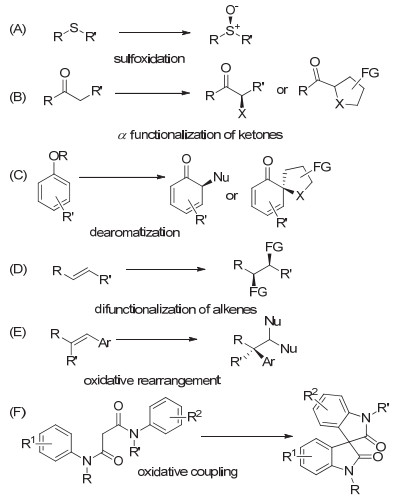
图 1.
手性高价碘促进的几类主要不对称转化
Figure 1.
Asymmetric transformations by hypervalent iodine reagents

近年来, 高价碘化学成为有机化学中的重要领域.由于其反应条件温和、安全高效、环境友好、低耗费以及操作简单等优点, 高价碘试剂引起了有机化学家的广泛关注, 被应用于各类反应转化.特别是近年来, 使用催化量的芳基碘化物和化学计量的氧化剂现场生成高价碘, 促进反应进行, 可以避免预先制备高价碘试剂的不便, 从而使高价碘化学的研究更为深入, 在各类反应中应用更为广泛[1, 2].
随着高价碘化学研究的不断深入, 手性高价碘促进的不对称转化, 也成为不对称合成研究中的一个重要领域, 取得了许多重要进展[3].但相对高价碘化学的蓬勃发展而言, 手性高价碘促进的不对称反应研究进展仍然相对缓慢.特别是与过渡金属催化的不对称反应相比较, 手性高价碘促进的不对称反应类型相对受限, 对映选择性相对不高.这一情况在很大程度上是由于手性高价碘试剂或前体发展缓慢所致.而对于反应机理认识不足, 也对手性高价碘试剂或前体的改造优化造成了困扰.
关于高价碘化学以及在不对称反应中的应用, 文献中已有很多的报道, 相关的综述以及书籍进行了大量的总结. 2011年Liang和Ciufolini[4]在综述中, 简单总结了关于手性高价碘试剂的发展以及四类主要的不对称反应(图 1, A~D).近几年来, 文献中关于手性高价碘的报道迅速增多, 目前已报道的手性高价碘试剂或前体有数十种, 但反应类型并未广泛拓展.我们在Liang和Ciufolini的基础上稍加整理, 在本综述中, 主要将穿插介绍取得重要进展的六类反应(图 1): (A)硫醚到亚砜的不对称氧化; (B)羰基化合物的α位不对称官能化; (C)酚类化合物的不对称去芳构化; (D)烯烃的不对称双官能团化; (E)烯烃的不对称氧化官能化重排反应; (F)不对称氧化偶联反应.此外, 其它的一些不对称转化也有所涉及.
目前文献中关于手性高价碘促进的各类不对称反应已经有较多的综述总结.在本文中, 我们不会对这些不对称反应本身做详细的探讨和分析, 而主要将针对手性高价碘试剂或前体的发展历史, 根据其骨架结构特点及时间线, 做一个简单的总结.
碘原子具有多种氧化态, 多种类型的手性碘, 包括一价、三价、五价、七价碘等, 在手性高价碘化学中都有所发展和应用.在目前的报道中, 手性碘(Ⅴ)有少量报道, 而手性碘(Ⅰ/Ⅲ)使用较为广泛.特别是Ochiai和Kita等[5]通过使用外加氧化剂氧化芳基碘化物现场生成碘(Ⅲ)的方法, 使原本需要当量使用的手性碘(Ⅲ)试剂, 可以改为催化量的手性碘(Ⅰ), 极大地方便了不对称反应的研究.
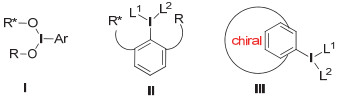
手性高价碘主要可以分为两类:一类是早期发展的手性高价碘试剂, 手性部分类似于过渡金属催化中的手性配体, 与碘原子相连(图 2, 式Ⅰ); 另一类则是整个分子骨架具有手性.后一种根据手性骨架的特点, 又可以分为具有柔性骨架和刚性骨架两类(图 2, 式Ⅱ和Ⅲ).下面我们将根据其结构特点, 具体的介绍这些手性高价碘化合物.
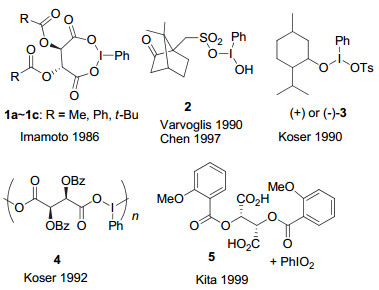
最早的手性高价碘试剂可以追溯到1907年, Pribram[6]制备了第一个手性高价碘试剂, 二苯基碘鎓酒石酸盐.但这一手性高价碘试剂并没有在不对称合成中得到关注和应用.直到1986年, Imamoto等[7]报道了氧化碘苯和L-酒石酸酐现场生成七元环的手性三价碘烷(图 3,1a~1c), 并应用到硫醚化合物转化为亚砜的不对称氧化, 才开启了手性高价碘促进的不对称反应研究大门.在Imamoto等的这一工作中, 能够以中等至良好的收率生成亚砜, 对映选择性达到53% ee(如图 4).
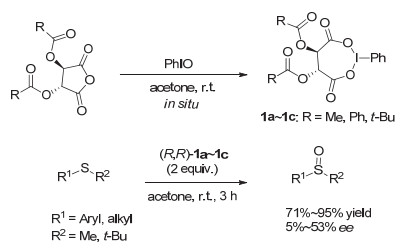
在Imamoto发展的这类芳基碘(Ⅲ)试剂中, 酒石酸部分类似于手性配体与碘苯结合, 控制不对称反应进行.反应结束后, 酒石酸部分与碘苯脱离.整个反应过程类似于过渡金属反应时配体与金属结合、脱除的过程.
之后其他研究小组如Varvoglis[8], Chen[9], Koser[10, 11]等先后报道了几类类似的手性芳基碘(Ⅲ)试剂(图 3, 2~4), 都是基于同样的手性配体形式的高价碘化物.这些手性芳基碘试剂在硫醚不对称氧化成亚砜的反应中, 都具有很好的反应活性, 但反应的对映选择性或非对映选择性不尽人意.与Imamoto的催化剂相比, 并无提高.
在早期发展的这些手性高价碘化合物中, 手性元素主要来源于“手性池”中的手性分子, 包括酒石酸、樟脑磺酸、薄荷醇等.由于手性取代基直接与高价碘原子相连, 这类高价碘试剂在反应结束后, 含碘部分被还原, 将从整个手性骨架上脱离下来.
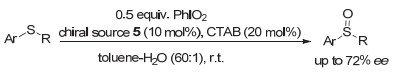
Kita等[12]选择酒石酸与五价碘试剂PhIO2作用, 现场生成手性的ⅠV试剂.在硫醚的不对称氧化反应中, 可以良好到优秀的收率得到亚砜产物, 对映选择性最高可以达到72% ee(图 5).这一反应中, 所采用的PhIO2为0.5 equiv., 手性源酒石酸衍生物为10 mol%.这是第一个催化的手性高价碘促进的不对称转化, 具有重要的意义.而且在这一反应中, 作者对可能的中间活化物种进行了探讨, 认为芳环上的邻位甲氧基对形成的手性高价碘物种可能存在配位作用, 从而有利于反应对映选择性的提高.这一推断, 对后来进一步的手性高价碘试剂或前体的设计, 显然具有重要的启发作用.
早期的手性高价碘试剂, 手性部分像配体一样与高价碘相连, 随着反应进行, 高价碘结构被还原, 从而从手性配体部分脱离.而在20世纪90年代以后, 对手性高价碘试剂或前体的认识不断深入, 基于手性骨架的高价碘试剂或前体得以发展.在本节中, 我们将首先介绍基于中心手性柔性骨架的高价碘试剂及前体.
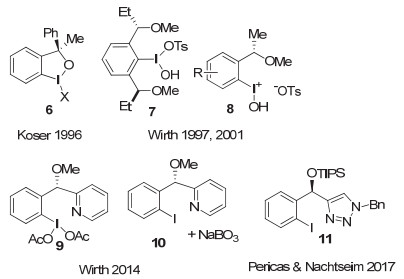
Koser[13], Wirth[14]等在1996和1997年先后报道了一类手性高价碘试剂, 代表性结构为图 6中的6~8.在这类试剂中, 手性的醇或醚取代基连接在碘原子的邻位芳基上.这一改变具有显著的优点:一方面, 手性中心靠近碘原子的位置, 使手性控制更为容易; 另一方面, 在反应完成后, 芳基碘片段仍然连接在手性骨架上, 这就为进一步重新氧化为高价碘试剂提供了可能.也为后来使用手性芳基碘化物作为催化剂, 外加氧化剂来现场生成手性高价碘物种, 促进不对称转化奠定了基础.
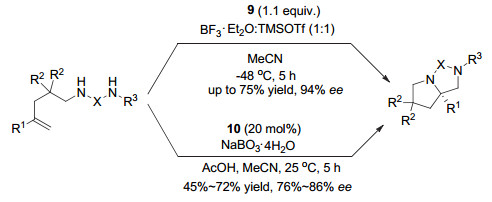
Wirth发展了一系列这类手性高价碘(Ⅰ/Ⅲ)试剂, 代表性例子包括手性高价碘(Ⅲ)试剂7和8.其中化合物7是一类具有C2-对称的手性单芳基碘(Ⅲ)试剂. Wirth等利用他们发展的手性芳基碘(Ⅲ)试剂, 在羰基的α位不对称官能团化、双键的不对称双官能团化反应中, 做了大量的工作.但大多情况下, 对映选择性不高.例如, 在酮的α-位磺酸酯化反应中, 对映选择性只能达到30% ee左右, 而在烯烃的双磺酸酯化反应中, 对映选择性最高也只能达到50% ee左右.一直到2014年, 他们[15]发展了另一个类似的手性碘(Ⅲ)试剂9, 在烯基化合物的分子内双胺化环化反应中, 才获得高的对映选择性, 产物最高获得94% ee.当使用前体芳基碘(Ⅰ)化合物10作为催化剂, NaBO3作为氧化剂, 他们首次实现了催化的烯烃分子内不对称双胺化过程, 可以中等的收率得到产物, 对映选择性最高达到86% ee(图 7).
2017年, Pericàs和Nachtsheim等[16]以这类结构为基础, 构建了一系列基于三氮唑的Koser类手性芳基碘化合物, 代表性结构为化合物11.他们采用这类芳基碘化物, 以m-CPBA作为氧化剂, 实现了萘酚类化合物的氧化Kita螺环内酯化反应.当使用当量的芳基碘化物时, 可以中等到良好的收率得到产物, 对映选择性最高达到86% ee, 通过简单重结晶, 可以获得99% ee.使用催化量(15 mol%)芳基碘化物时, 产物的收率和对映选择性有所降低(图 8).这一结果虽然不如下面将介绍的一些C2-对称手性芳基碘(Ⅰ/Ⅲ)促进的Kita螺环内酯化反应, 但在目前报道的C1-对称的手性芳基碘化物促进的同类反应中, 对映选择性最高.
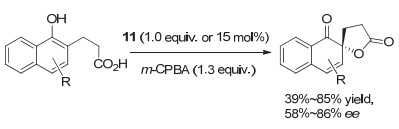
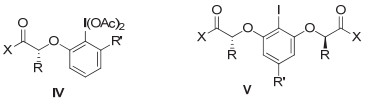
早期所发展的手性芳基碘试剂或催化剂, 在不对称转化中, 取得了重要的进展.但与过渡金属催化的各类反应相比, 对映选择性多差强人意.但从2007年以后, Fujita和Ishihara等基于乳酸, 发展了新类型的手性芳基碘试剂或前体, 其结构通式如图 9中的Ⅳ和Ⅴ结构.这类手性碘试剂和前体, 在不对称转化中取得了巨大的成功.本节中我们进行具体介绍.
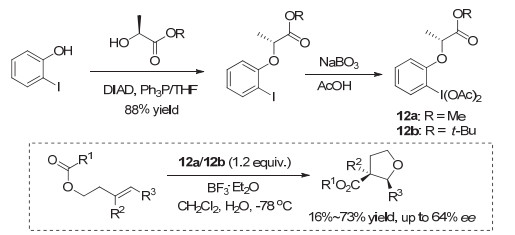
2007年, Fujita等[17]最先通过邻碘苯酚与乳酸的Mitsunobu反应, 在芳基碘的邻位引入乳酸手性片段, 发展了基于乳酸的新的手性碘(Ⅲ)试剂12a~12b.他们将这类手性碘(Ⅲ)试剂用于高烯丙醇酯的双键分子内不对称双官能团化环化反应, 取得了中等程度的对映选择性, 最高达到64% ee.但大多情况下, 由于副反应存在, 反应的收率较低(图 10).
尽管Fujita以乳酸为手性源发展的芳基碘(Ⅲ)试剂在不对称反应中的初次表现似乎并不比以往的手性芳基碘(Ⅰ/Ⅲ)更为出色, 但引起了Ishihara等的关注. Ishihara对Fujita发展的手性高价碘化合物做了进一步的改进, 最终迎来了手性高价碘化学领域的重大突破.
2010年, Ishihara等[18]利用乳酸为手性源, 发展了一类柔性的C2-对称的手性芳基碘化物, 通式结构为图 11中的结构13.作者在设计这一类手性芳基碘化物时, 既考虑了碘化物制备的简单性, 即三个片段之间通过简单化学反应结合的问题.也考虑了在形成的手性碘(Ⅲ)中间体中, 如何营造好的手性环境.他们认为三价碘与两个羰基氧之间形成的n-σ*相互作用以及碘(Ⅲ)上的配体与芳基碘化物之间形成的氢键相互作用, 为反应提供了很好的手性环境, 从而产生高对映选择性.这类芳基碘化物作为催化剂与m-CPBA共同作用, 在萘酚的不对称去芳构化螺环内酯化反应中, 产物可以达到90% ee左右, 通过简单重结晶, 可以获得99% ee值(图 11).而采用Fujita发展的单取代的芳基碘, 对映选择性只有30% ee左右.但这一反应收率受芳环取代基影响较大, 仍然存在较大的底物局限性.特别是带有给电子基团如甲氧基时, 收率较低, 可能是由于底物被氧化为二醌等副反应导致.
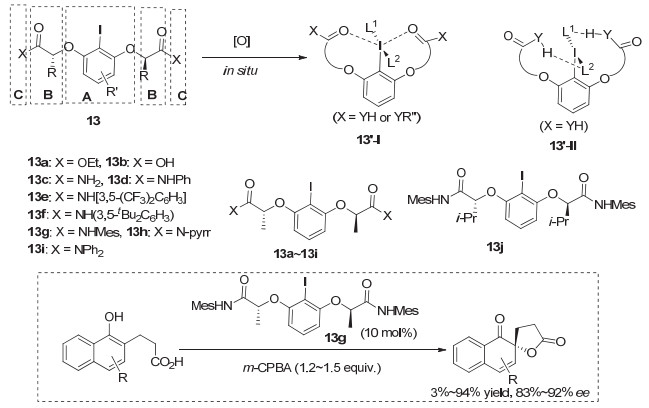
此前所发展的手性高价碘促进的不对称反应, 与过渡金属促进的反应相比, 对映选择性都相对较低, 因而实用性受到很大限制.而Ishihara发展的这一类基于乳酸的手性芳基碘化物, 真正展现了手性有机碘化物在不对称合成中隐藏的巨大的潜力.因此也引起了广泛的关注, 不少课题组发展了基于乳酸结构的手性碘(Ⅰ/Ⅲ)化合物, 并应用到多种类型的不对称转化.
Fujita等[19~21]在他们原来单取代的芳基碘(Ⅲ) 12a/12b的基础上, 进行进一步的改进.例如在碘的另一个邻位引入取代基, 形成2, 6-位双取代的手性碘(Ⅲ)试剂, 包括C2对称的手性碘(Ⅲ)试剂14等.他们运用这一类手性碘(Ⅲ)试剂, 在多种类型的烯烃双官能团化反应中, 取得了重要的进展(图 12).例如: 2010年, 他们[19]报道了邻羧基苯乙烯类化合物的内酯环化-对甲苯磺酸酯化反应, 对映选择性最高达到97% ee. 2011年, 他们[20]在烯烃的不对称分子间双官能团化反应中, 产物对映选择性高达96% ee. 2016年, 在烯烃的氧化环化芳基化反应中, 实现了高对映选择性地构建复杂的并杂环体系[21a].但在这些转化中, 由于多种副反应的存在, 大多情况下, 都只能得到中等的收率.
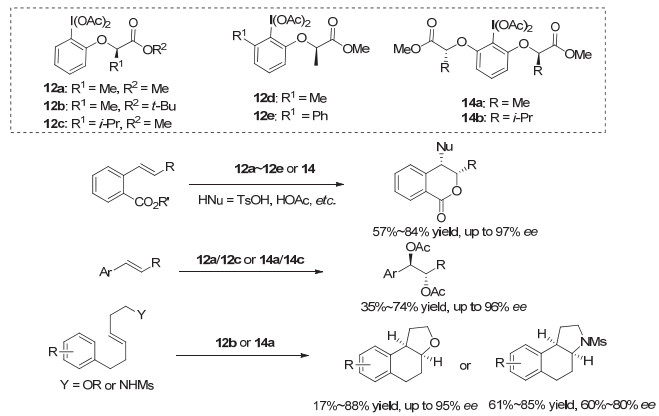
Muñiz等[22~24]采用这类C2对称的芳基碘(Ⅰ/Ⅲ)试剂, 高效地实现了一系列烯烃的双官能团化反应. 2011年, 他们[22]采用化学计量的芳基碘(Ⅲ)试剂14a, 高对映选择性地实现了苯乙烯类化合物的双胺化反应, ee值最高达到95%.对于苯丙烯, 反应生成两个手性中心, 同样可以获得高对映选择性.但对于烷基烯烃, 反应几乎没有对映选择性(图 13a). 2017年, 他们[23]进一步改进这类C2对称的芳基碘, 采用催化量的芳基碘(Ⅰ)化合物15a, 用m-CPBA作为氧化剂, 实现了高对映选择性的不对称催化双胺化反应, 对于苯乙烯和苯丙烯类底物, 产物均获得高达98% ee, 但大多情况下只有中等收率(图 13b).此外, 2016年, 他们[24]还采用催化量的芳基碘(Ⅰ)化物15b以及16, 实现了苯乙烯类化合物的高对映选择性双乙酸酯化反应, 产物收率中等到良好, 对映选择性最高达到94% ee(图 13c).
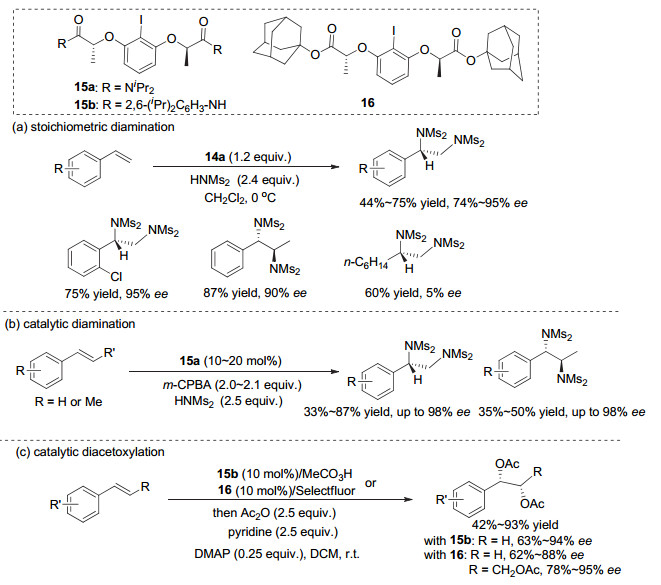
2012年, Wirth等[25]同样利用这类有机碘(Ⅲ)试剂17, 实现了烯烃分子内的不对称N, O双官能团化成环反应, 生成具有四取代的并环手性中心产物, ee值最高可以达到99%, 但仅限于分析量的小反应.在更大量的反应中, 反应收率在大多情况下不高, 并且对映选择性有所降低, 但仍然可以达到90% ee以上(图 14a).最近, Masson等[26]采用Ishihara的催化剂13g, 在烯烃不对称内酯环化反应中, 也取得了中等程度的收率以及较好的对映选择性, 最高达到94% ee(图 14b).
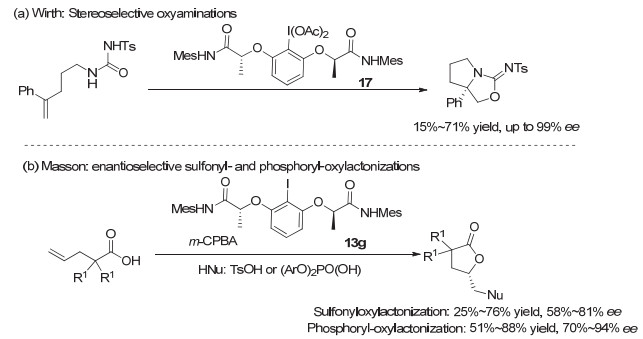
2013年, Nevado等则使用这类骨架发展了氧化的氟试剂, 代表性结构为化合物18a.他们[27a]采用手性芳基碘(Ⅲ)化合物18a, 首次发展了非活化烯烃的分子内高对映选择性不对称胺基化氟化反应, 以中等到良好的收率和对映选择性生成手性哌啶以及高哌啶产物.通过简单重结晶, 可以获得99% ee(图 15a).他们在苯乙烯类化合物的分子间胺基化氟化反应中, 同样可以高效率地得到2-氟-2-芳基乙胺类化合物, 但遗憾的是, 未能实现不对称反应.
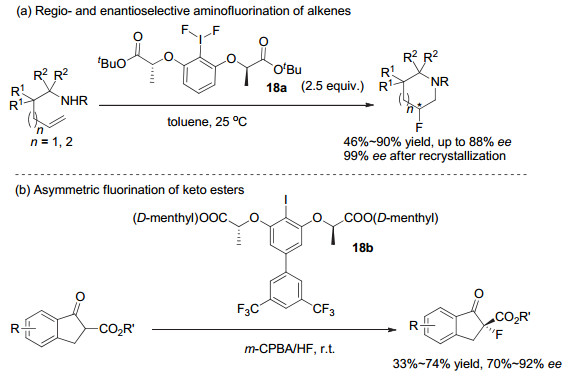
最近, Falivene和Rueping等[27b]在研究基于茚酮结构的β-酮酯化合物的不对称氟化反应时, 同样利用了Ishihara等发展的这类骨架, 发展了芳基碘化物18b, 可以中等的收率和较高的对映选择性实现不对称氟代反应, 反应可以在室温和空气中进行, 产物ee值大多在70%~92%之间(图 15b).他们对反应机理及过渡态进行了计算, 对酯基部位、甲基部位以及芳环上的取代基等都进行了探讨.
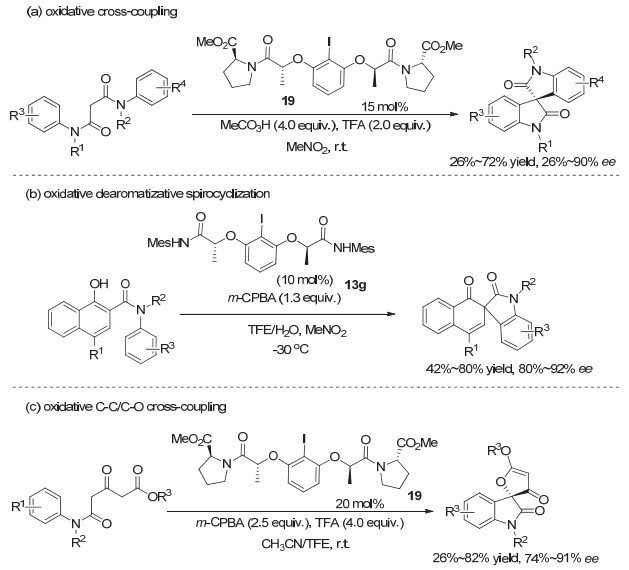
特别值得注意的是, 随着Ishihara类手性碘(Ⅰ/Ⅲ)化合物的广泛应用, 新的催化剂以及新的不对称反应类型也有了发展.以往基于乳酸酰胺所发展的Ishihara类型手性高价碘试剂, 大多为二级酰胺, 可以通过酰胺氮上的氢原子与底物或溶剂等形成氢键, 从而调控反应的对映选择性.但在2014年, 龚流柱等[28]研究催化的不对称氧化C—C偶联反应时, 发现使用三级乳酸酰胺芳基碘化物, 效果远比二级酰胺芳基碘化物要好.其中采用脯氨酸的三级乳酸酰胺芳基碘化物19作为催化剂, m-CPBA作为氧化剂时, 效果最佳.反应产物可以获得较高的对映选择性, ee值最高达到90%.从而实现了螺二氧化吲哚类化合物的高对映选择性构建.但由于副反应等存在, 大多情况下收率只能达到中等(图 16a). 2015年, 他们[29]又采用二级酰胺的有机碘化物13g, 实现了萘酚的不对称去芳构化反应构建手性螺环结构, 产物收率中等到良好, 对映选择性最高达到92% ee(图 16b). 2016年, 杜云飞等[30]采用和龚流柱同样的手性芳基碘化物19, 应用于串联氧化的C—O/C—C键形成, 实现了高对映选择性构建氧化吲哚类的螺环化合物, 产物基本在80%~90% ee左右(图 16c).
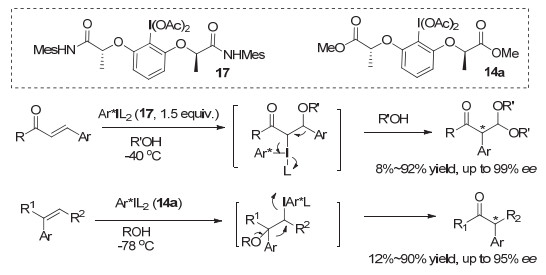
除了氧化的交叉偶联反应, 这类乳酸的手性芳基碘(Ⅰ/Ⅲ)试剂, 在新的不对称氧化重排反应中也取得了突破性进展. 2013年, Wirth等[31]首次报道了手性高价碘(Ⅲ)促进的β-芳基-α, β-不饱和酮的高对映选择性氧化重排反应, 生成α-芳基酮类化合物, 最高可达99% ee. 2016年, 他们[32]进一步针对1, 1-双取代烯烃发展了不对称氧化重排反应, 生成手性α-芳基酮, 大多数情况下, 都获得了中等到良好的收率和对映选择性(图 17).
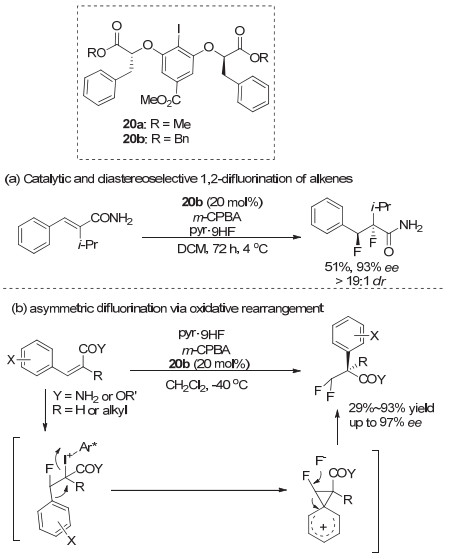
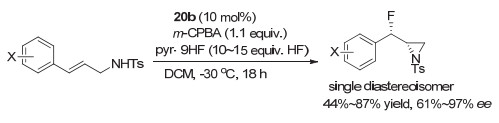
而在2016年, Jacobsen等[33]在使用20a/20b研究芳基碘化物催化烯烃的二氟化反应时, 发现当控制温度在4 ℃时, α, β-不饱和酰胺可以高立体选择性生成1, 2-二氟化产物(图 18a).而当反应温度控制在-40 ℃时, α, β-不饱和酰胺或酯底物的催化二氟化反应发生了氧化重排生成含二氟甲基手性中心的化合物(图 18b), 产物大多获得高对映选择性, 最高达到96% ee[34].使用未活化的普通烯烃时, 同样可以获得中等到良好的对映选择性.他们所使用的芳基碘化物20a/20b, 同样是根据Ishihara等的芳基碘化物改进而来, 在手性中心位置引入位阻更大的苄基取代甲基, 可以获得高对映选择性.同时把酯基部分改为苄酯, 可以提高反应活性, 使反应可以在低温下进行, 从而提升对映选择性.最近, Houk和薛小松等[35]对这一反应的化学选择性和立体选择性进行了计算, 进一步明确了这类重排反应对映选择性的机理.
最近, Jacobsen等[36]又利用这类芳基碘(Ⅰ)化合物, 实现了烯烃的催化不对称氟化/胺基化反应, 可以高对映选择性构建带两个手性中心的含氟氮杂三元环等化合物, 产物只有单一立体异构体, 对映选择性非常优异, 大多在90% ee以上, 最高可以达到97% ee(图 19).
Jacobsen的工作中, 对于手性碘的改造非常有意思, 他们不仅对乳酸酯部分进行了改造, 以期改进反应活性和对映选择性.同时, 他们在芳环上加入了吸电子的酯基, 尽管他们并未说明这一基团的加入对于反应的影响, 但吸电子基团的加入, 对于碘化物的电性显然有所影响, 值得进一步研究.
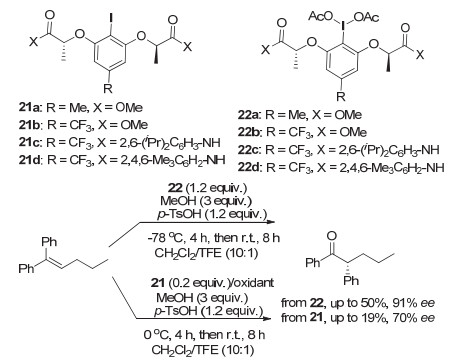
2017年, Wirth等[37]在芳环上引入不同的取代基, 如给电子的甲基以及吸电子的三氟甲基, 发展了一系列新结构的手性芳基碘(Ⅰ/Ⅲ)试剂21~22, 来研究高价碘试剂中芳环的电性对于氧化重排反应的活性以及对映选择性的影响(图 20).从他们的研究结果来看, 加入给电子基或吸电子基对于氧化重排反应的活性和对映选择性没有明显的影响.但这一结果在其它手性高价碘促进的反应中, 并不一定完全符合.手性碘试剂的电性对于反应活性以及对映选择性的影响, 在研究中仍然是一个非常值得思考和深入研究的方面.
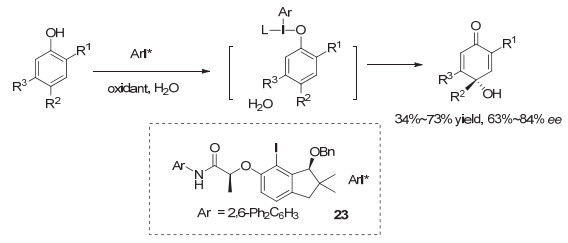
在最近的一个报道中, Maruoka等[38]采用手性茚醇为骨架, 发展芳基碘化物, 用于酚的不对称氧化去芳构化.最终, 为了获得高的对映选择性, 他们仍然选择在碘的另一个邻位引入了手性乳酸骨架, 代表性结构为23, 才能获得高的对映选择性, 但仍然只能达到中等收率(图 21).
以乳酸为手性源发展的这一类手性芳基碘化合物, 经过多个课题组的发展, 在多种反应类型中, 都展现出比其它高价碘试剂或催化剂更好的效果, 目前, 对这类芳基碘化物的研究还在继续深入, 可以预见, 将会有更多激动人心的报道出现.
基于柔性骨架中心手性的高价碘试剂或前体, 除了基于Koser类的结构(6~11)以及Fujita和Ishihara发展的乳酸类芳基碘结构外, 其它类型的结构也非常多.各类手性片段, 大多来源于“手性池”中的结构, 如氨基酸、氨基醇等, 通过成酯、酰胺等方式, 与芳基碘片段连接, 发展了多种类型的高价碘试剂或前体(图 22).
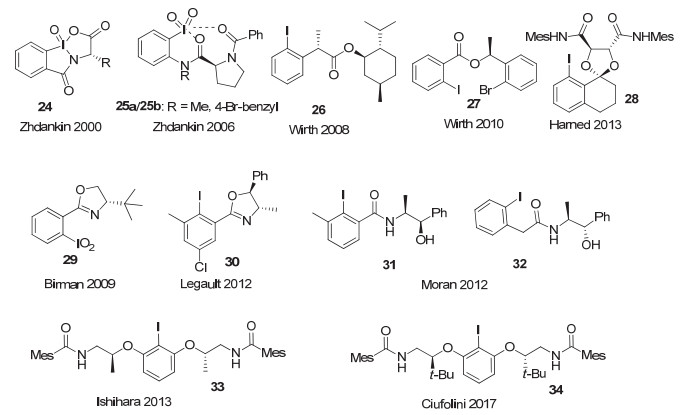
例如, 2000年, Zhdankin等[39]报道了一类手性芳基碘(Ⅴ)试剂24, 在这一结构中, 手性片段来源于“手性池”中的氨基酸分子.这类手性高价碘结构, 在硫醚的不对称氧化中, 表现并不好, ee值仅有11%~16%.而他们[40]在2006年又报道了另一种手性芳基碘(Ⅴ)试剂25, 手性元素来源于“手性池”中的脯氨酸.作者认为分子中的碘原子与来自两个酰胺基团的氧原子之间可能存在假键合或配位, 这将允许形成七元环, 从而有可能实现较好的手性控制.但这一试剂在硫醚的不对称氧化中, 同样表现不佳, 仅有29% ee.
Wirth等[41]报道了一系列酯类的芳基碘化物, 代表性结构如26和27.他们[42]使用27在苯丙酮的α-位不对称对甲苯磺酸酯化反应中, 最高获得26% ee, 而在5-羰基酸的羰基α-位内酯化反应中, 则几乎没有观察到对映选择性.
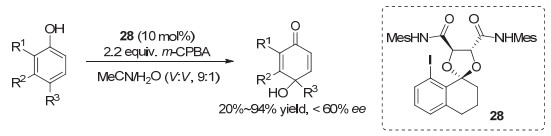
2013年, Harned等[43]基于Ishihara类碘化物的特点, 采用酒石酸为手性源, 发展了另一类的手性芳基碘化物, 代表性结构为化合物28.应用于酚类化合物的不对称去芳构化, 但收率和对映选择性并不令人满意(图 23).
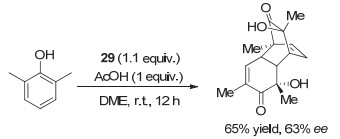
氨基醇作为手性源也被用于各类手性芳基碘化物中. 2009年, Birman等[44]发展了与Zhdankin类似的手性碘(Ⅴ)试剂.手性元素来源于“手性池”中的氨基醇, 通过转化生成邻位带手性噁唑啉取代基的芳基碘(Ⅴ)化合物, 代表性化合物为29.这类手性碘(Ⅴ)试剂被用于邻位烷基酚的不对称去芳构化/[4+2]D-A二聚反应, 反应收率很好, 但令人遗憾的是, 只有中等程度的对映选择性(图 24).
2012年, Legault等[45]报道了以类似的邻位带手性噁唑啉取代基的芳基碘化物作为催化剂, 代表性结构为30.他们用碘化物30为催化剂, 使用m-CPBA作为氧化剂, 实现了羰基化合物α-位的对甲苯磺酸酯化反应, 但对映选择性也仅在50% ee左右.
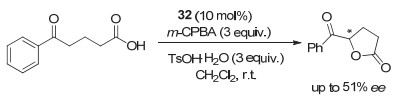
而几乎同时由Moran等[46]报道了采用手性氨基醇作为手性源的类似化合物31和32, 在m-CPBA作为氧化剂, 化合物31催化苯丙酮的羰基α-位对甲苯磺酰化反应中, 同样表现不佳, 对映选择性较低, 仅有18% ee.但在分子内形成五元环的反应中, 采用手性碘化物32为催化剂, 取得了中等的对映选择性, 最高达到51% ee, 是这类不对称内酯环化反应中最好的结果(图 25).
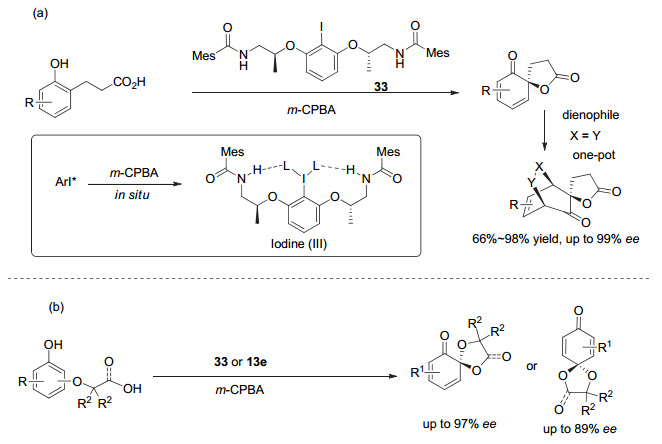
Fujita和Ishihara基于手性乳酸结构发展的手性芳基碘(Ⅰ/Ⅲ)试剂在不对称合成中得到了广泛的发展和应用.在多种类型的不对称转化中, 展现出比其它类型的手性碘试剂更好的对映选择性, 为手性高价碘化学打开了一扇大门.但值得注意的是, 他们2010年发展的这类手性芳基碘, 尽管在1-萘酚的不对称Kita氧化去芳构螺环化反应中取得了很好的对映选择性, 但对于2-萘酚以及苯酚类化合物的去芳构化反应, 其反应活性及对映选择性都不高.因此在他们2010年工作的基础上, 2013年, Ishihara等[47]采用氨基醇为手性源, 又进一步发展了第二代C2对称的柔性手性芳基碘化物, 其代表性结构为化合物33.在苯酚类化合物的不对称去芳构化螺环内酯化反应中, 第一次实现了非常高的对映选择性, 最高达到99% ee.并且作为催化剂前体的芳基碘化物, 只需要1~10 mol%, 转化效率也进一步提高.他们提出了该反应的作用模式, 提出了溶剂和氢键作用对反应对映选择性以及转化效率的影响等(图 26a).他们[48, 49]利用这一类芳基碘, 实现了包括β-萘酚、α-萘酚和苯酚等化合物的多类不对称去芳构螺环化反应(图 26b), 都取得了较好的收率以及高对映选择性, 与他们发展的第一代基于乳酸的手性芳基碘比较, 往往更胜一筹.
最近, Ciufolini等[50]也利用类似的芳基碘化物34, 与m-CPBA一起作用, 实现了其它类型的萘酚不对称去芳构螺环化反应, 取得了很高的对映选择性(图 27).
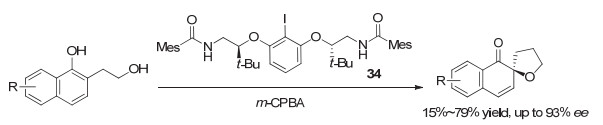
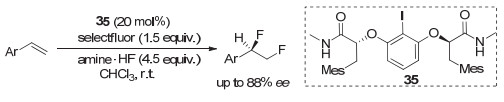
2016年, Gilmour等[51]在研究苯乙烯类化合物的不对称1, 2-二氟化反应时, 采用了Ishihara发展的第二代芳基碘化物33, 但对映选择性不好, 仅能获得约22% ee. 2018年, 他们[52]最终采用第一代乳酸类似的芳基碘化物35, 实现了较高对映选择性的1, 2-二氟化反应, 产物最高可获得88% ee(图 28).但反应中存在1, 1-二氟代副产物, 大多情况下只有中等收率.
在前面我们所讨论的手性芳基碘化物或其氧化物, 其手性基本为中心手性, 大多来源于“手性池”中的各类结构.这些柔性结构的手性芳基碘化物, 在不对称转化中得到了广泛的发展和应用.
除此之外, 一些在过渡金属催化中常用的刚性手性骨架, 也被用来发展新的手性高价碘化物或手性芳基碘化物.在这一节中, 我们将对此进行介绍.
在过渡金属催化的反应中, 手性联萘或联苯基团是常用的一类骨架.在手性芳基碘试剂发展中, 早在1990年, Ochiai等[53]采用联萘基团为手性骨架, 报道了第一类带有轴手性结构的手性芳基碘(Ⅲ)试剂, 代表性结构为36~37.他们研究认为化合物构型稳定性不高.
此后, 采用轴手性联萘或联苯骨架发展的芳基碘试剂, 在不对称合成中逐渐得以发展. 图 29中列出了一系列代表性的手性芳基碘试剂或前体, 在多种类型的不对称转化中得到了广泛发展及应用.
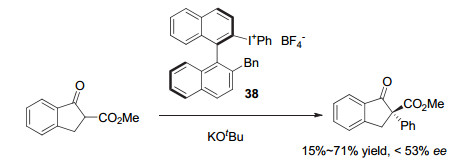
1999年, Ochiai等[54]采用手性联萘芳基碘做成二芳基碘鎓盐38, 首次用于环状β-酮酯的不对称α-芳基化反应.反应的对映选择性达到中等, 产物可以获得53% ee, 但收率较低(图 30).
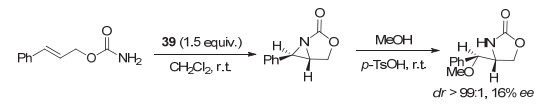
2011年, 徐镇江和支志明等[55]也采用手性联萘碘(Ⅲ)化合物39, 在烯烃的分子内氮杂环丙烷化反应中进行尝试, 但效果欠佳, 对映选择性仅达到16% ee(图 31).
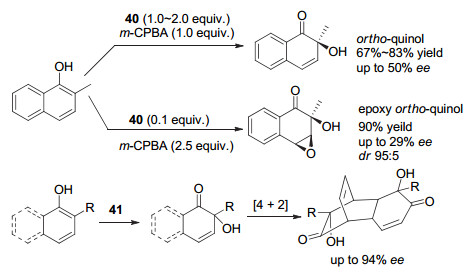
Quideau等发展了一系列的联萘或联苯骨架的手性芳基碘试剂, 应用于酚的不对称去芳构化反应等(图 32). 2009年, Quideau等[56]报道了以m-CPBA为共氧化剂, 与手性联萘碘代芳烃40促进苯酚类化合物的不对称去芳构化羟基化反应, 产率高达90%, 对映选择性最高可达到50% ee. 2014年他们[57]又报道了联苯骨架的手性三价及五价碘试剂, 其中手性五价碘41在苯酚类化合物的不对称去芳构羟基化/[4+2]二聚反应中表现优异, 对映选择性最高可以达到94% ee.
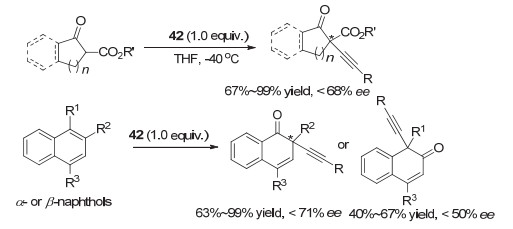
2017年, Quideau等[58]进一步发展了基于联苯骨架的炔基化试剂42, 用于β-酮酯的α-位不对称炔基化反应时, 大多情况下收率较高, 对映选择性中等, 最高为68% ee.他们也尝试利用这类试剂在萘酚底物中实现不对称炔基化反应, 同样, 大多数情况下收率较好, 但对映选择性只能达到中等(图 33).
2013年, Berthiol和Einhorn等[59]报道了3, 3'-二碘-联萘酚并马来酰亚胺作为骨架的芳基碘化物, 并在亚胺上引入带手性取代基的苯环结构, 代表性化合物为结构43.他们利用这类催化剂研究羰基化合物的不对称α-磺酸酯化反应, 但这类复杂的催化剂结构并未给反应带来高的对映选择性.
2017年, Dohi和Kita等[60]报道了使用联萘骨架手性芳基碘化物44和m-CPBA促进的萘酚化合物的不对称去芳构化螺环内酯化反应, 当使用当量的芳基碘化物时, 对映选择性最高可以达到76% ee, 而使用催化量的芳基碘化物, 对映选择性最高为64% ee. Masson等[61]则发展了非C2对称的联苯手性芳基碘化合物45, 催化羰基的α-磺酸酯化和膦酰酯化反应, 对映选择性也在60%~70% ee左右.
总体而言, 虽然联萘或联苯骨架的手性配体在过渡金属催化的反应中, 具有广泛的应用.但基于联萘或联苯骨架发展的手性芳基碘试剂或前体, 在不对称转化中的表现差强人意.这也反映出手性高价碘化学与传统的过渡金属化学在反应控制等方面具有很大不同, 仍然需要对反应机理等更深入的了解.
由周其林等[62]发展的手性螺二氢茚配体在过渡金属催化的反应中得到了广泛的应用.螺二氢茚刚性骨架结构极具特色, 在不对称合成领域引起了极大的研究兴趣.基于手性螺二氢茚骨架发展的高价碘在过去十年间也得以发展, 成为一类重要的手性芳基碘试剂(图 34).
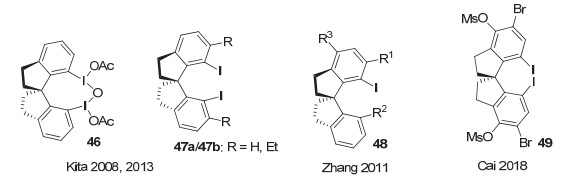
2008年, Kita等[63]报道了螺二氢茚骨架的手性芳基碘(Ⅲ)试剂46促进萘酚的首次不对称去芳构螺环内酯化反应(图 35), 这一反应也被称为Kita氧化去芳构螺环化反应.产物对映选择性高达86% ee.而以m-CPBA为共氧化剂, 以相应的手性芳基碘(Ⅰ)化合物47a作为催化剂, 催化的同一反应, 对映选择性虽然不如使用碘(Ⅲ)试剂, 但同样表现较好, 对映体选择性达到69% ee. 2013年, 他们[64]通过进一步在螺环上引入取代基, 发展了一系列螺二茚骨架的芳基碘化物.并以邻位乙基取代的手性碘化物47b为催化剂, 将萘酚的不对称去芳构螺环内酯化反应ee值提高到了90%以上.但在大多数情况下, 反应的收率都只能达到中等.这一类催化剂可与后来由Ishihara等基于乳酸手性发展的芳基碘化物相媲美, 是两类各有特色的手性芳基碘化合物, 在萘酚的不对称去芳构螺内酯化反应中取得了重大突破.
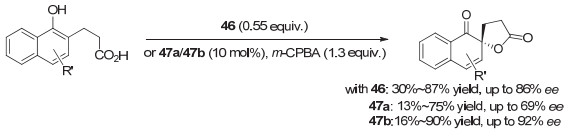
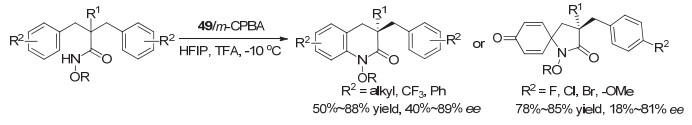
这类基于手性螺环的芳基碘化物也被其他研究小组运用到其它类型的不对称反应研究.例如, 2011年, 张弛等[65]报道了一系列螺二氢茚骨架的手性芳基单碘化物的合成, 并尝试用于羰基化合物的α-磺酸酯化, 但对映选择性最高只有50% ee左右. 2018年, 蔡倩等[66]报道了螺二氢茚骨架的手性芳基碘化物催化的不对称去对称化C—N氧化偶联反应, 代表性结构为化合物49.根据芳环取代基的不同, 可以生成手性四氢喹啉或带中心手性的螺环内酰胺, 产率中等到良好, 对映选择性最高分别达到89% ee和81% ee(图 36).
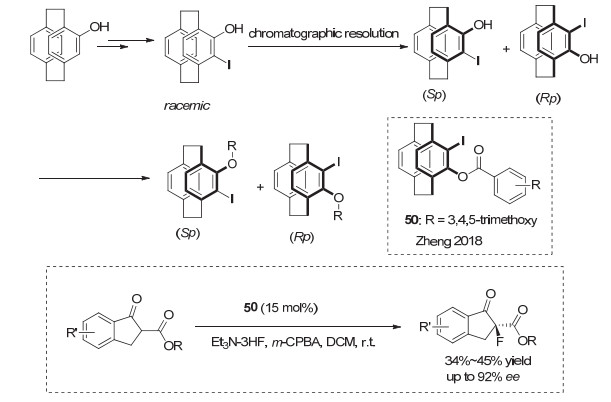
最近, 南京大学的郑文华等[67]报道了一类基于环蕃结构的新型平面手性碘代芳烃试剂, 使用[2.2]二聚对二甲苯为母核, 通过手性柱拆分分离技术, 构建了一系列手性芳基碘化合物, 其代表性结构为化合物50(图 37).他们利用这类手性芳基碘化物, 应用于β-酮酯的亲核氟化反应, 对映选择性最高可达到92% ee.但美中不足的是, 由于副反应影响, 这一反应收率较低.但作为首例平面手性芳基碘催化的高对映选择性反应, 这一类手性芳基碘化物的发展, 具有开创性意义.
除了上述基于联萘、联苯骨架、螺环骨架、环蕃骨架发展的刚性手性芳基碘试剂外, 文献中还有一些具有刚性结构的手性芳基碘试剂值得加以注意.
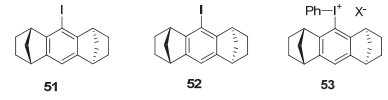
2012年, Ibrahim等[68]报道了一类桥环八氢蒽手性芳基碘化物, 这类结构中仍为中心手性, 但由于骨架为刚性结构, 我们在本节进行介绍.其代表性结构为化合物51~53(图 38).这类结构可以促进一系列反应, 如羰基的α-官能团化等, 但大多对映选择性不高.在不对称Kita氧化去芳构螺环内酯化反应中, 表现一般, 最高仅获得67% ee.
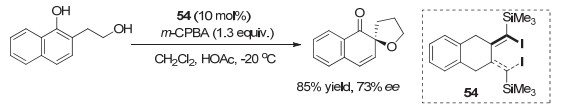
2017年, Ogasawara和Dohi等[69]报道了一类非常有意思的阻旋手性有机碘试剂, 代表性结构为45, 这是一类旋转受阻的二碘代1, 3-二烯类化合物.这是首例应用于不对称反应研究的非芳基有机碘化合物, 这类手性烯基碘化物在m-CPBA作用下, 催化萘酚的Kita氧化不对称去芳构螺环化反应, 对映选择性可以达到73% ee(图 39).
经过几十年的发展, 高价碘化学已成为有机合成中重要的合成工具.手性有机碘化物在不对称合成中的研究和应用也日渐广泛.在这篇综述中, 我们针对不同的骨架回顾了手性芳基碘试剂的发展以及它们在各类反应的一些特点.早期的手性芳基碘试剂是基于配体形式的高价碘试剂, 由碘原子与手性部位如手性樟脑磺酸、酒石酸等直接相连.这类芳基碘试剂主要用于硫醚不对称氧化成亚砜的反应, 但通常显示较低的对映选择性.之后在多个研究小组的努力下, 基于手性骨架发展的芳基碘试剂或前体得以发展和应用.其中, 基于柔性骨架中心手性发展的手性高价碘试剂或前体, 取得了巨大的成功.特别是Fujita和Ishihara等利用乳酸发展的手性高价碘试剂或前体, 在不对称去芳构化、羰基α-位官能团化、烯烃双官能团化、烯烃氧化重排、氧化偶联等多种类型的不对称转化中都取得了高的对映选择性.此外, 基于刚性骨架发展的手性高价碘试剂或前体, 也得到了很好的发展, 特别是Kita等基于螺二茚骨架的手性高价碘试剂和前体, 以及郑文华等基于环蕃结构发展的平面手性芳基碘, 在不对称去芳构螺环化、氧化偶联、羰基α-位氟化等反应中, 展现出优良的对映选择性.
但是, 对于手性高价碘促进的不对称转化而言, 目前还处于比较初始的阶段, 具有很大的研究空间.分析文献报道的各类手性高价碘试剂及反应, 我们认为至少在以下几个方面值得深入研究:
(1) 对于反应机理以及手性控制的微妙因素的认识还不够深入, 需要进一步的理论研究和实验验证.比如, 在反应活性以及对映选择性控制方面, 手性高价碘中芳环上的电性以及碘原子周围的化学环境对反应活性和对映选择性都具有重要影响, 有必要加以深入研究.与过渡金属配体不同, 由于碘原子半径较大, 邻位位阻太大, 可能影响底物与碘结合, 如何选择合适的邻位基团是需要考虑的重要因素.此外, 高价碘促进的反应, 往往存在多种类型的副反应, 反应收率欠佳.因此, 如何进行区域选择性控制以及副反应控制等, 也存在极大的研究空间.
(2) 反应类型及底物范围还有待进一步扩展.高价碘促进的非手性的反应非常广泛, 而对于不对称转化, 从早期对硫醚的不对称氧化开始, 目前已经有多种类型的反应可以通过手性高价碘试剂得以实现.但与过渡金属催化的不对称反应相比, 反应类型仍然非常局限, 有很大的拓展空间.此外, 不少手性高价碘促进的反应, 底物范围受到严重限制, 如何拓展底物适用范围, 也是一个需要发展的重要方面.
(3) 具有新骨架的手性高价碘试剂发展有待进一步加强.尽管目前已有数十种手性碘试剂, 但真正在不对称转化中能获得高对映选择性的还很少.新的手性高价碘试剂, 对于拓展反应类型以及实现高对映选择性转化, 最为关键.因此有必要继续设计、发展新骨架的手性高价碘.
手性高价碘化学作为一个新的研究领域, 已经取得了巨大的发展, 特别是近十年来, 进展显著.我们相信, 随着研究的不断深入, 新骨架手性高价碘试剂的发展以及新不对称转化类型的拓展, 将取得更令人鼓舞的成果, 在绿色不对称合成中发挥越来越重要的作用.
For books, see: (a) Chemistry of Hypervalent Compounds, Ed.: AKiba, K. Y., Wiley-VCH, New York, 1999. (b) Zhdankin, V. V. Hypervalent Iodine Chemistry: Preparation, Structure and Syn-thetic Application of Polyvalent Iodine Compounds, John Wiley & Sons Ltd., New York, 2014. (c) Iodine Chemistry And Applications, Ed.: Kaiho, T., John Wiley & Sons Ltd., New York, 2015. (d) Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, Ed.: Wirth, T., Springer, 2003.
For recent reviews, see: (a) Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328. (b) Duan, Y.; Jiang, S.; Han, Y.; Sun, B.; Zhang, C. Chin. j. Org. Chem. 2016, 36, 1973(in Chinese). (段亚南, 姜山, 韩永超, 孙博, 张弛, 有机化学, 2016, 36, 1973. ) (c) Ma, J.; Chen, L.; Yuan, Z.; Cheng, H. Chin. j. Org. Chem. 2018, 38, 1586(in Chinese). (马姣丽, 陈立成, 袁中文, 程辉成, 有机化学, 2018, 38, 1586. )
For selected recent reviews, see: (a) Flores, A.; Cots, E.; Bergès, J.; Muñiz, K. Adv. Synth. Catal. 2019, 361, DOI: 10.1002/adsc. 201800521. (b)MartínRomero,R.; Wöste,T. H.; Muñiz,K. Chem. AsianJ. 2014,9,972. (c)Singh,F. V.; Wirth,T. Chem. AsianJ. 2014,9,950. (d)Harned,A. M. TetrahedronLett. 2014,55,4681. (e)Parra,A.; Reboredo, S. Chem. Eur. J. 2013,19,17244.
Liang, H.; Ciufolini, M. A. Angew. Chem. Int. Ed. 2011, 50, 11849. doi: 10.1002/anie.v50.50
Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. j. Am. Chem. Soc. 2005, 127, 12244. (b) Dohi, T.; Maruyama, A.; Yoshimura, M.; Morimoto, K.; Tohma, H.; Kita, Y. Angew. Chem. Int. Ed. 2005, 44, 6193.
Pribram, R. Justus Liebigs Ann. Chem. 1907, 351, 481. doi: 10.1002/(ISSN)1099-0690
Imamoto, T.; Koto, H. Chem. Lett. 1986, 967.
Hatzigrigoriou, E.; Varvoglis, A.; Bakola-Christianopoulou, M. j. Org. Chem. 1990, 55, 315. doi: 10.1021/jo00288a053
Xia, M.; Chen, Z.-C. Synth. Commun. 1997, 27, 1321. doi: 10.1080/00397919708006060
Ray Ⅲ, D. G.; Koser, G. F. j. Am. Chem. Soc. 1990, 112, 5672. doi: 10.1021/ja00170a059
Ray Ⅲ D. G.; Koser, G. F. j. Org. Chem. 1992, 57, 1607. doi: 10.1021/jo00031a054
Tohma, H.; Takizawa, S.; Watanabe, H.; Fukuoka, Y.; Maegawa, T.; Kita, Y. j. Org. Chem. 1999, 64, 3519. doi: 10.1021/jo982295t
Rabah, G. A.; Koser, G. F. Tetrahedron Lett. 1996, 37, 6453. doi: 10.1016/0040-4039(96)01436-0
(a) Wirth, T.; Hirt, U. H. Tetrahedron Asymmetry 1997, 8, 23. (b) Hirt, U. H.; Spingler, B.; Wirth, T. j. Org. Chem. 1998, 63, 7674. (c) Hirt, U. H.; Schuster, M. F. H.; French, A. N.; Wiest, O. G.; Wirth, T. Eur. j. Org. Chem. 2001, 1569.
Mizar, P.; Laverny, A.; EI-Sherbini, M.; Farid, U.; Brown, M.; Malmedy, F.; Wirth, T. Chem. Eur. j. 2014, 20, 9910. doi: 10.1002/chem.201403891
Hempel, C.; Maichle-Mössmer, C.; Pericàs, M. A.; Nachtsheim, B. j. Adv. Synth. Catal. 2017, 359, 2941.
Fujita, M.; Okuno, S.; Lee, H. J.; Sugimura, T.; Okuyama, T. Tetrahedron Lett. 2007, 48, 8691. doi: 10.1016/j.tetlet.2007.10.015
(a) Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem. Int. Ed. 2010, 49, 2175. (b) Uyanik, M.; Yasui, T.; Ishihara, K. Tetrahedron 2010, 66, 5841.
(a) Fujita, M.; Yoshida, Y.; Miyata, K.; Wakisaka, A.; Sugimura, T. Angew. Chem. Int. Ed. 2010, 49, 7068. (b) Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Org. Lett. 2012, 14, 1294. (c) Shimogaki, M.; Fujita, M.; Sugimura, T. Eur. j. Org. Chem. 2013, 7128. (d) Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. Org. Lett. 2014, 16, 4634.
Fujita, M.; Wakita, M.; Sugimura, T. Chem. Commun. 2011, 47, 3983. doi: 10.1039/c1cc10129c
(a) Shimogaki, M.; Fujita, M.; Sugimura, T. Angew. Chem. Int. Ed. 2016, 55, 15797. (b) Shimogaki, M.; Fujita, M.; Sugimura, T. j. Org. Chem. 2017, 82, 11836.
Röben, C.; Souto, j. A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Angew. Chem. Int. Ed. 2011, 50, 9478. doi: 10.1002/anie.v50.40
Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. j. Am. Chem. Soc. 2017, 139, 4354. doi: 10.1021/jacs.7b01443
(a) Haubenreisser, S.; Wöste, T. H.; Martínez, C.; Ishihara, K.; Muñiz, K. Angew. Chem. Int. Ed. 2016, 55, 413. (b) Wöste, T. H.; Muñiz, K. Synthesis 2016, 48, 816.
(a) Farid, U.; Wirth, T. Angew. Chem. Int. Ed. 2012, 51, 3462. (b) Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Chem. Eur. J. 2016, 22, 1614.
Gelis, C.; Dumoulin, A.; Bekkaye, M.; Neuville, L.; Masson, G. Org. Lett. 2017, 19, 278. doi: 10.1021/acs.orglett.6b03631
(a) Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Angew. Chem. Int. Ed. 2013, 52, 2469. (b) Pluta, R.; Krach, P. E.; Cavallo, L.; Falivene, L.; Rueping, M. ACS Catal. 2018, 8, 2582.
Wu, H.; He, Y.-P.; Xu, L.; Zhang, D.-Y.; Gong, L.-Z. Angew. Chem. Int. Ed. 2014, 53, 3466. doi: 10.1002/anie.201309967
Zhang, D.-Y.; Xu, L.; Wu, H.; Gong, L.-Z. Chem. Eur. j. 2015, 21, 10314. doi: 10.1002/chem.201501583
Cao, Y.; Zhang, X.; Lin, G.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2016, 18, 5580. doi: 10.1021/acs.orglett.6b02816
Farid, U.; Malmedy, F.; Claveau, R.; Albers, C.; Wirth, T. Angew. Chem. Int. Ed. 2013, 52, 7018. doi: 10.1002/anie.201302358
Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T. Chem. Eur. j. 2016, 22, 4030. doi: 10.1002/chem.201504844
Banik, S. M.; Medley, j. W.; Jacobsen, E. N. j. Am. Chem. Soc. 2016, 138, 5000. doi: 10.1021/jacs.6b02391
Banik, S. M.; Medley, j. W.; Jacobsen, E. N. Science 2016, 353, 51. doi: 10.1126/science.aaf8078
Zhou, B.; Haj, M. K.; Jacobsen, E. N.; Houk, K. N.; Xue, X.-S. j. Am. Chem. Soc. 2018, 140, 15206. doi: 10.1021/jacs.8b05935
Mennie, K. M.; Banik, S. M.; Reichert, E. C.; Jacobsen, E. N. j. Am. Chem. Soc. 2018, 140, 4797. doi: 10.1021/jacs.8b02143
Qurban, J.; Elsherbini, M.; Wirth, T. j. Org. Chem. 2017, 82, 11872. doi: 10.1021/acs.joc.7b01571
Hashimoto, T.; Shimazaki, Y.; Omatsu, Y.; Maruoka, K. Angew. Chem. Int. Ed. 2018, 57, 7200. doi: 10.1002/anie.v57.24
Zhdandin, V. V.; Smart, j. T.; Zhao, P.; Kiprof, P. Tetrahedron Lett. 2000, 41, 5299. doi: 10.1016/S0040-4039(00)00836-4
Ladziata, U.; Carlson, J.; Zhdankin, V. V. Tetrahedron Lett. 2006, 47, 6301. doi: 10.1016/j.tetlet.2006.06.103
Altermann, S. M.; Richardson, R. D.; Page, T. K.; Schmidt, R. K.; Holland, E.; Mohammed, U.; Paradine, S. M.; French, A. N.; Richter, C.; Bahar, A. M.; Witulski, B.; Wirth, T. Eur. j. Org. Chem. 2008, 5315.
Farooq, U.; Schäfer, S.; Ali Shah, A.-U.-H.; Freudendahl, D. M.; Wirth, T. Synthesis 2010, 1023.
Volp, K. A.; Harned, A. M. Chem. Commun. 2013, 49, 3001. doi: 10.1039/c3cc00013c
Boppisetti, j. K.; Birman, V. B. Org. Lett. 2009, 6, 1221.
Guilbault, A.-A.; Basdevant, B.; Wanie, V.; Legault, C. Y. j. Org. Chem. 2012, 77, 11283. doi: 10.1021/jo302393u
Rodríguez, A.; Moran, W. j. Synthesis 2012, 44, 1178. doi: 10.1055/s-0031-1290590
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem. Int. Ed. 2013, 52, 9215. doi: 10.1002/anie.201303559
Uyanik, M.; Sasakura, N.; Mizuno, M.; Ishihara, K. ACS Catal. 2017, 7, 872. doi: 10.1021/acscatal.6b03380
Uyanik, M.; Yasui, Y.; Ishihara, K. j. Org. Chem. 2017, 82, 11946. doi: 10.1021/acs.joc.7b01941
Jain, N.; Xu, S.; Ciufolini, M. A. Chem. Eur. j. 2017, 23, 4542. doi: 10.1002/chem.201700667
Molnár, I. G.; Gilmour, R. j. Am. Chem. Soc. 2016, 138, 5004. doi: 10.1021/jacs.6b01183
Scheidt, F.; Schäfer, M.; Sarie, j. C.; Doniliuc, C. G.; Molloy, j. J.; Gilmour, R. Angew. Chem. Int. Ed. 2018, 57, 16431. doi: 10.1002/anie.201810328
Ochiai, M.; Takaoka, Y.; Masaki, Y. j. Am. Chem. Soc. 1990, 112, 5677. doi: 10.1021/ja00170a063
Ochiai, M.; Kitagawa, Y.; Takayama, N.; Takaoka, Y.; Shiro, M. j. Am. Chem. Soc. 1999, 121, 9234.
Deng, Q.-H.; Wang, j.-C.; Xu, Z.-J.; Zhou, C.-Y.; Che, C.-M. Synthesis 2011, 18, 2959.
Quideau, S.; Lyvinec, G.; Marguerit, M.; Bathany, K.; Ozanne-Beaudenon, A.; Buffeteau, T.; Cavagnat, D.; Chénedé, A. Angew. Chem. Int. Ed. 2009, 48, 4605. doi: 10.1002/anie.v48:25
Bosset, C.; Coffinier, R.; Peixoto, P. A.; Assal, M. E.; Miqueu, K. M.; Sotiropoulos, j.-M. Pouységu, L.; Quideau, S. Angew. Chem. Int. Ed. 2014, 53, 9860. doi: 10.1002/anie.201403571
Companys, S.; Peixoto, P. A.; Bosset, C.; Chassaing, S.; Miqueu, K.; Sotiropoulos, j.-M.; Pouységu, L.; Quideau, S. Chem. Eur. j. 2017, 23, 13309. doi: 10.1002/chem.v23.54
(a) Brenet, S.; Berthiol, F.; Einhorn, j. Eur. j. Org. Chem. 2013, 8094. (b) Brenet S.; Minozzi, C.; Clarens, B.; Amiri, L.; Berthiol, F. Synthesis 2015, 47, 3859
Dohi, T.; Sasa, H.; Miyazaki, K.; Fujitake, M.; Takenaga, N.; Kita, Y. j. Org. Chem. 2017, 82, 11954. doi: 10.1021/acs.joc.7b02037
Levitre, G.; Dumoulin, A.; Retailleau, P.; Panossian, A.; Leroux, F. R.; Masson, G. j. Org. Chem. 2017, 82, 11877. doi: 10.1021/acs.joc.7b01597
谢建华, 周其林, 化学学报, 2014, 72, 778. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344524.shtmlXue, j.-H.; Zhou, Q.-L. Acta Chim. Sinica 2014, 72, 778(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344524.shtml
Dohi, T.; Maruyama, A.; Takenaga, N.; Senami, K.; Minamitsuji, Y.; Fujioka, H.; Caemmerer, S. B.; Kita, Y. Angew. Chem. Int. Ed. 2008, 47, 3787. doi: 10.1002/(ISSN)1521-3773
Dohi, T.; Takenaga, N.; Nakae, T.; Toyoda, Y.; Yamasaki, M.; Shiro, M.; Fujioka, H.; Maruyama, A.; Kita, Y. j. Am. Chem. Soc. 2013, 135, 4558. doi: 10.1021/ja401074u
Yu, J.; Cui, J.; Hou, X.-S.; Liu, S.-S.; Gao, W.-C.; Jiang, S.; Tian, J.; Zhang, C. Tetrahedron: Asymmetry 2011, 22, 2039. doi: 10.1016/j.tetasy.2011.12.003
Ding, Q.; He, H.; Cai, Q. Org. Lett. 2018, 20, 4554. doi: 10.1021/acs.orglett.8b01849
Wang, Y.; Yuan, H.; Lu, H.; Zheng, W.-H. Org. Lett. 2018, 20, 2555. doi: 10.1021/acs.orglett.8b00711
Murray, S. J.; Müller-Bunz, H.; Ibrahim, H. Chem. Commun. 2012, 48, 6268. doi: 10.1039/c2cc32280c
Ogasawara, M.; Sasa, H.; Hu, H.; Amano, Y.; Nakajima, H.; Takenaga, N.; Nakajima, K.; Kita, Y.; Takahashi, T.; Dohi, T. Org. Lett. 2017, 19, 4102. doi: 10.1021/acs.orglett.7b01876

图 1 手性高价碘促进的几类主要不对称转化
Figure 1 Asymmetric transformations by hypervalent iodine reagents

图 2 不同类型的手性高价碘试剂结构特点
Figure 2 Different types of chiral hypervalent iodine reagents and their general structures

图 4 手性高价碘促进的硫醚不对称氧化为亚砜
Figure 4 Asymmetric oxidation of sulfides to sulfoxides by hypervalent iodine reagents

图 5 硫醚到手性亚砜的催化不对称氧化
Figure 5 Catalytic asymmetric oxidation of sulfides to sulfoxides by hypervalent iodine reagents

图 8 基于三氮唑的手性芳基碘促进的不对称Kita螺环内酯化反应
Figure 8 Enantioselective Kita spirocyclizations by triazole-based chiral iodoarenes

图 9 基于乳酸的手性芳基碘结构通式
Figure 9 General structures of chiral aryl iodine reagents (Ⅰ/Ⅲ) developed from lactic acid

图 10 基于乳酸的手性高价碘试剂及烯烃的分子内不对称环化反应
Figure 10 Asymmetric intramolecular cyclization of alkene by lactic acid-based chiral hypervalent iodine reagents

图 11 Ishihara组发展的C2对称的柔性骨架芳基碘及Kita氧化去芳构螺环化反应研究
Figure 11 Lactic acid-based C2-symmetric chiral hypervalent iodine reagents by Ishihara et al. and the applications in enantioselective Kita oxidative spirocyclization

图 12 Fujita组在烯烃不对称双官能团化研究中的进展
Figure 12 Asymmetric bifunctionalization of alkenes by Fujita et al

图 13 Muñiz组关于苯乙烯类化合物的不对称双官能团化研究
Figure 13 Asymmetric bifunctionalization of styrenes by Muñiz et al

图 14 其它的烯烃不对称双官能团化反应
Figure 14 Other examples for asymmetric bifunctionalization of alkenes

图 15 手性碘氟化试剂及不对称氟化反应
Figure 15 Chiral aryliodine (Ⅰ/Ⅲ) reagents for enantioselective fluorinations

图 16 不对称氧化交叉偶联反应构建螺环结构
Figure 16 Asymmetric oxidative coupling for the formation of spirocycles

图 17 Wirth组对烯烃的不对称氧化重排反应研究
Figure 17 Asymmetric oxidative rearrangements by Wirth et al

图 18 烯烃不对称1, 2-二氟化及氧化重排反应
Figure 18 Diastereoselective 1, 2-difluorination and oxidative rearrangement of alkenes by Jacobsen et al

图 19 烯烃的催化不对称氟化/胺基化
Figure 19 Catalytic diastereo- and enantioselective fluoroamination of alkenes

图 20 芳基碘(Ⅰ/Ⅲ)试剂的电性对于氧化重排反应活性和对映选择性的影响研究
Figure 20 The influences of electron property of chiral aryliodine reagents to the reactivity and enantioselectivity in oxidative rearrangement reactions

图 22 基于柔性骨架中心手性的其它高价碘试剂或前体
Figure 22 Other chiral hypervalent reagents or precursors based on conformationally flexible skeletons with central chirality

图 24 酚的不对称去芳构/[4+2]串联反应
Figure 24 Asymmetric dearomatization/[4+2] tandem reactions of phenols

图 25 羰基α-位的分子内不对称内酯环化反应
Figure 25 Asymmetric lactonization of 5-oxo-5-phenylpentanoic acid

图 26 Ishihara发展的第二代柔性结构C2-对称的手性芳基碘
Figure 26 The second generation of C2-symmetric and conformationally flexible chiral iodoarenes by Ishihara

图 29 基于手性联萘或联苯骨架发展的芳基碘试剂和前体
Figure 29 Chiral aryliodine reagents or precursors based on chiral binaphthalene or biphenyl skeletons

图 30 手性碘鎓盐在羰基α-位芳基化反应中的研究
Figure 30 Chiral diaryliodonium salts for asymmetric α-arylation of β-keto ester enolates

图 35 基于螺环结构的手性芳基碘在Kita氧化去芳构螺环内酯化反应中的研究
Figure 35 Spirobiindane-based chiral aryliodine reagents and the applications in Kita oxidative spirocyclizations

图 38 Ibrahim组发展的手性高价碘试剂和前体
Figure 38 Chiral hypervalent iodines and precursors by Ibrahim

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:














 下载:
下载: