Table 1.
Selected Bond Lengths (Å) and Bond Angles (°)
Citation:
Zhi-Min HU, Rong-Min YU, Can-Zhong LU. Crystal Structure and Photophysical Property of 9-(4-(5-(6-Methylpyridin-2-yl)-3-phenyl-1H-1, 2, 4-triazol-1-yl)phenyl)-9H-carbazole[J]. Chinese Journal of Structural Chemistry,
2020, 39(10): 1871-1876.
doi:
10.14102/j.cnki.0254–5861.2011–2924

Crystal Structure and Photophysical Property of 9-(4-(5-(6-Methylpyridin-2-yl)-3-phenyl-1H-1, 2, 4-triazol-1-yl)phenyl)-9H-carbazole
English
Crystal Structure and Photophysical Property of 9-(4-(5-(6-Methylpyridin-2-yl)-3-phenyl-1H-1, 2, 4-triazol-1-yl)phenyl)-9H-carbazole
Abstract:
X-ray single-crystal structure analysis, UV-Vis, and photoluminescent properties of organic compound, 9-(4-(5-(6-methylpyridin-2-yl)-3-phenyl-1H-1, 2, 4-triazol-1-yl)phenyl)-9H-carbazole (compound 1) were studied. Compound 1 crystallizes in monoclinic system, space group P21/c with a = 16.0434(9), b = 17.920(1), c = 8.3453(5) Å, β = 94.142(5)°, V = 2393.0(2), Z = 4, Mr = 477.55, Dc = 1.326 g/cm3, F(000) = 1000, μ = 0.080 mm–1, GOOF = 1.025, the final R = 0.0505 and wR = 0.1226 for 4921 observed reflections with I > 2σ(I). Compound 1 is a donor-acceptor type molecule, in which the carbazolyl group (cz) acts as an electron donor, and the group of 2-(1, 3-diphenyl-1H-1, 2, 4-triazol-5-yl)-6-methylpyridine (MePyTz) as an electron acceptor. In methylene chloride, compound 1 exhibits deep blue luminescence with maximum at 440 nm, lifetime of 4.2 ns under N2 and quantum yield of ф = 0.18 at room temperature. The experimental and computational results show that the emission of compound 1 originates from the lowest singlet excited state (S1) with charge transfer character.
-
Key words:
- organic donor-acceptor compound
- / crystal structure
- / photoluminescence
- / DFT calculation
-
1. INTRODUCTION
Over the last decade, OLED (organic light-emitting diode) has become one of the research hotspots due to its low power consumption, self-illumination and so on, and been regarded as a new generation of flat panel display technology with broad application prospects in TV, computer, communication terminal and instrument display, vehiclemounted display, military and aerospace fields[1-5]. Emissive materials play important roles in OLED. Recently, purely organic emissive compounds of donor-acceptor type are of tremendous interest for their promising applications in OLED, among which carbazole is used extensively as electron donor because it has a unique rigid planar structure, a large conjugated system and a good ability to transfer electrons inside the molecule[6-8]. In a previous paper, we reported the synthesis of a carbazole-containing donoracceptor compound, 9-(4-(5-(6-methylpyridin-2-yl)-3-phenyl-1H-1, 2, 4-triazol-1-yl)phenyl)-9H-carbazole, and its usage as a chelating diimine ligand in the synthesis of a highly emissive cuprous complex[15]. Herein, we report the structure and photophysical properties of this carbazolecontaining ligand.
2. EXPERIMENTAL
2.1 Material and instrument
All reactions were performed under N2 atmosphere unless specified. Chemicals were purchased from commercial sources and used without further purification. The title compound was synthesized according to the literature[14, 15]. 1H NMR spectra were recorded on a Bruker Avance III 400MHz NMR spectrometer. Elemental analyses (C, H, N) were carried out with an Elemental Vario EL III elemental analyzer. Photoluminescence spectra were recorded on a HORIBA Jobin-Yvon FluoroMax-4 SPECTROMETER. The UV-Vis absorption spectra were recorded with a PerkinElmer Lambda 45 UV/vis spectrophotometer. The lifetimes of the samples at room temperatures were carried out by a HORIBA Jobin-Yvon FluoroMax-4 instrument with a Multi-channel scaling (MCS) peripheral equipment and a spectra LED (373 nm). The PL quantum yields, which were defined as the number of photons emitted per photon absorbed by the system, were measured by FluoroMax-4-equipped with an integrating sphere.
2.2 Crystal structure determination and refinement
Crystals suitable for X-ray single-crystal analysis were obtained from crystallization of the title compound in a mixture solvent of methylene chloride/ethanol. A colorless crystal of compound 1 with dimensions of 0.50 × 0.40 × 0.20 mm3 was used for X-ray diffraction analysis. Diffraction data of the complex were collected on a SuperNova, Dual, Cu at zero, Atlas diffractometer equipped with graphite-monochromated MoKα radiation (λ = 0.71073 Å). A total of 13915 reflections were collected at 100 K in the range of 2.27≤θ≤30.22º by using an ω-scan mode, of which 6132 were unique with Rint = 0.0305 and 4921 were observed with I > 2σ(I). The structure was solved by direct methods with SHELXS-2014 and refined by full-matrix least-squares methods with SHELXL-2014 program package[9]. All of the non-hydrogen atoms were located with successive difference Fourier synthesis. Hydrogen atoms were added in idealized positions. The non-hydrogen atoms were refined anisotropically. The final R = 0.0505, wR = 0.1226 (w = 1/[σ2(Fo2) + (0.0607P)2 + 1.0984P], where P = (Fo2 + 2Fc2)/3) for the observed reflections with I > 2σ(I), and R = 0.0643, wR = 0.1344 for all data with S = 1.025, (Δ/σ)max = 0.001, (Δρ)max = 0.287 and (Δρ)min = –0.334 e/Å3. Selected bond lengths and bond angles are listed in Table 1.
Table 1

 DownLoad:
CSV
DownLoad:
CSV
Bond Dist. Angle (°) N(1)–C(2) 1.335(2) C(2)–N(1)–C(6) 118.3(1) N(1)–C(6) 1.338(2) C(8)–N(3)–C(4) 102.4(1) N(2)–C(7) 1.328(2) C(7)–N(2)–C(8) 103.5(1) N(2)–C(8) 1.358(2) N(3)–N(4)–C(15) 118.8(1) N(3)–C(8) 1.328(2) N(3)–N(4)–C(7) 109.7(1) N(3)–N(4) 1.368(2) N(2)–C(7)–N(4) 109.6(1) N(4)–C(15) 1.432(2) N(5)–C(19)–C(24) 108.9(1) N(5)–C(18) 1.420(2) C(18)–N(5)–C(30) 125.4(1) N(5)–C(19) 1.400(2) C(19)–N(5)–C(30) 108.2(1) N(5)–C(30) 1.398(2) C(18)–N(5)–C(19) 124.4(1) 2.3 Computational methodology
The computations were carried out using Gaussian 09W[10]. The input geomerical data are from X-ray crystal structure. Geometrical optimization was performed by using density functional theory (DFT) with the hybrid Becke three-parameter Lee-Yang-Parr (B3LYP) functional level[11, 12]. In this calculation, all-electron basis set of 6-31G* was used for N, C, and H atoms. Visualization of the optimized structures and frontier molecular orbitals were performed by GaussView. The partition orbital composition was analyzed by using the Multiwfn 2.4 program[13].
3. RESULTS AND DISCUSSION
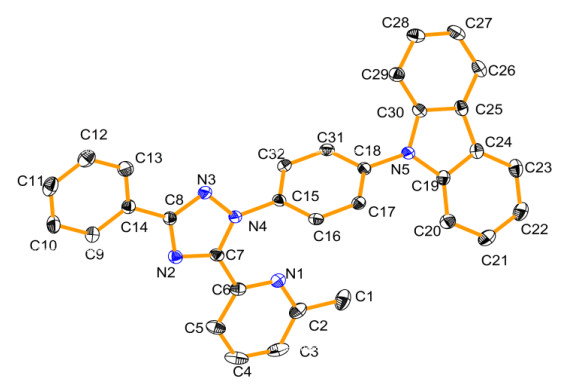
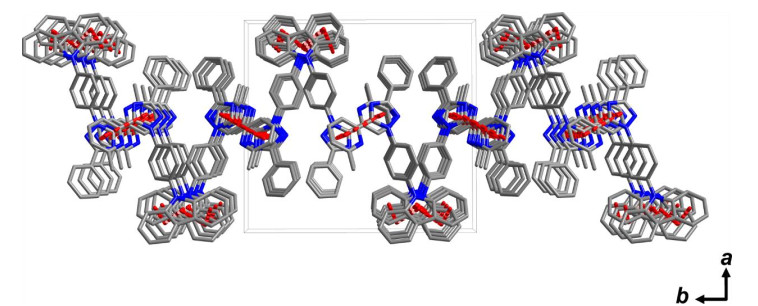
Compound 1 has been previously synthesized, but its structure and photophysical properties have not been studied in details[14, 15]. Fig. 1 shows its molecular structure in ORTEP diagram. Selected bonding distances and angles are summarized in Table 1. For clarity, the molecule is considered to contain three parts, including the carbazole moiety, the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine moiety, and the benzene ring that links the former moieties. The bonding distances between carbon atoms in the aromatic rings fall in the normal range. The carbazole moiety and the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine moiety are planar, respectively. The bonding distances of the C–N bonds in the carbazole moiety are about 1.40 Å, longer than those of the triazole-pyridine moiety. The bond lengths of C–N between the bridging benzene ring and the carbazole or 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine groups are about 1.43 Å, obviously shorter than that of a typical C–N single bond, suggesting that electronic delocalization and conjugation may extend partially over the whole molecule. The bridging benzene ring is not in the same plane to neither the carbazole group nor the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine group. The dihedral angles formed by the planes defined by the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine moiety and the carbazole group with the intermediate benzene ring are 48.42(4)° and 65.28(4)°, respectively. Additionally, in the X-ray structure, the carbazole and the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine group are not coplanar, with their dihedral angle to be 22.42(5)º. Fig. 2 shows a relatively weak intermolecular π-π stacking interaction between two pyridine rings, between a pyridine and a pyrazole rings, between a nitrogen-containing five-membered ring in the carbazole portion and a benzene ring in carbazole portion and between two benzene rings in carbazole portion to form a two-dimensional structure with the distances of 3.513, 3.858, 3.741 and 4.057 Å, respectively. The defined ring and relative parameters of π-π interactions in compound 1 are summarized in Table 2.
Figure 1
 Figure 1. ORTEP diagram (above) of compound 1. Thermal ellipsoids are drawn at 50% probability
Figure 1. ORTEP diagram (above) of compound 1. Thermal ellipsoids are drawn at 50% probabilityFigure 2
 Figure 2. π-π stacking interaction diagram for compound 1
Figure 2. π-π stacking interaction diagram for compound 1Table 2
Table 2. Defined Ring and Relative Parameters of π-π Interactions in Compound 1DownLoad:
CSV
Cg(1): N2-- > C7-- > N4-- > N3-- > C8-- > ; Cg(2): N5-- > C19-- > C24-- > C25-- > C30-- > ; Cg(3): N1-- > C2-- > C3-- > C4-- > C5-- > C6-- > ;
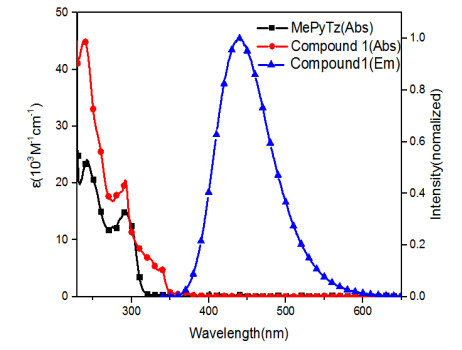
Cg(4): C19-- > C20-- > C21-- > C22-- > C23-- > C24-- > ; Cg(5): C25-- > C26-- > C27-- > C28-- > C29-- > C30-- >Cg(I) Cg(J) Cg-Cg Alpha CgI_Perp CgJ_Perp Slippage Cg(1) Cg(3)#1 3.8577(8) 5.76(7) 3.3194(5) 3.4874(6) 1.649 Cg(2) Cg(4)#2 4.0573(9) 2.71(7) 3.5968(6) 3.6533(6) 1.765 Cg(3) Cg(3)#3 3.5126(8) 0.00(7) 3.3138(6) 3.3139(6) 1.165 Cg(4) Cg(5)#4 3.7405(9) 3.88(7) 3.5945(6) 3.6541(6) 0.799 Symmetry codes: #1: 1 – x, 1 – y, 1 – z; #2: x, 3/2 – y, 1/2 + z; #3: 1 – x, 1 – y, –z; #4: x, 3/2 – y, –1/2 + z Fig. 3 shows the UV-vis absorption of the free 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine (MePyTz) and compound 1 in methylene chloride, and emission spectra of 1 in methylene chloride at room temperature. Compound 1 exhibits multiple intense absorption bands with peaks at 238, 292 nm, a broad peak at about 337 nm, and a shoulder at 371 nm. The intense absorption bands of compound 1 are very similar to those of the free MePyTz. Therefore, the intense absorption bands at 238 and 292 nm are assigned to the spinallowed π-π* transitions inside the 2-methyl-6-(3-phenyl-1H-1, 2, 4-triazol-5-yl)pyridine group, while the other bands to the intramolecular charge transfer (ICT) transitions with electron transfer from the donor to the acceptor moieties.
Figure 3
 Figure 3. UV-vis absorption and emission spectra of compound 1 in CH2Cl2. Compound 1 displays a deep blue emission with maximum at 440 nm. The PLQY of it is 18%, and its lifetime is 4.2 ns under N2
Figure 3. UV-vis absorption and emission spectra of compound 1 in CH2Cl2. Compound 1 displays a deep blue emission with maximum at 440 nm. The PLQY of it is 18%, and its lifetime is 4.2 ns under N2In methylene chloride solution at room temperature, compound 1 displays a deep blue emission with maximum at 440 nm. The emission spectrum is broad and unstructured, suggesting the charge-transfer character of the emission. The photoluminescence quantum yield of the emission is 18%. The transient PL decay spectra at room temperature under N2 demonstrate a lifetime of 4.2 ns. Based on the spectral profile and the lifetime of the emission, it is logic to assume that the emission of compound 1 is from a S1 excited state that has charge transfer character[16, 17]. This is consistent with the results of DFT and TDDFT calculations.
The electronic structure, the type and character of electronic transition and the composition of the involved orbitals can be elucidated by DFT and TDDFT calculations carried out on the optimized S0 geometry of compound 1. 1 is composed of a carbazole moiety and an MePyTz moiety, and is in a twisted D-A conformation with a dihedral angle of 54.9° between the electron-donor unit (cz) and the electron-acceptor unit (MePyTz) on its optimized S0 geometry, as compared to 48.42(4)° in single crystal structure. It is known that a largely twisted geometry will lead to an efficient spatial separation between the HOMO and LUMO for a D-A molecule. As expected, DFT calculations show that the HOMO of compound 1 is located on the donor moiety (82.0% in cz), and the LUMO is on the acceptor moiety (98.5% in MePyTz). For a D-A molecule, a well spatial separation of HOMO and LUMO often results in a small ∆EST value and facilitates the thermally activated delayed fluorescence (TADF)[18-21]. However, compound 1 has a large ∆EST value of 0.43 eV from calculations, which is consistent to the fact that no TADF was observed for compound 1.
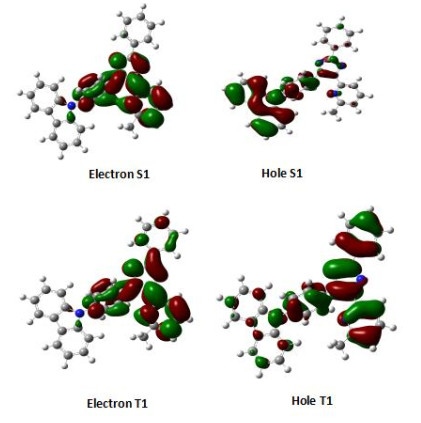
TD-DFT calculations also revealed that the S1 and T1 involve very different electronic transitions. From calculations, the S0 → S1 transition is predominantly composed of the HOMO→LUMO (98%) transition, but the S0 → T1 mainly consists of HOMO-2 → LUMO (45%) transition. Natural transition orbital (NTO) analyses were performed to reveal the origins of excited states. Fig. 4 displays the contour surface diagrams of electron and hole orbitals for S1 and T1. The maps of S1 and T1 are shown in Fig. 3. The electron and hole maps of S1 state are essentially the same to those of HOMO and LUMO, respectively. The hole of T1 in 1 is composed of a major contribution from the MePyTz segment (89.2%) and a little contribution from carbazole (10.8%). The electron distribution lies dominantly on the MePyTz segment (98.5%). Therefore, the calculation results allow us to assign the S1 as a charge transfer (CT) state, while T1 as an LE state which majorly localizes in the MePyTz segment.
Figure 4
 Figure 4. Electron and hole orbitals for S1 and T1 in the excited state for compound 1
Figure 4. Electron and hole orbitals for S1 and T1 in the excited state for compound 1In conclusion, the structure and photophysical properties of a donor-acceptor type compound 1 were investigated experimentally and theoretically. The twisted structure of the donor-acceptor molecule results in a well spatial separation between the HOMO and HOMO orbitals. The S1 state is contributed solely by the transition from HOMO to LUMO with obvious charge transfer character. The T1 state is confined in the acceptor moiety and has LE character. The emission of the compound at room temperature is originated from S1. The big ∆EST value of 0.43 eV suggests that the compound should not be a good TADF candidate for OLED.
-
-
[1]
Bin, Z. Y.; Liu, Z. Y.; Duan, L. Organic radicals outperform LiF as efficient electron-injection materials for organic light-emitting diodes. J. Phys. Chem. Lett. 2017, 8, 4769–4773. doi: 10.1021/acs.jpclett.7b02125
-
[2]
Zhao, C. G.; Duan, L. Review on photo- and electrical aging mechanisms for neutral excitons and ions in organic light-emitting diodes. J. Mater. Chem. C 2020, 8, 803–820. doi: 10.1039/C9TC05373E
-
[3]
Heimel, P.; Mondal, A.; May, F. Unicolored phosphor-sensitized fluorescence for efficient and stable blue OLEDs. Nat. Commun. 2018, 9, 4990–4990. doi: 10.1038/s41467-018-07432-2
-
[4]
Jia, J. H.; Liang, D.; Yu, R. M.; Chen, X. L.; Meng, L. Y.; Chang, J. F.; Liao, J. Z.; Yang, M. X.; Li, X. N.; Lu, C. Z. Coordination-induced thermally activated delayed fluorescence: from non-TADF donor-acceptor-type ligand to TADF-active Ag-based complexes. Chem. Mater. 2020, 32, 620–629. doi: 10.1021/acs.chemmater.9b04585
-
[5]
Zampetti, A.; Minotto, A.; Cacialli, F. Near-infrared (NIR) organic light-emitting diodes (OLEDs): challenges and opportunities. Adv. Funct. Mater. 2019, 29, 1807623. doi: 10.1002/adfm.201807623
-
[6]
Ledwon, P. Recent advances of donor-acceptor type carbazole-based molecules for light emitting applications. Org. Electron. 2019, 75, 105422. doi: 10.1016/j.orgel.2019.105422
-
[7]
Wex, B.; Kaafarani, B. R. Perspective on carbazole-based organic compounds as emitters and hosts in TADF applications. J. Mater. Chem. C 2017, 5, 8622–8653. doi: 10.1039/C7TC02156A
-
[8]
Wong, M. Y.; Zysman-Colman, E. Purely organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 2017, 29, 1605444. doi: 10.1002/adma.201605444
-
[9]
Sheldrick, G. M. SHELXL-2014/7, Program for Refinement of Crystal Structures. Institute for Inorganic Chemistry. University of Göttingen: Göttingen, Germany 2014.
-
[10]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A. Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; AleLaham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian Inc., Wallingford, CT 2009, Gaussian 09, Revision D. 01.
-
[11]
Zhao, Y.; Truhlar, D. G. Hybrid Meta density functional theory methods for thermochemistry, thermochemical Kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. doi: 10.1021/jp048147q
-
[12]
Lu, T.; Chen, F. W. A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. doi: 10.1002/jcc.22885
-
[13]
Huang, P. C.; Parthasarathy, K.; Cheng, C. H. Copper-catalyzed intramolecular oxidative C–H functionalization and C–N formation of 2-aminobenzophenones: unusual pseudo-1, 2-shift of the substituent on the aryl ring. Chem. Eur. J. 2013, 19, 460–464. doi: 10.1002/chem.201203859
-
[14]
Shan, G. G.; Li, H. B.; Zhu, D. X.; Su, Z. M.; Liao, Y. Intramolecular π-stacking in cationic iridium(III) complexes with a triazole-pyridine type ancillary ligand: synthesis, photophysics, electrochemistry properties and piezochromic behavior. J. Mater. Chem. 2012, 22, 12736–12739. doi: 10.1039/c2jm30480e
-
[15]
Zhang, Q.; Chen, X. L.; Chen, J.; Wu, X. Y.; Yu, R. M.; Lu, C. Z. Photo- and electro-luminescence of four cuprous complexes with sterically demanding and hole transmitting diimine ligands. Dalton. Trans. 2015, 44, 10022–10029. doi: 10.1039/C5DT01108F
-
[16]
Thangaraji, V.; Rajamalli, P.; Jayakumar, J.; Huang, M. J.; Chen, Y. M.; Cheng, C. H. Quinolinylmethanone-based thermally activated delayed fluorescence emitters and the application in OLEDs: effect of intramolecular H-bonding. ACS. Appl. Mater. Interfaces 2019, 11, 17128–17133. doi: 10.1021/acsami.8b22704
-
[17]
Yang, Z. Y.; Mao, Z.; Xie, Z. L.; Zhang, Y.; Liu, S. W.; Zhao, J.; Xu, Z. R.; Chi, Z. G.; Aldred, M. P. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 2017, 46, 915–1016. doi: 10.1039/C6CS00368K
-
[18]
Endo, A.; Ogasawara, M.; Takahashi, A.; Yokoyama, D.; Kato, Y.; Adachi, C. Thermally activated delayed fluorescence from Sn4+-porphyrin complexes and their application to organic light emitting diodes — a novel mechanism for electroluminescence. Adv. Mater. 2009, 21, 4802–4806. doi: 10.1002/adma.200900983
-
[19]
Kawasumi, K.; Wu, T.; Zhu, T. Y.; Chae, H. S.; VanVoorhis, T.; Baldo, M. A.; Swager, T. M. Thermally activated delayed fluorescence materials based on homoconjugation effect of donor-acceptor triptycenes. J. Am. Chem. Soc. 2015, 137, 11908–11911. doi: 10.1021/jacs.5b07932
-
[20]
Klessinger, M. Conical intersections and the mechanism of singlet photoreactions. Angew. Chem. Int. Ed. 1995, 34, 549–551. doi: 10.1002/anie.199505491
-
[21]
Chen, T.; Zheng, L.; Yuan, J.; An, Z. F.; Chen, R. F.; Tao, Y.; Li, H. H.; Xie, X. J.; Huang, W. Understanding the control of singlet-triplet splitting for organic exciton manipulating: a combined theoretical and experimental approach. Sci. Rep. 2015, 5, 10923–10934. doi: 10.1038/srep10923
-
[1]
-
Figure 1 ORTEP diagram (above) of compound 1. Thermal ellipsoids are drawn at 50% probability
Figure 3 UV-vis absorption and emission spectra of compound 1 in CH2Cl2. Compound 1 displays a deep blue emission with maximum at 440 nm. The PLQY of it is 18%, and its lifetime is 4.2 ns under N2
Table 1. Selected Bond Lengths (Å) and Bond Angles (°)
Bond Dist. Angle (°) N(1)–C(2) 1.335(2) C(2)–N(1)–C(6) 118.3(1) N(1)–C(6) 1.338(2) C(8)–N(3)–C(4) 102.4(1) N(2)–C(7) 1.328(2) C(7)–N(2)–C(8) 103.5(1) N(2)–C(8) 1.358(2) N(3)–N(4)–C(15) 118.8(1) N(3)–C(8) 1.328(2) N(3)–N(4)–C(7) 109.7(1) N(3)–N(4) 1.368(2) N(2)–C(7)–N(4) 109.6(1) N(4)–C(15) 1.432(2) N(5)–C(19)–C(24) 108.9(1) N(5)–C(18) 1.420(2) C(18)–N(5)–C(30) 125.4(1) N(5)–C(19) 1.400(2) C(19)–N(5)–C(30) 108.2(1) N(5)–C(30) 1.398(2) C(18)–N(5)–C(19) 124.4(1)  下载: 导出CSV
下载: 导出CSV
Table 2. Defined Ring and Relative Parameters of π-π Interactions in Compound 1
Cg(1): N2-- > C7-- > N4-- > N3-- > C8-- > ; Cg(2): N5-- > C19-- > C24-- > C25-- > C30-- > ; Cg(3): N1-- > C2-- > C3-- > C4-- > C5-- > C6-- > ;
Cg(4): C19-- > C20-- > C21-- > C22-- > C23-- > C24-- > ; Cg(5): C25-- > C26-- > C27-- > C28-- > C29-- > C30-- >Cg(I) Cg(J) Cg-Cg Alpha CgI_Perp CgJ_Perp Slippage Cg(1) Cg(3)#1 3.8577(8) 5.76(7) 3.3194(5) 3.4874(6) 1.649 Cg(2) Cg(4)#2 4.0573(9) 2.71(7) 3.5968(6) 3.6533(6) 1.765 Cg(3) Cg(3)#3 3.5126(8) 0.00(7) 3.3138(6) 3.3139(6) 1.165 Cg(4) Cg(5)#4 3.7405(9) 3.88(7) 3.5945(6) 3.6541(6) 0.799 Symmetry codes: #1: 1 – x, 1 – y, 1 – z; #2: x, 3/2 – y, 1/2 + z; #3: 1 – x, 1 – y, –z; #4: x, 3/2 – y, –1/2 + z
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 615
- HTML全文浏览量: 5
 下载:
下载:
