下, 催化剂用量为0.2 mol%, 反应时间为22 min, 球磨转速为600 r/min, 产物全部为二取代的苯并咪唑衍生物 (Eq. 13).该方法产物后处理简单, 不需要溶剂参与, 催化剂需要量少且可以回收.与其他反应方法相比, 呈现出很好的环境友好性, 有很好的工业应用前景.
2.1.7 吡喃并吡唑类化合物的合成
2014年, Das等[63]利用未被修饰SnO2量子点催化多取代的吡喃并吡唑类和螺环吲哚类衍生物的合成取得了很好的效果.量子点是三个维度尺寸都在100 nm以下的纳米材料, 量子点具有许多独特的纳米性质.催化剂的量少并且产物产率高, 对不同取代基团的原料适用性好.还考察了油酸修饰的SnO2量子点, SnO2纳米花等不同形貌的纳米SnO2形貌对催化反应的影响, 结果表明未修饰的SnO2量子点催化效率最高.催化剂重复使用6次而不影响其催化活性 (Eq. 22)
2.1.1 苯并噁唑的合成
由纳米氧化物合成苯并噁唑类化合物的方法被广泛报道, 以邻氨基苯酚为原料, 2013年, Vahdat等[36]应用纳米SnO2作为一种绿色可回收催化剂, 用乙醇作为溶剂, 催化邻氨基苯酚类衍生物分别与芳香醛类化合物和原甲酸甲酯在室温条件下反应, 分别得到芳基取代的苯并噁唑衍生物和烷基取代的苯并噁唑衍生物 (Eq. 10).这种合成方法产率高, 催化剂用量少且可多次回收使用, 合成时间仅为30 min左右, 有广泛的应用前景. 2014年, Banerjee等[37]利用纳米ZnO催化剂在室温下催化合成了一系列有生物活性的2-芳基-1, 3-苯并噁唑的衍生物.当使用2-氨基苯硫酚为原料时, 该方法同时适用于合成1, 3-苯并噁唑类化合物, 加入催化剂后, 反应时间大大缩短到8 min, 产率高达90%以上 (Eq. 11). 2015年, Maleki等[38]设计了一种Ag负载在TiO2上的纳米粒子 (Ag@TiO2) 催化剂, 应用于一锅法水相合成苯并噁唑类衍生物, 该反应中由于催化剂的使用, 大大拓展了反应原料的范围, 邻氨基苯酚可以分别与不同的芳香醛、芳香酰胺、羧酸、原甲酸酯、芳香酰氯类化合物反应制得目标产物, 并且反应在室温条件下进行, 反应时间为5 min左右 (Eq. 12).
2.1.6 咪唑并吡啶类化合物的合成
2015年, Foroumadj等[61]引入γ构型的Fe2O3纳米粒子催化剂γ-Fe2O3-Hap-(CH2)3-NHSO3H, 在无溶剂条件下, 利用三组分一锅高效合成了一类喹啉与咪唑并吡啶连接的稠杂环的化合物 (Eq. 20).
2016年, Shrivastava等[62]成功设计可回收的非均相纳米ZnO催化剂, 在乙醇为溶剂回流的条件下, 合成了一类和吡唑相连的咪唑并吡啶类稠杂环化合物, 这两类合成途径操作简单, 反应时间少, 催化剂重复利用催化活性没有明显减少, 反应条件温和无毒, 高效构建了稠杂环的咪唑并吡啶类化合物的衍生物 (Eq. 21).
2.1.4 喹喔啉类化合物的合成
有很多报道关于纳米氧化颗粒催化喹喔啉类化合物的合成, 原料通常为邻苯二胺和1, 2-二羰基化合物, 报道过的催化剂有Fe3O4@SiO2/席夫碱类纳米粒子[53]、纳米ZrO2催化剂[54]、淀粉为负载的纳米ZrO2和磁铁矿颗粒 (ZrOL2@SMNP)[55]、纳米Co粒子[56]、负载在SiO2聚甲基丙烯腈酯上的纳米Fe3O4颗粒 (Fe3O4@SiO2-imid-PMA)[57]等 (Eq. 17).
2014年, Lee等[58]利用纳米CeO2为催化剂高效催化三组分合成2-氨基喹喔啉类化合物, 反应原料为邻苯二胺、芳香醛、异腈类衍生物, 反应取得很高产率, 并且用水为溶剂, 反应过程绿色环保.值得注意的是, 当适用靛红为醛类化合物参与三组分反应时, 并没有获得相应的螺环吲哚类化合物, 而是得到了并环的吲哚并喹喔啉类化合物.醛基在纳米氧化物的作用下活化, 得到亚胺离子与异腈发生亲核取代反应生成中间体通过环加成反应, 最终被氧化生成喹喔啉类化合物.可以总结由纳米粒子参与的过程大都生成了亚胺中间体 (Scheme 8).
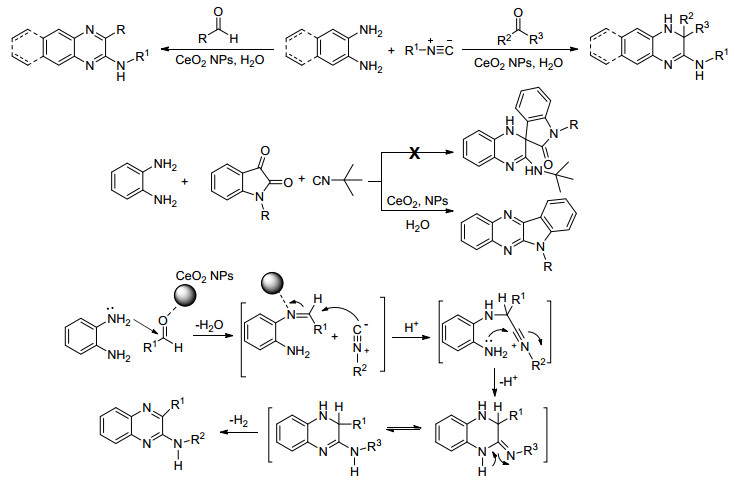 图 图式8
纳米CeO2粒子催化合成喹喔啉类化合物机理图
Figure 图式8.
The possible mechanism for the formation of quinaxolin-2-amine derivatives catalyzed by CeO2 NPs
图 图式8
纳米CeO2粒子催化合成喹喔啉类化合物机理图
Figure 图式8.
The possible mechanism for the formation of quinaxolin-2-amine derivatives catalyzed by CeO2 NPs
2015年, Wang等[59]设计合成一种有孔隙的Fe3O4@Cu2O纳米粒子作为合成喹喔啉类化合物的高效可回收催化剂, 这类催化剂为合成喹喔啉化合物提供了新的途径, 采用原料为邻苯二胺和端炔类化合物, 加入添加剂二甲氨基吡啶 (DMAP) 和碳酸铯 (Cs2CO3), 在甲苯为溶剂条件下回流反应.催化剂可回收多次使用且选择性高, 当邻苯二胺上带有吸电子性的取代基团时, 产物产率为零 (Eq. 18).
3 结论
综上所述, 纳米金属氧化物因其材料结构及比表面积的特殊性, 良好的催化活性和可重复利用等优点被不断开发和应用于多种含氮的稠杂环化合物的合成中, 近年来纳米金属氧化物不但自身可以作为催化剂, 还可以负载其他贵金属等作为催化剂, 使催化范围大大增加.随着人们对新药的不断开发, 对催化合成方法的不断探索, 对催化剂的进一步修饰, 得到高效、经济、绿色的合成方法仍将是今后研究的热点.因此纳米金属氧化物催化剂将在催化合成方面有巨大的应用前景.
-
-
[1]
Shafiee, A. Eur. J. Med. Chem. 2015, 97, 334. doi: 10.1016/j.ejmech.2015.03.043
-
[2]
Rani, R.; Granchi, C. Eur. J. Med. Chem. 2015, 97, 505. doi: 10.1016/j.ejmech.2014.11.031
-
[3]
Yang, R.; Gao, Z.-F.; Zhao, J.-Y.; Li, W.-B.; Zhou, L.; Miao, F. J. Agric. Food Chem. 2015, 63, 1906. doi: 10.1021/jf505609z
-
[4]
Manivannan, E.; Chaturvedi, S. C. Bioorg. Med. Chem. 2011, 19, 4520. doi: 10.1016/j.bmc.2011.06.019
-
[5]
Giardina, D.; Martarelli, D.; Sagratini, G.; Angeli, P.; Ballinari, D.; Gulini, U.; Melchiorre, C.; Poggesi, E.; Pompei, P. J. Med. Chem. 2009, 52, 4951. doi: 10.1021/jm8016046
-
[6]
Wermuth, C. G.; Schlewer, G.; Bourguignon, J. J.; Maghioros, G.; Bouchet, M. J.; Moire, C.; Kan, J. P.; Worms, P.; Biziere, K. J. Med. Chem. 1989, 32, 528. doi: 10.1021/jm00123a004
-
[7]
Rueda, L.; Castellote, I.; Castro-Pichel, J.; Chaparro, M. J.; de la Rosa, J. C.; Garcia-Perez, A.; Gordo, M.; Jimenez-Diaz, M. B.; Kessler, A.; Macdonald, S. J. F.; Martinez, M. S.; Sanz, L. M.; Gamo, F. J.; Fernandez, E. ACS Med. Chem. Lett. 2011, 2, 840. doi: 10.1021/ml2001517
-
[8]
Wang, J.; Tan, H.; Li, Y.; Ma, Y.; Li, Z.; Guddat, L. W. J. Agric. Food Chem. 2011, 59, 9892. doi: 10.1021/jf2021607
-
[9]
胡泉子, 秦瑗, 席海涛, 孙小强, 合成化学, 2011, 19, 480.Hu Q. Z.; Qin Y; Xi H. T; Sun X. Q. Chin. J. Synth. Chem. 2011, 19, 480 (in Chinese).
-
[10]
Yang, W.; Chen, J.; Huang, X.; Ding, J.; Liu, M.; Wu, H. Org. Lett. 2014, 16, 5418. doi: 10.1021/ol5026553
-
[11]
丁正伟, 谭启涛, 刘秉新, 张可, 许斌, 化学学报, 2015, 73, 1302. doi: 10.6023/A15040263Ding, Z. W.; Tan, Q. T.; Liu, B. X.; Zhang, K.; Xu, B. Acta Chim. Sinica 2015, 73, 1302 (in Chinese). doi: 10.6023/A15040263
-
[12]
赵金钵, 张前, 化学学报, 2015, 73, 1235. doi: 10.6023/A15010063Zhao, J. B.; Zhang, Q. Acta Chim. Sinica 2015, 73, 1235 (in Chinese). doi: 10.6023/A15010063
-
[13]
张钊瑞, 郑晓霖, 郭长彬, 有机化学, 2016, 36, 1241. doi: 10.6023/cjoc201511004Zhang, Z. R.; Zheng, X. L.; Guo, C. B. Chin. J. Org. Chem. 2016, 36, 1241 (in Chinese). doi: 10.6023/cjoc201511004
-
[14]
季益刚, 吴磊, 范青华, 化学学报, 2014, 72, 798. doi: 10.6023/A14040325Ji, Y. G.; Wu, L.; Fan, Q. H. Acta Chim. Sinica 2014, 72, 798 (in Chinese). doi: 10.6023/A14040325
-
[15]
Elwahy, A. H. M.; Shaaban, M. R. RSC Adv. 2015, 5, 75659. doi: 10.1039/C5RA11421G
-
[16]
Teimouri, A.; Chermahini, A. N. Chin. J. Chem. 2012, 30, 372. doi: 10.1002/cjoc.v30.2
-
[17]
Ma, F. P.; Li, P. H.; Li, B. L.; Mo, L. P.; Liu, N.; Kang, H. J.; Liu, Y. N.; Zhang, Z. H. Appl. Catal. A:Gen. 2013, 457, 34. doi: 10.1016/j.apcata.2013.03.005
-
[18]
Li, B.; Hu, H.; Mo, L.; Zhang, Z. RSC Adv. 2014, 4, 12929. doi: 10.1039/c3ra47855f
-
[19]
Li, B.; Zhang, M.; Hu, H.; Du, X.; Zhang, Z. New J. Chem. 2014, 38, 2435. doi: 10.1039/c3nj01368e
-
[20]
Sabbaghan, M.; Ghalaei, A. J. Mol. Liq. 2014, 193, 116. doi: 10.1016/j.molliq.2013.12.018
-
[21]
Hajinasiri, R.; Hossaini, Z.; Sheikholeslami-Farahani, F. Comb. Chem. High Throughput Screening 2015, 18, 42. doi: 10.2174/1386207317666141203123133
-
[22]
Azgomi, N.; Mokhtary, M. J. Mol. Catal. A:Chem. 2015, 398, 58. doi: 10.1016/j.molcata.2014.11.018
-
[23]
Sobhani, S.; Honarmand, M. Appl. Catal. A:Gen. 2013, 467, 456. doi: 10.1016/j.apcata.2013.08.006
-
[24]
Eshghi, H.; Khojastehnezhad, A.; Moeinpour, F.; Rezaeian, S.; Bakavoli, M.; Teymouri, M.; Rostami, A.; Haghbeen, K. Tetrahedron 2015, 71, 436. doi: 10.1016/j.tet.2014.12.010
-
[25]
Teimouri, A.; Chermahini, A. N. J. Mol. Catal. A:Chem. 2011, 346, 39. doi: 10.1016/j.molcata.2011.06.007
-
[26]
Safari, J.; Zarnegar, Z. J. Chem. Sci. 2013, 125, 835. doi: 10.1007/s12039-013-0462-2
-
[27]
Zarnegar, Z.; Safari, J. RSC Adv. 2014, 4, 20932. doi: 10.1039/c4ra03176h
-
[28]
Reddy, B.; Vijayakumar, V.; Arasu, M.; Al-Dhabi, N. Molecules 2015, 20, 19221. doi: 10.3390/molecules201019221
-
[29]
Mitra, S.; Bagdi, A. K.; Majee, A.; Hajra, A. Tetrahedron Lett. 2013, 54, 4982. doi: 10.1016/j.tetlet.2013.07.050
-
[30]
Zakerinasab, B.; Nasseri, M. A.; Hassani, H.; Samieadel, M. M. Res. Chem. Intermed. 2015, 42, 3169.
-
[31]
Sangshetti, J. N.; Dharmadhikari, P. P.; Chouthe, R. S.; Fatema, B.; Lad, V.; Karande, V.; Darandale, S. N.; Shinde, D. B. Bioorg. Med. Chem. Lett. 2013, 23, 2250. doi: 10.1016/j.bmcl.2013.01.041
-
[32]
Murty, M. S. R.; Penthala, R.; Buddana, S. K.; Prakasham, R. S.; Das, P.; Polepalli, S.; Jain, N.; Bojja, S. Med. Chem. Res. 2014, 23, 4579. doi: 10.1007/s00044-014-1025-x
-
[33]
Kamal, A.; Swapna, P. RSC Adv. 2013, 3, 7419. doi: 10.1039/c3ra22485f
-
[34]
Salam, N.; Sinha, A.; Mondal, P.; Roy, A. S.; Jana, N. R.; Islam, S. M. RSC Adv. 2013, 3, 18087. doi: 10.1039/c3ra43184c
-
[35]
Bhardwaj, M.; Jamwal, B.; Paul, S. Catal. Lett. 2016, 146, 629. doi: 10.1007/s10562-015-1672-7
-
[36]
Vahdat, S. M.; Raz, S. G.; Baghery, S. J. Chem. Sci. (Bangalore, India) 2014, 126, 579. doi: 10.1007/s12039-013-0544-1
-
[37]
Banerjee, S.; Payra, S.; Saha, A.; Sereda, G. Tetrahedron Lett. 2014, 55, 5515. doi: 10.1016/j.tetlet.2014.07.123
-
[38]
Maleki, B.; Baghayeri, M.; Vahdat, S. M.; Mohammadzadeh, A.; Akhoondi, S. RSC Adv. 2015, 5, 46545. doi: 10.1039/C5RA06618B
-
[39]
Shiraishi, Y.; Sugano, Y.; Tanaka, S.; Hirai, T. Angew. Chem., Int. Ed. 2010, 49, 1656. doi: 10.1002/anie.200906573
-
[40]
Karami, B.; Nikoseresht, S.; Khodabakhshi, S. Chin. J. Catal. 2012, 33, 298. doi: 10.1016/S1872-2067(11)60329-X
-
[41]
Alinezhad, H.; Salehian, F.; Biparva, P. Synth. Commun. 2012, 42, 102. doi: 10.1080/00397911.2010.522294
-
[42]
Santra, S.; Majee, A.; Hajra, A. Tetrahedron Lett. 2012, 53, 1974. doi: 10.1016/j.tetlet.2012.02.021
-
[43]
Inamdar, S. M.; More, V. K.; Mandal, S. K. Tetrahedron Lett. 2013, 54, 579. doi: 10.1016/j.tetlet.2012.11.091
-
[44]
Yang, D.; Zhu, X.; Wei, W.; Sun, N.; Yuan, L.; Jiang, M.; You, J.; Wang, H. RSC Adv. 2014, 4, 17832. doi: 10.1039/C4RA00559G
-
[45]
Shelkar, R.; Sarode, S.; Nagarkar, J. Tetrahedron Lett. 2013, 54, 6986. doi: 10.1016/j.tetlet.2013.09.092
-
[46]
Hajipour, A. R.; Khorsandi, Z.; Mortazavi, M.; Farrokhpour, H. RSC Adv. 2015, 5, 107822. doi: 10.1039/C5RA22207A
-
[47]
Jamatia, R.; Saha, M.; Pal, A. K. RSC Adv. 2014, 4, 12826. doi: 10.1039/c3ra47860b
-
[48]
Maleki, A.; Ghamari, N.; Kamalzare, M. RSC Adv. 2014, 4, 9416. doi: 10.1039/c3ra47366j
-
[49]
Sharma, H.; Kaur, N.; Singh, N.; Jang, D. O. Green Chem. 2015, 17, 4263. doi: 10.1039/C5GC00536A
-
[50]
Azizi, K.; Karimi, M.; Heydari, A. Tetrahedron Lett. 2015, 56, 812. doi: 10.1016/j.tetlet.2014.12.110
-
[51]
Amoozadeh, A.; Azadeh, R. A.; Rahmani, S.; Salehi, M.; Kubicki, M.; Dutkiewicz, G. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1874. doi: 10.1080/10426507.2015.1031753
-
[52]
Bahrami, K.; Khodaei, M. M.; Naali, F. J. Exp. Nanosci. 2016, 11, 148. doi: 10.1080/17458080.2015.1038659
-
[53]
Esmaeilpour, M.; Sardarian, A. R. Green Chem. Lett. Rev. 2014, 7, 301. doi: 10.1080/17518253.2014.948078
-
[54]
Jafarpour, M.; Rezapour, E.; Ghahramaninezhad, M.; Rezaeifard, A. New J. Chem. 2014, 38, 676. doi: 10.1039/C3NJ00655G
-
[55]
Jafarpour, M.; Rezaeifard, A. Transition Met. Chem. 2015, 41, 205.
-
[56]
Rajabi, F.; Alves, D.; Luque, R. Molecules 2015, 20, 20709. doi: 10.3390/molecules201119731
-
[57]
Javidi, J.; Esmaeilpour, M. Mater. Res. Bull. 2016, 73, 409. doi: 10.1016/j.materresbull.2015.10.002
-
[58]
Edayadulla, N.; Lee, Y. R. RSC Adv. 2014, 4, 11459. doi: 10.1039/c4ra00717d
-
[59]
Wang, Z.; Hu, G.; Liu, J.; Liu, W.; Zhang, H.; Wang, B. Chem. Commun. 2015, 51, 5069. doi: 10.1039/C5CC00250H
-
[60]
Azarifar, A.; Nejat-Yami, R.; Azarifar, D. J. Iran Chem. Soc. 2012, 10, 297.
-
[61]
Mouradzadegun, A.; Ma'mani, L.; Mahdavi, M.; Rashid, Z.; Shafiee, A.; Foroumadi, A.; Dianat, S. RSC Adv. 2015, 5, 83530. doi: 10.1039/C5RA12307K
-
[62]
Swami, S.; Devi, N.; Agarwala, A.; Singh, V.; Shrivastava, R. Tetrahedron Lett. 2016, 57, 1346. doi: 10.1016/j.tetlet.2016.02.045
-
[63]
Paul, S.; Pradhan, K.; Ghosh, S.; De, S. K.; Das, A. R. Tetrahedron 2014, 70, 6088. doi: 10.1016/j.tet.2014.02.077
-
[64]
Santra, S.; Bagdi, A. K.; Majee, A.; Hajra, A. RSC Adv. 2013, 3, 24931. doi: 10.1039/c3ra43917h
-
[65]
Farrokhi, A.; Ghodrati, K.; Yavari, I. Catal. Commun. 2015, 63, 41. doi: 10.1016/j.catcom.2014.09.046
-
[66]
Maleki, A.; Aghaei, M.; Ghamari, N. Chem. Lett. 2015, 44, 259. doi: 10.1246/cl.141074
-
[67]
Zhang, J.; Ren, D. C.; Ma, Y. M.; Wang, W. T.; Wu, H. Tetrahedron 2014, 70, 5274. doi: 10.1016/j.tet.2014.05.059
-
[68]
Zhang, J.; Zhao, J. W.; Wang, L. P.; Liu, J.; Ren, D. C.; Ma, Y. M. Tetrahedron 2016, 72, 936. doi: 10.1016/j.tet.2015.12.055
-
[69]
Zhang, J.; Liu, J.; Ma, Y. M.; Ren, D. C.; Cheng, P.; Zhao, J. W.; Zhang, F.; Yao, Y. Bioorg. Med. Chem. Lett. 2016, 26, 2273. doi: 10.1016/j.bmcl.2016.03.052
-
[70]
张金, 程佩, 马养民, 刘佳, 苗智, 范超, 有机化学, 2016, 36, 1368. doi: 10.6023/cjoc201512008Zhang, J.; Cheng, P.; Ma, Y. M.; Liu, J.; Miao, Z.; Fan, C. Chin. J. Org. Chem. 2016, 36, 1368 (in Chinese). doi: 10.6023/cjoc201512008
-
[71]
张金, 刘佳, 马养民, 杨秀芳, 程佩, 范超, 卢萍, 高等学校化学学报, 2016, 37, 1629. http://www.cjcu.jlu.edu.cn/CN/abstract/abstract27834.shtmlZhang, J.; Liu, J.; Ma, Y. M.; Yang, X. F.; Cheng, P.; Fan, C.; Lu, P. Chem. J. Chin. Univ. 2016, 37, 1629 (in Chinese). http://www.cjcu.jlu.edu.cn/CN/abstract/abstract27834.shtml
-
[1]
-

图式2 纳米氧化锌催化生成吡咯类化合物机理图
Scheme 2 Possible mechanism for the formation of substituted pyrroles catalyzed by ZnO nanorods

图式3 MNPs@SiO2-IL催化合成噻唑类化合物机理图
Scheme 3 Possible mechanism for the formation of thiazole derivatives catalyzed by MNPs@SiO2-IL

图式4 纳米In2O3催化咪唑类化合物合成机理
Scheme 4 The possible mechanism for the formation of substituted imidazoles catalyzed by In2O3NPs

图式5 室温水相下纳米Cu (0)-Fe3O4@SiO2/NH2催化三唑类化合物的合成
Scheme 5 Synthesis of triazole derivatives catalyzed by Cu (0)-Fe3O4@SiO2/NH2 NPs in water at room temperature

图式6 不同纳米金属氧化物催化苯并咪唑类化合物的合成
Scheme 6 One-pot synthesis of benzimidazole derivatives using different nanoparticles

图式7 不同纳米金属氧化物催化苯并咪唑和1, 5-吩噻嗪类衍生物的合成
Scheme 7 Synthesis of benzimidazoles and 1, 5-benzodiaze-pines using different NPs

图式8 纳米CeO2粒子催化合成喹喔啉类化合物机理图
Scheme 8 The possible mechanism for the formation of quinaxolin-2-amine derivatives catalyzed by CeO2 NPs
-


 下载:
下载:








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 2670
- HTML全文浏览量: 292
 下载:
下载:
