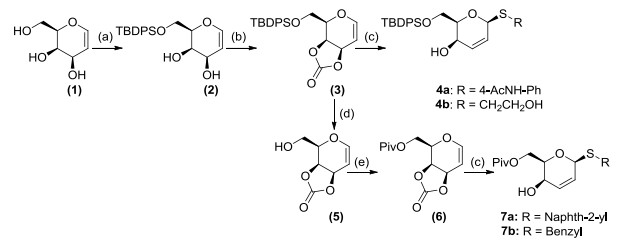
Scheme 1.
Synthetic routes for the β-thiogalactoside
Synthesis and Absolute Configuration of ((2R, 3R, 6S)-3-Hydroxy-6-(naphthalen-2-ylthio)-3, 6-dihydro-2H-pyran-2-yl)methyl Pivalate
Yang JIAO , Jia-Ding ZHANG , Shu-Zhang WANG , Hui YAO , Ming-Guo LIU , Nian-Yu HUANG

Over the last few decades, nucleoside analogues create a significant class of antimetabolic agents for the treatment of malignant tumors[1], numerous viral diseases[2] and bacterial infections[3] with the corresponding mechanism of stopping cancer cell proliferation, inhibiting viral replication and preventing cell-wall biosynthesis. Sulfur is prevalent in biologically active natural products and commercial drugs for variable medical conditions including depression, arthritis, diabetes, cancer, and acquired immune deficiency syndrome (AIDS) because of its critical role in primary metabolism[4], which also shows unique chemical reactivity in organic chemistry as the building blocks for the preparation of sulfur-containing heterocyclic rings or aliphatic chain, such as the β-lactam antibiotics penicillins[5], sulfonamide antimicrobial agents[6], cytotoxic thioamycolamides A–E[7], and anti-infective glycolipid KRN7000[8]. Thioglycosides are also common glycomimetics (e.g., S-linked galactosylceramides[9]) and enhance hydrolytic and enzymatic degradation relative to the natural O-linked congeners while retaining similar conformational preferences[10, 11].
Thioglycosides have attracted considerable interest owing to the synthetic challenges and their potential bioactivity, and extensive researches including cross-coupling reaction[12, 13] and Ferrier-type reaction[14, 15] were reported to the construction of these molecules in recent years. Inspired by such studies, we designed and synthesized the pivalyl protected β-thiogalactoside under the catalysis of palladium in this work (Scheme 1).
All chemical reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and used without further purification. Solvents were dried and purified using standard techniques. D-Galactal (1) was purchased from J & K Chemical Co., Ltd., China. Reactions were monitored by thin layer chromatography (TLC) on silica gel GF254 pre-coated plates. 1D and 2D NMR spectra were recorded at a Bruker ultrashiedTM 400 MHz plus spectrometer. Chemical shifts (δ) were reported in ppm, using residual solvent as an internal standard. Melting points were tested with an uncorrected X-4 digital melting point apparatus. HR-ESI-MS was obtained using a Waters Q-TOF premierTM mass spectrometer. Characterization data for known compounds were checked in comparison with literature for consistency and not presented in this report.
D-Galactal (1, 1.46 g, 10 mmol) and imidazole (0.82 g, 12 mmol) were dissolved in anhydrous N, N΄-dimethylformamide (DMF, 40 mL), and tert-butyldiphenylchlorosilane (TBDPSCl, 2.90 g, 10.5 mmol) was slowly added to the mixture over 20 min. The mixture was stirred at room temperature for 6 hours until the completion of the reaction monitored by TLC. The reaction was quenched by 5% aqueous NaHCO3 and the crude product was extracted by ethyl acetate (30 mL × 3). After removal of the solvent, pure (2R, 3R, 4R)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3, 4-dihydro-2H-pyran-3, 4-diol (2) was obtained by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 1/1, v/v) as colorless syrup[16] (3.41 g, yield 89%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.83~7.62 (m, 4H), 7.55~7.34 (m, 6H), 6.41 (dd, J = 6.2, 1.5 Hz, 1H), 4.75 (dt, J = 6.2, 1.9 Hz, 1H), 4.46~4.29 (m, 1H), 4.17 (dd, J = 6.3, 2.0 Hz, 1H), 4.01 (dd, J = 11.9, 6.9 Hz, 1H), 3.96~3.88 (m, 2H), 2.99 (d, J = 5.3 Hz, 1H), 2.60 (d, J = 10.2 Hz, 1H), 1.09 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 144.4, 135.6, 135.5, 132.6, 132.4, 130.0, 127.8, 127.8, 103.4, 65.8, 64.4, 63.8, 26.7, 19.1.
The mixture of intermediate (2, 2.68 g, 7 mmol), N, N΄-carbonyldiimidazole (1.17 g, 7.2 mmol) and catalytic amount of imidazole (10 mg) was dissolved in anhydrous tetrahydrofuran (THF, 30 mL) under N2 atmosphere. The solution was stirred at room temperature for 8 hours until completion of the acylating process by TLC monitoring. The mixture was quenched by ice water (50 mL), and crude product was extracted by ethyl acetate (30 mL × 3). After separating the organic layer and removal of the solvent, the oily crude was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 10/1, v/v) to give 1, 5-anhydro-6-O-(tert-butyldiphenylsilyl)-3, 4-O-carbonate-2-deoxy-D-lyxo-hex-1-enopyranose (3) as colorless syrup[17] (2.61 g, yield 91%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.76~7.68 (m, 4H), 7.54~7.43 (m, 6H), 6.68 (d, J = 6.3 Hz, 1H), 5.25 (dd, J = 7.7, 3.1 Hz, 1H), 5.10 (d, J = 7.7 Hz, 1H), 4.99 (ddd, J = 6.2, 3.1, 1.1 Hz, 1H), 4.06 (d, J = 7.8 Hz, 1H), 4.03~3.99 (m, 2H), 1.13 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.2, 149.1, 135.5, 135.5, 134.8, 132.7, 132.5, 130.1, 130.0, 127.9, 127.9, 127.7, 98.0, 73.8, 72.9, 68.9, 61.7, 26.8, 26.6, 19.2.
The mixture of D-galactal carbonate (3, 410 mg, 1.0 mmol), thiol (1.0 mmol), tris(dibenzylideneacetone)dipalladium (Pd2(dba)3, 9.1 mg, 0.01 mmol) and 9, 9-dimethyl-4, 5-bis(diphenylphosphino)xanthene (xantphos, 11.6 mg, 0.02 mmol) was dissolved in anhydrous dichloromethane (10 mL) under nitrogen atmosphere, which was stirred at room temperature for 12 hours until the completion of reaction (monitored by TLC). The mixture was quenched by ice water (5 mL), and crude product was extracted by dichloromethane (8 mL × 3). After removal of the solvent, the crude product was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 5/1, v/v) to give the target product (4).
N-(4-(((2S, 5R, 6R)-6-(((tert-Butyldiphenylsilyl)oxy)methyl)-5-hydroxy-5, 6-dihydro-2H-pyran-2-yl)thio)phenyl)acetamide (4a). Colorless oil (458 mg, yield 86%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.76~7.63 (m, 4H), 7.49~7.34 (m, 10H), 6.04 (ddd, J = 10.0, 5.8, 2.0 Hz, 1H), 5.90 (dd, J = 10.0, 1.6 Hz, 1H), 5.41 (d, J = 1.9 Hz, 1H), 3.93~3.79 (m, 3H), 3.72 (ddd, J = 12.7, 6.3, 1.7 Hz, 1H), 2.15 (s, 3H), 1.05 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 138.3, 135.6, 135.1, 133.4, 133.3, 131.0, 129.7, 129.6, 127.7, 127.7, 126.4, 119.6, 81.8, 78.6, 63.3, 61.5, 26.8, 24.6, 19.2. HRMS (ESI) m/z: calcd. for C30H35NO4SSiNa+ (M + Na)+ 556.1953; found 556.1950.
(2R, 3R, 6S)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-6-((2-hydroxyethyl)thio)-3, 6-dihydro-2H-pyran-3-ol (4b). Colorless oil (359 mg, yield 81%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.79~7.71 (m, 4H), 7.49~7.42 (m, 6H), 6.26 (ddd, J = 10.0, 5.9, 2.2 Hz, 1H), 5.83 (dd, J = 10.0, 1.6 Hz, 1H), 5.35 (d, J = 1.9 Hz, 1H), 4.04~3.94 (m, 2H), 3.91~3.78 (m, 3H), 3.61 (ddd, J = 14.5, 9.7, 2.8 Hz, 1H), 3.53 (s, 1H), 3.08 (d, J = 8.4 Hz, 1H), 2.94 (ddd, J = 15.0, 9.7, 3.8 Hz, 1H), 2.75 (ddd, J = 15.0, 4.2, 2.9 Hz, 1H), 1.10 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm): 135.5, 133.3, 133.1, 130.7, 129.9, 129.7, 129.6, 127.6, 127.6, 79.4, 78.7, 63.1, 62.4, 61.4, 32.9, 26.8, 19.2. HRMS (ESI) m/z: calcd. for C24H32O4SSiNa+ (M + Na)+ 467.1688; found 467.1684.
Compound 3 (2.05 g, 5 mmol) was dissolved in anhydrous THF (20 mL) under N2 atmosphere, and tetra-n-butylammonium fluoride trihydrate (TBAF, 1.6 g, 5.1 mmol) in THF (20 mL) was added with a syringe to the previous solution in an ice-water bath. The reaction mixture was stirred at 0 oC for 1.5 hours until the complete consumption of starting material (monitored by TLC). The reaction was quenched with water (20 mL) and the product was extracted by ethyl acetate (30 mL × 3). The organic phase was concentrated under reduced pressure and pure D-glalactal-3, 4-O-carbonate (5) was obtained by flash column chromatography (eluent: petroleum ether/ethyl acetate = 5/1, v/v) in silica gel as as colorless syrup[16] (0.76 g, yield 88%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.74 (d, J = 6.3 Hz, 1H), 5.24 (dd, J = 7.7, 3.2 Hz, 1H), 5.01 (ddd, J = 6.3, 3.2, 1.2 Hz, 1H), 4.98~4.91 (m, 1H), 4.10~3.99 (m, 2H), 3.93 (d, J = 9.6 Hz, 1H), 2.24 (s, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.0, 149.1, 98.1, 73.9, 73.1, 69.0, 61.5.
D-glalactal-3, 4-O-carbonate (5, 0.69 g, 4 mmol) and triethylamine (Et3N, 1.66 mL, 12 mmol) were dissolved in anhydrous CH2Cl2 (30 mL) under N2 atmosphere, and pivaloyl chloride (Piv-Cl, 0.5 mL, 4.1 mmol) was added at 0 ℃. The mixture was stirred for 2 hours and monitored by TLC until the completion of the reaction. The reaction was quenched by 5% aqueous NaHCO3 and the crude product was extracted by ethyl acetate (20 mL × 3). After removal of the solvent, the crude product obtained under reduced pressure was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 10/1, v/v) to give the ((3aR, 4R, 7aR)-2-oxo-4, 7a-dihydro-3aH-[1, 3]dioxolo[4, 5-c] pyran-4-yl)methyl pivalate (6) as yellow syrup (706 mg, yield 87%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.71 (d, J = 6.3 Hz, 1H), 5.20 (dd, J = 7.7, 3.2 Hz, 1H), 5.00 (ddd, J = 6.3, 3.2, 1.1 Hz, 1H), 4.87 (d, J = 7.7 Hz, 1H), 4.44 (dd, J = 11.6, 6.9 Hz, 1H), 4.34 (dd, J = 11.6, 5.8 Hz, 1H), 4.17 (dd, J = 11.3, 5.7 Hz, 1H), 1.23 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 177.9, 153.7, 149.1, 98.0, 73.0, 71.5, 68.7, 62.1, 38.7, 27.0.
The mixture of D-galactal carbonate (6, 256 mg, 1.0 mmol), thiol (1.0 mmol), Pd2(dba)3 (9.1 mg, 0.01 mmol) and 9, 9-dimethyl-4, 5-bis(diphenylphosphino)xanthene (xantphos, 11.6 mg, 0.02 mmol) was dissolved in anhydrous dichloromethane (4 mL) under nitrogen atmosphere, and the reaction mixture was stirred at room temperature for 12 hours until the completion of reaction (TLC monitoring). The mixture was quenched by ice water (5 mL), obtaining the crude product extracted by dichloromethane (5 mL × 3). After removing the solvent, the crude product was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 5/1, v/v) to give the target product (7).
((2R, 3R, 6S)-3-Hydroxy-6-(naphthalen-2-ylthio)-3, 6-dihydro-2H-pyran-2-yl)methyl pivalate (7a). White solids (316 mg, yield 85%, m.p. 107~108 ℃). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 1.1 Hz, 1H), 7.83~7.77 (m, 3H), 7.64 (dd, J = 8.5, 1.8 Hz, 1H), 7.51~7.48 (m, 2H), 6.06 (ddd, J = 10.0, 5.5, 1.9 Hz, 1H), 6.00 (dd, J = 10.0, 1.4 Hz, 1H), 5.60 (d, J = 1.8 Hz, 1H), 4.37~4.28 (m, 2H), 3.84 (td, J = 6.3, 1.8 Hz, 1H), 3.81~3.76 (m, 1H), 1.18 (s, 9H). 13C NMR (101 MHz, CDCl3) δ (ppm): 178.3, 133.3, 132.7, 132.6, 131.0, 130.3, 129.5, 129.4, 128.3, 127.7, 127.6, 126.6, 82.2, 76.2, 63.4, 61.5, 38.7, 27.1. HRMS (ESI) m/z: calcd. for C21H24O4SNa+ (M + Na)+ 395.1293; found 395.1291.
((2R, 3R, 6S)-6-(Benzylthio)-3-hydroxy-3, 6-dihydro-2H-pyran-2-yl)methyl pivalate (7b). Colorless oil (272 mg, yield 81%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.34~7.25 (m, 5H), 6.14 (ddd, J = 10.0, 5.7, 2.2 Hz, 1H), 5.86 (dd, J = 10.1, 1.4 Hz, 1H), 5.15 (dd, J = 3.7, 1.8 Hz, 1H), 4.37 (dd, J = 11.6, 5.7 Hz, 1H), 4.27 (dd, J = 11.6, 6.8 Hz, 1H), 4.00 (d, J = 13.3 Hz, 1H), 3.86~3.74 (m, 3H), 1.90 (d, J = 10.2 Hz, 1H), 1.24 (s, 9H).13C NMR (100 MHz, CDCl3) δ (ppm): 178.5, 137.5, 130.8, 129.2, 129.1, 128.5, 127.2, 78.2, 76.1, 63.5, 61.9, 38.8, 33.9, 27.1. HRMS (ESI) m/z: calcd. for C18H24O4SNa+ (M + Na)+ 359.1288; found 359.1287.
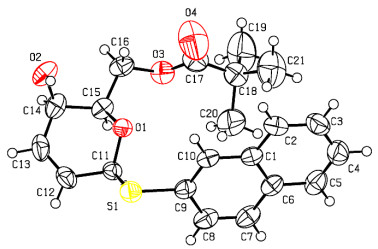
Compound 7a was crystallized after slow evaporation from a saturated chloroform solution as colorless blocks with dimensions of 0.12mm × 0.10mm × 0.08mm in the monoclinic system. Diffraction data of the single crystal were collected at 120 K on a Bruker SMART APEX-II CCD diffractometer with graphite-monochromated CuKα radiation (λ = 1.54184 Å) using the Bruker Collect software. A total of 6645 reflections were collected in the range of 4.95≤θ≤73.79º by using an ω-scan mode, of which 3621 were unique with Rint = 0.0390 and 3326 were observed with I > 2σ(I). After the initial corrections and data reduction, intensities of reflections were used to solve (by direct methods) and refine the structures (on F2) using the WINGX program. A weighting scheme based upon P = (Fo2 + 2Fc2)/3 was employed. All the hydrogen atoms were located from difference Fourier maps and included in the refinements as riding. Empirical absorption corrections were applied. The structures were solved by direct methods using SHELXS-97 programs[18]. All of the non-hydrogen atoms were located from difference Fourier maps, and then refined anisotropically with SHELXL-97 via a full-matrix least-square procedure[19]. The final R = 0.0478, wR = 0.1384, (Δ/σ)max = 0.000, S = 1.040, (Δρ)max = 0.345 and (Δρ)min = –0.214 e/Å3. The Flack parameter was 0.019(15) (Fig. 1).
Human gastric cancer HGC-27 cell lines and human gastric mucosa epithelial GES-1 cell lines were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Cell viability was assessed by 3-(4, 5)-dimethylthiazol-2-yl-2, 5-diphenyltetrazolium bromide (MTT) cell staining as previously described[20]. The cell lines (1.0 × 104 cells/well) were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin at humidified 5% CO2 atmosphere in a 96-well plate. The cell lines were exposed to the drugs with different concentrations (100.0, 50.0 and 10.0 μg/mL) for 48 hours, and paclitaxel (10.0 μg/mL) was as the positive control. MTT (50 μL of a 5 mg/mL in PBS; Sigma-Aldrich) was added to each well and the cell lines were incubated in a CO2 incubator at 37 ℃ for 5 hours. Following media removal, the MTT-formazan formed by metabolically viable cells was dissolved in 200 μL of DMSO (Sigma-Aldrich) and the absorbance was measured in a plate reader at 492 nm. The survival (%) was calculated by the following formula: No. of viable cells (dye excludeed cells)/No. of the total.
Since the pivalate protected molecules could enhance the cytotoxicity as prodrugs[21], the key intermediate D-galactal carbonate (6) was prepared from commercial D-galactal (1) through a four-step reaction with a total yield of 62% (Scheme 1). Conditions optimization for the S-glycosylation reaction was conducted by the palladium catalyst in the presence of phosphorus ligands (P-ligands) including 1, 4-bis(diphenylphosphino)butane (DPPB), 1, 1΄-bis(diphenylphosphino)ferrocene (DPPF), tricyclohexylphosphine (P(Cy)3) and 4, 5-bis(diphnylphosphino)-9, 9-dimethylxanthene (xantphos) in various solvents (Table 1). The palladium(II) salt and DPPB ligand failed to catalyze the S-glycosylation reaction in THF (entries 1 and 2), and subsequently coor-dinated palladium catalysts seemed to promote the reaction, although it is in a poor yield (entry 3~5). Changing the polar solvent (DMF, DMSO) or non-polar toluene also resulted in failure (entry 6~8). To our surprise, less-polar solvent including dichloromethane and carbon tetrachloride showed some positive results (entry 9~11), and the optimal condition for the S-glycosylation reaction was finally confirmed as the 2.5 mol% Pd2(dba)3 catalyst and the 5 mol% xantphos in dichloromethane at 35 ℃ for 12 hours after the ligands and temperature screening (entry 12~15). Due to the potential steric effect and better cytotoxities for some aromatic substituents (phenyl, benzyl or naphthyl) of sugars[22, 23], three aromatic groups (4a, 7a and 7b) and one aliphatic group (4b) substituted thioglycosides were smoothly prepared under these conditions.
 DownLoad:
CSV
DownLoad:
CSV
| Entrya | Catalyst | Ligand | Solvent | Yieldb |
| 1 | PdCl2 | DPPB | THF | 0 |
| 2 | Pd(OAc)2 | DPPB | THF | 0 |
| 3 | Pd(PPh3)4 | DPPB | THF | 3 |
| 4 | Pd(acac)2 | DPPB | THF | 9 |
| 5 | Pd2(dba)3 | DPPB | THF | 15 |
| 6 | Pd2(dba)3 | DPPB | DMF | 0 |
| 7 | Pd2(dba)3 | DPPB | DMSO | 0 |
| 8 | Pd2(dba)3 | DPPB | Toluene | 0 |
| 9 | Pd2(dba)3 | DPPB | CH3CN | 0 |
| 10 | Pd2(dba)3 | DPPB | CH2Cl2 | 30 |
| 11 | Pd2(dba)3 | DPPB | CCl4 | 25 |
| 12 | Pd2(dba)3 | DPPF | CH2Cl2 | 0 |
| 13 | Pd2(dba)3 | PCy3 | CH2Cl2 | 0 |
| 14 | Pd2(dba)3 | Xantphos | CH2Cl2 | 68 |
| 15c | Pd2(dba)3 | Xantphos | CH2Cl2 | 85 |
| a Unless otherwise specified, all reactions were carried out with 0.2 mmol of intermediate 5, 0.2 mmol of 2-naphthalenethiol, 2.5 mol% Pd catalyst and 5 mol% P-ligand in 2 mL solvent, which were stirred at 20 ℃ for 12 hours under N2 atmosphere. b Isolated yield. cThe reaction was conducted under 35 ℃. | ||||
The target thioglycosides (4a/4b and 7a/7b) were characterized by NMR and HR-ESI-MS. All of the proton and carbon signals were in accord with the characteristic functional groups of the sugar skeleton. For example, the alkenyl protons at C(12)=C(13) bonds were located at 5.86~6.14 ppm as two sets of multiplets (ddd and dd) in 1H NMR spectroscopy, and the aliphatic carbon's signals were assigned to 61~83 ppm in the 13C NMR spectra. The protons of tert-butyl group were ascribable to 1.2 ppm approximately, and there was an additional carbon signal (33.9 ppm) of the methylene in benzyl group besides other aliphatic carbons'. The adduct ion [M + Na]+ in the HR-ESI-MS spectrum could be also observed for the target compound.
The selected bond lengths, bond angles and torsion angles of compound 7a are listed in Table 2, and all the bond lengths of C–C and C=C are in accordance with the standard compilations and the literature[21]. The bond angles containing unsaturated bonds in C(3)–C(2)–C(1), C(13)–C(12)–C(11) and O(4)–C(17)–O(3) range from 120° to 124°. The torsion angle of naphthalen-2-ylthio group C(9)–S(1)–C(11)–O(1) was equal to –68.2(2)°, and the π-conjugated aliphatic backbone C(11)–C(12)–C(13)–C(14) and C(16)–O(3)–C(17)– O(4) showed a planar conformation with the corresponding torsion angles of 2.8(6)° and –6.2(6)°, respectively. The aliphatic 6-membered sugar ring O(1)–C(11)–C(12)–C(13)– C(14)–C(15) exhibited a twist-boat form (Φ = 328.7397°, θ = 51.90°, puckering amplitude (Q) = 0.5196°).
DownLoad:
CSV
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| C(1)–C(2) | 1.425(5) | C(12)–C(13) | 1.321(6) | O(1)–C(11) | 1.421(4) | ||
| C(2)–C(3) | 1.363(6) | C(14)–C(13) | 1.492(6) | O(2)–C(14) | 1.426(4) | ||
| C(10)–C(1) | 1.418(5) | S(1)–C(9) | 1.770(4) | O(3)–C(17) | 1.324(5) | ||
| C(11)–C(12) | 1.495(5) | S(1)–C(11) | 1.811(3) | O(4)–C(17) | 1.205(5) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(3)–C(2)–C(1) | 120.0(4) | O(1)–C(15)–C(14) | 109.2(2) | C(9)–S(1)–C(11)–O(1) | –68.2(2) | ||
| C(9)–S(1)–C(11) | 100.90(14) | C(13)–C(12)–C(11) | 121.7(3) | C(11)–C(12)–C(13)–C(14) | 2.8(6) | ||
| C(11)–O(1)–C(15) | 110.6(2) | O(3)–C(17)–C(18) | 111.8(3) | C(16)–O(3)–C(17)–O(4) | –6.2(6) | ||
| O(1)–C(11)–S(1) | 108.9(2) | O(4)–C(17)–O(3) | 123.5(4) | C(13)–C(14)–C(15)–O(1) | –48.8(4) |
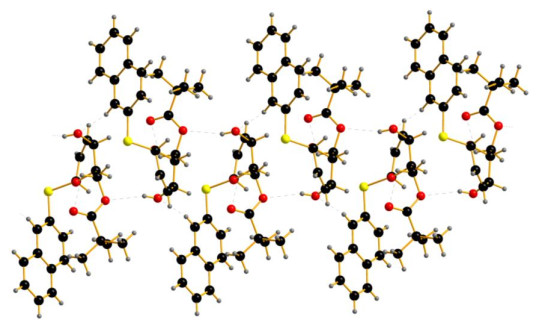
As listed in Table 3, intermolecular and intramolecular O–H···O and C–H···O interactions linked the molecules into a one-dimensional infinite chain running along the a axis, which helped to stabilize the supramolecular architecture (Fig. 2).
DownLoad:
CSV
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠(DHA) |
| O(2)–H(2)···O(3)a | 0.82 | 2.29 | 3.0830(2) | 162 |
| C(10)–H(10)···O(2)a | 0.93 | 2.53 | 3.4321(2) | 165 |
| C(16)–H(16B)···O(4) | 0.97 | 2.27 | 2.6954(1) | 105 |
| Symmetry code: (a) –1/2+x, 3/2–y, –z | ||||
As our continuous interest in search of natural products-derived biological small molecules[22-25], the target thioglycoside (4a, 4b, 7a and 7b) and its key synthetic intermediates (3, 5 and 6) were evaluated for in vitro anti-proliferative activities against human gastric carcinoma cell lines (HGC-27) for 48 hours at 37 ℃ by MTT assays. All data were presented as mean ± standard deviation and analyzed by SPSS[26]. Paclitaxel was chosen as a positive control with the IC50 of 4.16 μmol/L. The results indicated that both of the synthetic intermediates showed poor anti-proliferative effect against HGC-27 cell lines (IC50 > 100 μmol/L, Table 4). However, four of the thioglycosides (4a, 4b, 7a and 7b) had better cytotoxic effect with the IC50 value of 69~88 μmol/L, respectively. All of the tested compounds exhibited low toxicities against the normal human gastric mucosa epithelial GES-1 cell lines (IC50 > 500 μmol/L). Further exploration of the substrate scope for this reaction was under way in our research group.
DownLoad:
CSV
| Compound | Anti-proliferative results by MTT assays (IC50, μmol/L) | |
| Human gastric cancer cell lines (HGC-27) | Human gastric epithelial cell lines (GES-1) | |
| 3 | > 100 | > 500 |
| 5 | > 100 | > 500 |
| 6 | > 100 | > 500 |
| 4a | 78.6 | > 500 |
| 4b | 81.8 | > 500 |
| 7a | 69.5 | > 500 |
| 7b | 88.2 | > 500 |
| Paclitaxel a | 4.16 | > 500 |
| a Positive control | ||
In summary, two β-thiogalactosides were prepared through the palladium-catalyzed one-pot Tsuji-Trost reaction. The absolute configuration of 7a was confirmed with a Flack parameter of 0.019(15) by X-ray crystallography. The in vitro cytotoxity evaluation indicated that thioglycosides showed better anti-proliferative effect than the D-galactal carbonate intermediates, which encouraged us to further investigate the reaction with the aim of systematically assessing the biological activity for these thioglycosides.
Damaraju, V. L.; Damaraju, S.; Young, J. D.; Baldwin, S. A.; Mackey, J.; Sawyer, M. B.; Cass, C. E. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. doi: 10.1038/sj.onc.1206952
Seley-Radtke, K. L.; Yates, M. K. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antiviral Res. 2018, 154, 66–86. doi: 10.1016/j.antiviral.2018.04.004
Osada, H. Discovery and applications of nucleoside antibiotics beyond polyoxin. J. Antibiot. 2019, 72, 855–864. doi: 10.1038/s41429-019-0237-1
Feng, M.; Tang, B.; Liang, S. H.; Jiang, X. Sulfur containing scaffolds in drugs: synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. doi: 10.2174/1568026615666150915111741
Wright, A. J. The penicillins. Mayo Clin. Proc. 1999, 74, 290–307. doi: 10.4065/74.3.290
Vicente, D.; Pérez-Trallero, E. Tetracyclines, sulfonamides, and metronidazole. Enferm. Infecc. Microbiol. Clin. 2010, 28, 122–130. doi: 10.1016/j.eimc.2009.10.002
Pan, C.; Kuranaga, T.; Kakeya, H. Total synthesis of thioamycolamide A via a biomimetic route. Org. Biomol. Chem. 2020, 18, 8366–8370. doi: 10.1039/D0OB01942A
Kobayashi, E.; Motoki, K.; Uchida, T.; Fukushima, H.; Koezuka, Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol. Res. 1995, 7, 529–534.
Zhang, L.; Carthy, C. M.; Zhu, X. Synthesis of a glucosylated alpha-S-galactosylceramide as potential immunostimulant. Carbohydr. Res. 2017, 448, 43–47. doi: 10.1016/j.carres.2017.05.022
Lian, G.; Zhang, X.; Yu, B. Thioglycosides in carbohydrate research. Carbohydr. Res. 2015, 403, 13–22. doi: 10.1016/j.carres.2014.06.009
Codée, J. D.; Litjens, R. E.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Thioglycosides in sequential glycosylation strategies. Chem. Soc. Rev. 2005, 34, 769–782. doi: 10.1039/b417138c
Zhu, F.; Miller, E.; Zhang, S. Q.; Yi, D.; O'Neill, S.; Hong, X.; Walczak, M. A. Stereoretentive C(sp3)-S cross-coupling. J. Am. Chem. Soc. 2018, 140, 18140–18150. doi: 10.1021/jacs.8b11211
Zhu, M.; Alami, M.; Messaoudi, S. Electrochemical nickel-catalyzed Migita cross-coupling of 1-thiosugars with aryl, alkenyl and alkynyl bromides. Chem. Commun. 2020, 56, 4464–4467. doi: 10.1039/D0CC01126F
Procopio, A.; Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Nardi, M.; Oliverio, M.; Russo, B. A facile Er(OTf)3-catalyzed synthesis of 2, 3-unsaturated O- and S-glycosides. Carbohydr. Res. 2007, 342, 2125–2131. doi: 10.1016/j.carres.2007.05.034
Stevanović, D.; Pejović, A.; Damljanović, I.; Minić, A.; Bogdanović, G. A.; Vukićević, M.; Radulović, N. S.; Vukićević, R. D. Ferrier rearrangement promoted by an electrochemically generated zirconium catalyst. Carbohydr. Res. 2015, 407, 111–121. doi: 10.1016/j.carres.2015.02.001
Meng, S.; Zhong, W.; Yao, W.; Li, Z. Stereoselective phenylselenoglycosylation of glycals bearing a fused carbonate moiety toward the synthesis of 2-deoxy-β-galactosides and β-mannosides. Org. Lett. 2020, 22, 2981–2986. doi: 10.1021/acs.orglett.0c00732
Lai, M. N.; Abdulmajed Othman, K.; Yao, H.; Wang, Q. Y.; Feng, Y. K.; Huang, N. Y.; Liu, M. G.; Zou, K. Open-air stereoselective construction of C-aryl glycosides. Org. Lett. 2020, 22, 1144–1148. doi: 10.1021/acs.orglett.9b04665
Sheldrick, G. M. SHELXL 97, Program for Crystal Structure Determinations. University of Göttingen, Germany 1997.
Sheldrick, G. M. SHELXL 97, Program for the Refinement of Crystal Structure. University of Göttingen, Germany 1997.
Pascua-Maestro, R.; Corraliza-Gomez, M.; Diez-Hermano, S.; Perez-Segurado, C.; Ganfornina, M. D.; Sanchez, D. The MTT-formazan assay: complementary technical approaches and in vivo validation in Drosophila larvae. Acta Histochem. 2018, 120, 179–186. doi: 10.1016/j.acthis.2018.01.006
Rose, J. D.; Parker, W. B.; Someya, H.; Shaddix, S. C.; Montgomery, J. A.; Secrist, J. A. Enhancement of nucleoside cytotoxicity through nucleotide prodrugs. J. Med. Chem. 2002, 45, 4505–4512. doi: 10.1021/jm020107s
MacMillan, J. B.; Guang, X. Z.; Skepper, C. K.; Molinski, T. F. Phorbasides A−E, cytotoxic chlorocyclopropane macrolide glycosides from the marine sponge Phorbas sp. CD determination of C-methyl sugar configurations. J. Org. Chem. 2008, 73, 3699–3706. doi: 10.1021/jo702307t
Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Replacing D-glucosamine with its l-enantiomer in glycosylated antitumor ether lipids (GAELs) retains cytotoxic effects against epithelial cancer cells and cancer stem cells. J. Med. Chem. 2017, 60, 2142–2147. doi: 10.1021/acs.jmedchem.6b01773
Paolini, J. P. The bond order-bond length relationship. J. Comput. Chem. 1990, 11, 1160–1163. doi: 10.1002/jcc.540111007
Cao, C. Q.; Yan, X. M.; Yang, Q. L.; Luo, H. J.; Huang, N. Y. Synthesis and crystal structure of (Z)-2-methyl-5, 6-dihydrobenzo[d]thiazol-7(4H)-one O-prop-2-yn-1-yl oxime derivatives. Chin. J. Struc. Chem. 2014, 33, 1683−1688.
Lu, X. F.; Yang, Z.; Huang, N. Y.; He, H. B.; Deng, W. Q.; Zou, K. Synthesis and cytotoxic activities of 2-substituted (25R)-spirostan-1, 4, 6-triene-3-ones via ring-opening/elimination and "click" strategy. Bioorg. Med. Chem. Lett. 2015, 25, 3726–3729. doi: 10.1016/j.bmcl.2015.06.028
Yao, Y.; Xiong, C. P.; Zhong, Y. L.; Bian, G. W.; Huang, N. Y.; Wang, L.; Zou, K. Intramolecular and Ferrier rearrangement strategy for the construction of C1-β-D-xylopyranosides: synthesis, mechanism and biological activity study. Adv. Syn. Cat. 2019, 361, 1012–1017. doi: 10.1002/adsc.201801423
Wang, Y.; Yao, H.; Hua, M.; Jiao, Y.; He, H. B.; Liu, M. G.; Huang, N. Y.; Zou, K. Direct N-glycosylation of amides/amines with glycal donors. J. Org. Chem. 2020, 85, 7485–7493. doi: 10.1021/acs.joc.0c00975
West, B. T. Analyzing longitudinal data with the linear mixed models procedure in SPSS. Eval. Health Prof. 2009, 32, 207−228. doi: 10.1177/0163278709338554

Figure 1 ORTEP drawing of compound 6a showing thermal ellipsoids at the 50% probability level

Figure 2 Packing diagram of compound 7a. The O–H···O interactions are shown as dashed lines
Table 1. Condition Optimization for the S-Glycosylation Reaction
| Entrya | Catalyst | Ligand | Solvent | Yieldb |
| 1 | PdCl2 | DPPB | THF | 0 |
| 2 | Pd(OAc)2 | DPPB | THF | 0 |
| 3 | Pd(PPh3)4 | DPPB | THF | 3 |
| 4 | Pd(acac)2 | DPPB | THF | 9 |
| 5 | Pd2(dba)3 | DPPB | THF | 15 |
| 6 | Pd2(dba)3 | DPPB | DMF | 0 |
| 7 | Pd2(dba)3 | DPPB | DMSO | 0 |
| 8 | Pd2(dba)3 | DPPB | Toluene | 0 |
| 9 | Pd2(dba)3 | DPPB | CH3CN | 0 |
| 10 | Pd2(dba)3 | DPPB | CH2Cl2 | 30 |
| 11 | Pd2(dba)3 | DPPB | CCl4 | 25 |
| 12 | Pd2(dba)3 | DPPF | CH2Cl2 | 0 |
| 13 | Pd2(dba)3 | PCy3 | CH2Cl2 | 0 |
| 14 | Pd2(dba)3 | Xantphos | CH2Cl2 | 68 |
| 15c | Pd2(dba)3 | Xantphos | CH2Cl2 | 85 |
| a Unless otherwise specified, all reactions were carried out with 0.2 mmol of intermediate 5, 0.2 mmol of 2-naphthalenethiol, 2.5 mol% Pd catalyst and 5 mol% P-ligand in 2 mL solvent, which were stirred at 20 ℃ for 12 hours under N2 atmosphere. b Isolated yield. cThe reaction was conducted under 35 ℃. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected Bond Lengths (Å) and Bond Angles (°) for Compound 7a
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| C(1)–C(2) | 1.425(5) | C(12)–C(13) | 1.321(6) | O(1)–C(11) | 1.421(4) | ||
| C(2)–C(3) | 1.363(6) | C(14)–C(13) | 1.492(6) | O(2)–C(14) | 1.426(4) | ||
| C(10)–C(1) | 1.418(5) | S(1)–C(9) | 1.770(4) | O(3)–C(17) | 1.324(5) | ||
| C(11)–C(12) | 1.495(5) | S(1)–C(11) | 1.811(3) | O(4)–C(17) | 1.205(5) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(3)–C(2)–C(1) | 120.0(4) | O(1)–C(15)–C(14) | 109.2(2) | C(9)–S(1)–C(11)–O(1) | –68.2(2) | ||
| C(9)–S(1)–C(11) | 100.90(14) | C(13)–C(12)–C(11) | 121.7(3) | C(11)–C(12)–C(13)–C(14) | 2.8(6) | ||
| C(11)–O(1)–C(15) | 110.6(2) | O(3)–C(17)–C(18) | 111.8(3) | C(16)–O(3)–C(17)–O(4) | –6.2(6) | ||
| O(1)–C(11)–S(1) | 108.9(2) | O(4)–C(17)–O(3) | 123.5(4) | C(13)–C(14)–C(15)–O(1) | –48.8(4) |
下载: 导出CSV
Table 3. Hydrogen Bonds for Compound 7a
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠(DHA) |
| O(2)–H(2)···O(3)a | 0.82 | 2.29 | 3.0830(2) | 162 |
| C(10)–H(10)···O(2)a | 0.93 | 2.53 | 3.4321(2) | 165 |
| C(16)–H(16B)···O(4) | 0.97 | 2.27 | 2.6954(1) | 105 |
| Symmetry code: (a) –1/2+x, 3/2–y, –z | ||||
下载: 导出CSV
Table 4. Bioactivity Evaluation of the Compounds
| Compound | Anti-proliferative results by MTT assays (IC50, μmol/L) | |
| Human gastric cancer cell lines (HGC-27) | Human gastric epithelial cell lines (GES-1) | |
| 3 | > 100 | > 500 |
| 5 | > 100 | > 500 |
| 6 | > 100 | > 500 |
| 4a | 78.6 | > 500 |
| 4b | 81.8 | > 500 |
| 7a | 69.5 | > 500 |
| 7b | 88.2 | > 500 |
| Paclitaxel a | 4.16 | > 500 |
| a Positive control | ||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: