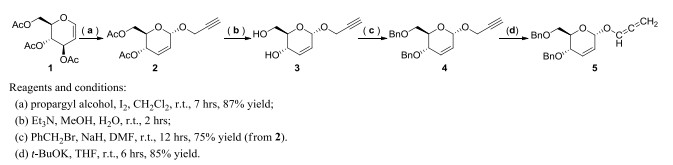
Figure 1.
Synthetic routes for the key intermediate 5
Synthesis and Absolute Configuration of(2R, 3S, 4Z, 6Z)-1, 3-bis(benzyloxy)-8-chloro-7-((E)-(2-(2, 4-dinitrophenyl)hydrazono)methyl)octa-4, 6-dien-2-ol
OTHMAN Karwan ABDULMAJED , Jin-Jin CAI , Rui XIE , Xin-Ran ZHOU , Hui YAO , Nian-Yu HUANG

Nowadays, the development of antitumor drugs for the treatment of malignant tumors has attracted wide attention of medicinal chemists[1]. For example, nitrogen mustard has been proven as an effective tumor alkylating agent for a long history of over 70 years, and the targeted modification of nitrogen mustards is still an important strategy for the discovery of anticancer drugs presently[2, 3]. Natural products are an important original source of many widely used anti-cancer agents[4, 5], and the widespread existence of nitrogen-containing compounds such as amino acids, alkaloids, and functional materials coupled with their use as useful synthons has prompted the exploration of new activation methods in chemistry[6, 7] and biology[8, 9].
As the stable mimics of O-glycosides, the C-glycosides exhibit better features in terms of pharmacodynamics and pharmacokinetics and have been developed as potential therapeutic agents, such as the antidiabetic agents dapagliflozin[10], Pro-Xylane[11] and KRN7000[12]. Owing to the potential bioactivity as well as the synthetic challenges, C-glycosides have attracted considerable interest, and extensive researches including cyclization, cross-coupling and intramolecular rearrangements reactions were reported to the construction of these molecules in recent years[13-15]. Inspired by these studies, we designed and synthesized the benzyl protected O-glucal (Scheme 1), and then treated it to C-glycoside via the [1, 3]-sigmatropic O→C rearrangement strategy[16] using Lewis acid catalyst in this work (Scheme 2).
Most of chemical reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and used without further purification. Solvents were dried and purified using standard techniques. Tri-O-acetyl-D-glucal (1) was purchased from J & K Chemical Co., Ltd., China. Reactions were monitored by thin layer chromatography on silica gel GF254 pre-coated plates. 1D and 2D nuclear magnetic resonance (NMR) spectra were recorded at a Bruker UltrashiedTM 400 MHz Plus spectrometer. Chemical shifts (δ) were reported in ppm, using residual solvent as an internal standard. Melting points were tested with an uncorrected X-4 digital melting point apparatus. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were obtained using a Waters Q-TOF premierTM mass spectrometer. Characterization data for known compounds were checked in comparison with literature for consistency and not presented in this report.
The propargyl alcohol (0.62 g, 11 mmol) and iodine (253 mg, 1 mmol) were added to a mixture of tri-O-acetyl-D-glucal (1, 2.72 g, 10 mmol) in dry dichloromethane (20 mL) at room temperature. The reaction mixture was stirred for 7 hours until the completion of the reaction monitored by thin-layer chromatography (TLC). Then the I2 catalyst was filtrated from the reaction and washed with dichloromethane. The organic phase was combined and condensed under vacuum to get crude product, which was purified by silica gel column chromatography (eluents: petroleum ether/ethyl acetate = 6/1, v/v) to get O-glucal (2, 2.33 g) as white solid in 87% yield. m.p. 57~58 ℃[17]. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.92 (d, J = 2.0 Hz, 1H), 5.84 (dt, J = 12.0, 4.0 Hz, 1H), 5.34 (dd, J = 8.8, 2.3 Hz, 1H), 5.23 (s, 1H), 4.20 (d, J = 2.0 Hz, 1H), 4.23~4.27 (m, 1H), 4.18 (dd, J = 12.0, 2.8 Hz, 1H), 4.06~4.11 (m, 1H), 2.45 (t, J = 1.2 Hz, 1H), 2.09 (d, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 170.7, 170.2, 129.7, 127.1, 126.0, 92.7, 67.1, 65.0, 62.7, 55.0, 20.9, 20.7.
The O-glucal (2, 2.33 g, 8.7 mmol) was dissolved in MeOH/H2O (v/v = 1:1, 30 mL), and triethyl amine (3 mL) was added and stirred for 2 hours at ambient temperature. After the completion of the hydrolysis reaction by TLC monitoring, the solvent was removed under reduced pressure. The resulting clear oily residue (glucal intermediate 3) was used without any further purification. The solution of glucal (3) in dry N, N΄-dimethylforamide (20 mL) was cooled down to 0 ℃, and sodium hydride (60 percent suspension in mineral oil, 0.70 g, 17.4 mmol) was added and stirred for 30 min. Benzyl bromide (3.42 g, 20 mmol) was added, the cooling bath was removed and the reaction mixture was stirred at room temperature until all substrate was fully consumed. The mixture was cooled down to 0 ℃ (ice bath), and water (50 mL) was added slowly, followed by methylene chloride (30 mL). Organic layer was separated, and then the water solution was extracted with methylene chloride (2 × 20 mL). Combined organic solutions were washed with water until neutral, then with brine, and dried over anhydrous sodium sulfate. Drying agents and solvents were removed and the product was purified by column chromatography (Silica Gel 60 Merck), using n-hexane and ethyl acetate as eluents (10:1, v/v). Fractions containing the product were pooled together, evaporated to dryness and dried under reduced pressure to give 2.37 g of the propargyl ether intermediate (4, 75% yield)[18]. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.23~7.34 (m, 1.0H), 6.10 (d, J = 6.0 Hz, 1H), 5.77 (dt, J = 8.0, 4.0 Hz, 1H), 5.23 (s, 1H), 4.58~4.66 (m, 2H), 4.42~4.52 (m, 2H), 4.30 (d, J = 4.0 Hz, 1H), 4.20 (dd, J = 12.0, 2.4 Hz, 2H), 3.94 (dt, J = 3.0 Hz, 1H), 3.72 (d, J = 2.0 Hz, 2H), 2.40 (t, J = 2.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 138.0, 137.9, 131.2, 128.3, 128.3, 128.3, 127.9, 127.8, 127.7, 127.7, 127.6, 127.6, 125.9, 92.9, 79.4, 77.9, 76.6, 74.4, 73.6, 71.0, 70.1, 69.4, 69.0, 68.6, 54.7, 54.4.
To a stirred solution of propargyl ether intermediate (4, 1.82 g, 5 mmol) in anhydrous tetrahydrofuran (THF, 30 mL) was added potassium tert-butoxide (t-BuOK, 0.62 g, 5.5 mmol), and the reaction was stirred at room temperature for 6 hours until the starting material was fully consumed (monitored by TLC). The reaction was diluted with water (5 mL) and CH2Cl2 (30 mL), and the mixture was quenched by brine (30 mL). The organic phase was separated, washed with water, dried by MgSO4, and concentrated under vacuum to give the allenyl ether intermediate (5) in 85% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.24~7.36 (m, 10H), 6.69 (t, J = 6.0 Hz, 1H), 6.14 (d, J = 10.2 Hz, 1H), 5.82 (dt, J = 10.2, 2.3 Hz, 1H), 5.32~5.42 (m, 2H), 5.28 (d, J = 0.96 Hz, 1H), 4.44~4.66 (m, 4H), 4.21 (dd, J = 9.4, 1.4 Hz, 1H), 3.97 (dt, J = 9.4, 2.9 Hz, 1H), 3.69~3.77 (m, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 201.2, 138.0, 137.9, 131.7, 128.3, 128.2, 127.8, 127.7, 127.7, 127.7, 127.5, 125.3, 118.4, 93.6, 89.5, 79.4, 73.2, 71.3, 70.0, 69.9, 68.5.
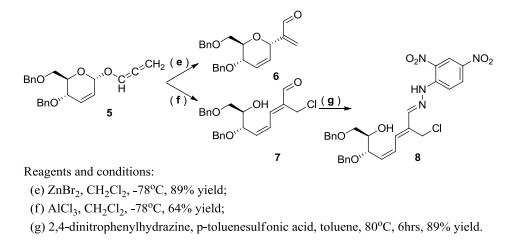
To a stirred solution of 5 (0.37 g, 1 mmol) in dry CH2Cl2 (8 mL) and ZnBr2 (22 mg, 0.1 mmol) was added at –78 ℃. The reaction mixture was stirred for 1 hour until all substrate was consumed. Saturated brine (10 mL) was added, and the mixture was extracted with CH2Cl2. The extract was washed several times with saturated NaHCO3 aqueous solution and dried over MgSO4. Evaporation of the solvent in vacuo gave an oil, which was purified on a flash silica gel column (eluents: n-hexane/ethyl acetate = 10:1, v/v) to give 6 (0.32 g, 89%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.60 (s, 1H), 7.24~7.31 (m, 10H), 6.60 (d, J = 1.1 Hz, 1H), 6.18 (s, 1H), 5.97~6.05 (m, 2H), 5.15 (s, 1H), 4.46~4.62 (m, 4H), 3.98~4.02 (m, 1H), 3.63~3.77 (m, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm): 193.1, 147.0, 138.1, 137.9, 134.9, 128.4, 128.3, 128.0, 127.9, 127.8, 127.7, 127.5, 127.0, 73.4, 72.1, 71.0, 70.1, 69.2, 68.8. HRMS calcd. for C23H25O4 [M+H]+: 365.1752, found: 365.1753.
The allenyl ether intermediate (5, 0.37 g, 1 mmol) was dissolved in dry CH2Cl2 (8 mL) at –78 ℃, and AlCl3 (13 mg, 0.1 mmol) was added. The reaction mixture was stirred for 0.5 hour under argon until completion. Saturated brine (10 mL) was added, and then organic phase was separated, washed with water, dried by MgSO4, and concentrated under vacuum to give yellow oily residue, which was isolated by column chromatography (eluents: n-hexane/ethyl acetate = 5:1, v/v) to give a sugar ring-opened C-glycoside 7 (0.26 g, 64%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.17 (s, 1H), 7.25~7.36 (m, 10H), 7.11 (d, J = 12.6 Hz, 1H), 6.90 (td, J = 11.9, 0.7 Hz, 1H), 6.16 (td, J = 10.5, 0.7 Hz, 1H), 4.62 (d, J = 11.8 Hz, 1H), 4.47~4.53 (m, 3H), 4.30~4.45 (m, 4H), 4.03 (q, J = 5.5 Hz, 1H), 3.62 (dd, J = 9.5, 5.8 Hz, 1H), 3.54 (dd, J = 9.5, 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 191.9, 144.7, 140.7, 138.1, 137.5, 137.2, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.7, 127.1, 74.2, 73.4, 71.8, 70.8, 70.2, 32.8. HRMS calcd. for C23H25ClO4Na [M+Na]+: 423.1339, found: 423.1338.
To a stirred solution of compound 7 (0.2 g, 0.5 mmol) in anhydrous toluene (6 mL) were successively added p-toluenesulfonic acid (17 mg, 0.1 mmol) and 2, 4-dinitrophenylhydrazine (99 mg, 0.5 mmol), which was heated to 80 ℃ for 6 hours under N2 protection until completion (TLC monitored). The mixture was quenched with H2O (10 mL), and the aqueous phase was extracted by ethyl acetate (8 mL) for three times. The combined organic phase was washed with saturated sodium chloride solution and dried over sodium sulfate. After the removal of solvent, the product (8) was isolated by column chromatography (eluents: cyclohexane/ethyl acetate = 3:1, v/v) to give red solids (0.26 g, 89% yield). 1H NMR (400 MHz, CDCl3) δ (ppm): 11.04 (s, 1H), 9.16 (d, J = 2.5 Hz, 1H), 8.35 (dd, J = 9.5, 2.4 Hz, 1H), 7.97 (d, J = 4.01, 1H), 7.25~7.38 (m, 10H), 7.12 (s, 1H), 6.85 (t, J = 11.6 Hz, 1H), 6.62 (d, J = 12.1 Hz, 1H), 5.98 (t, J = 10.2 Hz, 1H), 4.64 (d, J = 11.8 Hz, 1H), 4.50~4.55 (m, 4H), 4.42 (dd, J = 11.7, 4.9 Hz, 2H), 4.07~4.11 (m, 1H), 3.67 (dd, J = 9.3, 5.7 Hz, 1H), 3.51 (dd, J = 9.3, 6.3 Hz, 1H), 2.45 (d, J = 4.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 148.0, 144.4, 138.5, 137.9, 137.6, 136.0, 134.9, 134.1, 130.1, 129.5, 128.5, 128.4, 128.0, 127.9, 127.8, 127.5, 127.4, 123.3, 116.8, 77.2, 74.6, 73.2, 71.9, 70.8, 70.4, 35.2. HRMS calcd. for C29H29ClN4O7Na [M+Na]+: 603.1617, found: 603.1611.
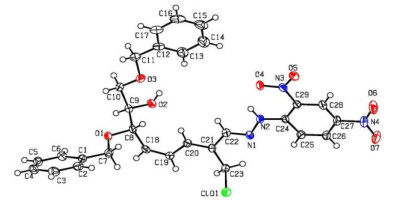
Compound 8 was crystallized after slow evaporation from a saturated chloroform solution as colorless blocks with dimensions of 0.13mm × 0.12mm × 0.11mm in the monoclinic system. Diffraction data of the single crystal were collected at 100 K on a Bruker SMART APEX-Ⅱ CCD diffractometer with graphite-monochromated CuKα radiation (λ = 1.54184 Å) using the Bruker Collect software. A total of 19039 reflections were collected in the range of 2.346≤θ≤72.98º by using an ω-scan mode, of which 5611 were unique with Rint = 0.0778 and 5169 were observed with I > 2σ(I). After the initial corrections and data reduction, intensities of reflections were used to solve (by direct methods) and refine the structures (on F2) using the WINGX program. A weighting scheme based upon P = (Fo2 + 2Fc2)/3 was employed. All the hydrogen atoms were located from difference maps and included in the refinements as riding. Empirical absorption corrections were applied. The structures were solved by direct methods using SHELXS-97 programs[19]. All of the non-hydrogen atoms were located from difference Fourier maps, and then refined anisotropically with SHELXL-97 via a full-matrix least-square procedure[20]. The final R = 0.0522, wR = 0.1358, (Δ/σ)max = 0.001, S = 1.060, (Δρ)max = 0.227 and (Δρ)min = –0.232 e/Å3. The Flack parameter was 0.00(2) (Fig. 1).
Human gastric cancer HGC-27 cell lines and human gastric mucosa epithelial GES-1 cell lines were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Cell viability was assessed by MTT cell staining as previously described[21]. The cell lines (1.0 × 104 cells/well) were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin at humidified 5% CO2 atmosphere in a 96-well plate. The cell lines were exposed to the drugs with different concentrations (100.0, 50.0 and 10.0 μg/mL) for 48 hours, and Paclitaxel (10.0 μg/mL) was as the positive control. MTT (50 μL of a 5 mg/mL in PBS; Sigma-Aldrich) was added to each well and the cell lines were incubated in a CO2 incubator at 37 ℃ for 5 hours. Following media removal, the MTT-formazan formed by metabolically viable cells was dissolved in 200 μL of DMSO (Sigma-Aldrich) and the absorbance was measured in a plate reader at 492 nm. The survival (%) was calculated by the following formula: No. of viable cells (dye excludeed cells)/No. of the total.
Optimization studies were performed on the [1, 3]-sigmatropic O→C rearrangement of D-glucal (Scheme 2) using various Lewis acid catalysts in solvents under –20 ℃ (Table 1). Iodine, silver(I) triflate or its combined catalysts failed to promote the C-glycosylation reaction (entry 1~3). Scandium(Ⅲ) triflate was observed to give the desired product in 31% yield (entry 4), and transition metal chloride included also showed poor catalytic effect (entry 5~8). To our surprise, the zinc bromide exhibited the best yield of 55% in dichloromethane for the rearrangement reaction (entry 9), and changing the solvent led to decrease in the yield (entry 10~14). Lowering the reaction temperature to –78 ℃ helped to improve the yield of C-glucoside efficiently (entry 15), and changing other haloalkane solvents including chloroform or 1, 2-dichloroethane could also promote the rearrangement reaction (entry 16, 17) in similar high yields to dichloromethane solvent (86%~89%). Considering the cost and ease of handling after the reaction, the best reaction condition was optimized to ZnBr2 as catalyst in CH2Cl2 under –78 ℃.
 DownLoad:
CSV
DownLoad:
CSV
| Entrya | Catalyst | Solvent | Yieldb |
| 1 | I2 | CH2Cl2 | N.D. c |
| 2 | AgOTf | CH2Cl2 | N.D. |
| 3 | Au(PPh3)Cl, AgOTf | CH2Cl2 | N.D. |
| 4 | Sc(OTf)3 | CH2Cl2 | 31% |
| 5 | FeCl3 | CH2Cl2 | 9% |
| 6 | ZnCl2 | CH2Cl2 | 18% |
| 7 | AlCl3 | CH2Cl2 | N.D. d |
| 8 | CuCl2 | CH2Cl2 | N.D. |
| 9 | ZnBr2 | CH2Cl2 | 55% |
| 10 | ZnBr2 | Tetrahydrofuran (THF) | 35% |
| 11 | ZnBr2 | Dimethyl sulfoxide (DMSO) | N.D. |
| 12 | ZnBr2 | Diethyl ether (Et2O) | 21% |
| 13 | ZnBr2 | Ethanol (EtOH) | N.D. |
| 14 | ZnBr2 | Toluene | 43% |
| 15 | ZnBr2 | CH2Cl2 | 89% d |
| 16 | ZnBr2 | CHCl3 | 88% d |
| 17 | ZnBr2 | CH2ClCH2Cl | 86% d |
| a All reactions were carried out with 0.1 mmol of 5 and 10 mol% catalyst in 2 mL of solvent at –20 ℃. b Isolated yield. c A sugar ring-opened product (7) was isolated. d The reaction was conducted under –78 ℃ | |||
The target C-glycosides 6~8 were characterized by NMR and HR-ESI-MS. 2D NMR spectroscopic techniques were also selectively used to determine the relative configuration of the proposed molecules. The proton and carbon signals were in accord with the characteristic functional groups of the sugar skeleton. For example, the proton's NMR signal of the aldehyde groups in compounds 6 and 7 could be clearly observed as singlet at 9.60 and 9.17 ppm, respectively. The anomeric proton of C-glycoside 6 was located at 5.15 ppm (broad singlet), which showed strong correlation peaks to the vinyl's protons of sugar ring (5.97~6.05 ppm, multiple peaks, 2H) and enal's group (6.60 ppm, doublets, J = 1.1 Hz, 1H) in H/H correlation spectroscopy (COSY). The nuclear Overhauser effect spectroscopy (NOESY) correlations could not be found between the anomeric proton and other aliphatic protons, which provided spectral evidence for relative alpha-confirmation of the proposed D-glucal structure for compound 6. In the sugar ring-opened C-glycoside 7 (73.4, 70.8, 70.2 and 32.8 ppm) and 8 (73.2, 70.8, 70.4 and 35.2 ppm), four sets of carbon signals for the methylene group were observed in 13C NMR and DEPT-135 spectra, and the most shielded carbon signal was assigned to the chloromethyl group. The adduct ion [M + H]+ or [M + Na]+ in the HR-ESI-MS spectrum could be also observed for the target compound.
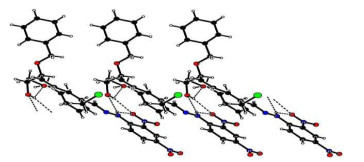
The selected bond lengths, bond angles and torsion angles are listed in Table 2, and all the bond lengths of C–C and C=C were in accordance with the standard compilations and the literature[22]. The bond angles containing unsaturated bonds in C(19)–C(18)–C(8), C(20)–C(21)–C(22) and C(22)–N(1)–N(2) range from 114° to 127°. The torsion angle of ring-opened sugar chain C(18)–C(8)–C(9)–O(2) was equal to –47.87°, and the π-conjugated aliphatic backbone C(18)–C(19)–C(20)– C(21) and C(21)–C(22)–N(1)–N(2) showed a planar conformation with the corresponding torsion angles of –178.02° and 179.82°, respectively. The three benzene rings of C(1)~C(6) (ring A), C(12)~C(17) (ring B) and C(24)~C(29) (ring C) formed dihedral angles of 61.82° (A to B), 64.97° (A to C) and 11.88° (B to C), respectively. As listed in Table 3, intermolecular and intramolecular O–H···O, C–H···O and N–H···O interactions linked the molecules into a one-dimensional infinite chain running along the a axis, which helped to stabilize the 3D supramolecular architecture (Fig. 2).
DownLoad:
CSV
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Cl(01)–C(23) | 1.807(4) | N(2)–N(1) | 1.377(4) | C(8)–C(18) | 1.499(5) | ||
| O(2)–C(9) | 1.428(5) | N(2)–C(24) | 1.351(5) | C(19)–C(18) | 1.339(6) | ||
| O(5)–N(3) | 1.232(5) | N(1)–C(22) | 1.277(5) | C(19)–C(20) | 1.442(6) | ||
| O(7)–N(4) | 1.229(5) | C(21)–C(22) | 1.450(5) | C(21)–C(20) | 1.351(5) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(2)–C(9)–C(8) | 109.9(3) | C(22)–N(1)–N(2) | 114.3(3) | C(18)–C(8)–C(9)–O(2) | –47.87 | ||
| C(19)–C(18)–C(8) | 126.7(4) | O(4)–N(3)–C(29) | 118.6(3) | C(18)–C(19)–C(20)–C(21) | –178.02 | ||
| C(20)–C(21)–C(22) | 117.9(3) | O(6)–N(4)–O(7) | 123.9(4) | C(20)–C(21)–C(22)–N(1) | –175.49 | ||
| C(20)–C(21)–C(23) | 124.0(4) | C(21)–C(23)–Cl(01) | 109.0(3) | C(21)–C(22)–N(1)–N(2) | 179.82 |
 DownLoad:
CSV
DownLoad:
CSV
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠(DHA) |
| O(2)–H(2)···O(3) | 0.82 | 2.19 | 2.6573(1) | 116 |
| O(2)–H(2)···O(4)(a) | 0.82 | 2.32 | 2.8364(1) | 121 |
| N(2)–H(2A)···O(2)(b) | 0.86 | 2.50 | 3.2817(1) | 152 |
| N(2)–H(2A)···O(4) | 0.86 | 1.99 | 2.6152(1) | 128 |
| C(10)–H(10B)···O(1) | 0.97 | 2.59 | 2.9485(1) | 102 |
| C(13)–H(13)···O(3) | 0.93 | 2.47 | 2.7895(1) | 100 |
| C(16)–H(16)···O(5)(c) | 0.93 | 2.53 | 3.3946(1) | 156 |
| C(22)–H(22)···O(2)(c) | 0.93 | 2.37 | 3.2436(1) | 156 |
| C(25)–H(25)···N(1) | 0.93 | 2.43 | 2.7525(1) | 101 |
| Symmetry codes: (a) 1 + x, y, z; (b) –1 + x, y, z; (c) –1 – x, 1/2 + y, 1/2 – z | ||||
As our continuous interest in search of natural productsderived biological small molecules[23-25], the target C-glyscosides 6~8 were evaluated for in vitro anti-proliferative activities against human gastric carcinoma cell lines (HGC-27) for 48 hours at 37 ℃ by MTT assays. All data were presented as mean ± standard deviation[26] and analyzed by SPSS[27]. Paclitaxel was chosen as a positive control with the IC50 of 4.16 μmol/L. The results indicated that the sugar ring-opened C-glycosides 7 and 8 exhibited poor anti-proliferative effect against HGC-27 cell lines (IC50 > 100 μmol/L). However, the C-glycoside 6 showed better cytotoxic effect with the IC50 value of 45.71 μmol/L (Table 4). Both compounds exhibited low toxicities against the normal human gastric mucosa epithelial GES-1 cell lines (IC50 > 500 μmol/L). Further exploration of the substrate scope for this reaction was under way in our research group.
DownLoad:
CSV
| Compound | Anti-proliferative results by MTT assays (IC50, μmol/L) | |
| Human gastric cancer cell lines (HGC-27) | Human gastric epithelial cell lines (GES-1) | |
| 6 | 45.71 | > 500 |
| 7 | > 100 | > 500 |
| 8 | > 100 | > 500 |
| Paclitaxel a | 4.16 | > 500 |
| a Positive control. | ||
In summary, three C-glycosides were prepared through the [1, 3]-sigmatropic O → C rearrangement of O-allenyl ether of D-glucal under the catalysis of ZnBr2 or AlCl3. The absolute configuration of the sugar ring-opened C-glycoside 8 was confirmed with a Flack parameter of 0.00(2) by X-ray crystallography. The in vitro cytotoxity evaluation indicated that C-pyranoside showed better anti-proliferative effect than the sugar ring-opened C-glycoside, which encouraged us to further investigate the reaction with the aim of systematically assessing the biological activity for the C-glycosides.
Olgen, S. Overview on anticancer drug design and development. Curr. Med. Chem. 2018, 25, 1704−1719. doi: 10.2174/0929867325666171129215610
Diethelm-Varela, B.; Ai, Y.; Liang, D.; Xue, F. Nitrogen mustards as anticancer chemotherapies: historic perspective, current developments and future trends. Curr. Top Med. Chem. 2019, 19, 691−712. doi: 10.2174/1568026619666190401100519
Singh, R. K.; Kumar, S.; Prasad, D. N.; Bhardwaj, T. R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401−433. doi: 10.1016/j.ejmech.2018.04.001
Hou, X. M.; Wang, C. Y.; Gerwick, W. H.; Shao, C. L. Marine natural products as potential anti-tubercular agents. Eur. J. Med. Chem. 2019, 165, 273−292. doi: 10.1016/j.ejmech.2019.01.026
Davison, E. K.; Brimble, M. A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1−8.
Le Goff, G.; Ouazzani, J. Natural hydrazine-containing compounds: biosynthesis, isolation, biological activities and synthesis. Bioorg. Med. Chem. 2014, 22, 6529−6544. doi: 10.1016/j.bmc.2014.10.011
Akhtar, M. J.; Yar, M. S.; Khan, A. A.; Ali, Z.; Haider, M. R. Recent advances in the synthesis and anticancer activity of some molecules other than nitrogen containing heterocyclic moeities. Mini. Rev. Med. Chem. 2017, 17, 1602−1632.
Parshikov, I. A.; Silva, E. O.; Furtado, N. A. Transformation of saturated nitrogen-containing heterocyclic compounds by microorganisms. Appl. Microbiol. Biotechnol. 2014, 98, 1497−1506. doi: 10.1007/s00253-013-5429-1
Yu, J.; Maliutina, K.; Tahmasebi, A. A review on the production of nitrogen-containing compounds from microalgal biomass via pyrolysis. Bioresour Technol. 2018, 270, 689−701. doi: 10.1016/j.biortech.2018.08.127
Paik, J.; Blair, H. A. Dapagliflozin: a review in type 1 diabetes. Drugs 2019, 79, 1877−1884. doi: 10.1007/s40265-019-01213-x
Bouloc, A.; Roo, E.; Moga, A.; Chadoutaud, B.; Zouboulis, C. C. A compensating skin care complex containing pro-xylane in menopausal women: results from a multicentre, evaluator-blinded, randomized study. Acta Derm. Venereol. 2017, 97, 541−542. doi: 10.2340/00015555-2572
Janssens, J.; Decruy, T.; Venken, K.; Seki, T.; Krols, S.; Van der Eycken, J.; Tsuji, M.; Elewaut, D.; Van Calenbergh, S. Efficient divergent synthesis of new immunostimulant 4΄΄-modified α-galactosylceramide analogues. ACS Med. Chem. Lett. 2017, 8, 642−647. doi: 10.1021/acsmedchemlett.7b00107
Liao, H. Z.; Ma, J. M.; Yao, H.; Liu, X. W. Recent progress of C-glycosylation methods in the total synthesis of natural products and pharmaceuticals. Org. Biomol. Chem. 2018, 16, 1791−1806. doi: 10.1039/C8OB00032H
Yang, Y.; Yu, B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 2017, 117, 12281−12356. doi: 10.1021/acs.chemrev.7b00234
Lai, M. N.; Othman, K. A.; Yao, H.; Wang, Q. Y.; Feng, Y. K.; Huang, N. Y.; Liu, M. G.; Zou, K. Open-air stereoselective construction of C-aryl glycosides. Org. Lett. 2020, 22, 1144−1148. doi: 10.1021/acs.orglett.9b04665
Kona, C. N.; Ramana, C. V. Gold(I)-catalysed [1, 3] O→C rearrangement of allenyl ethers. Chem. Commun. 2014, 50, 2152−2154. doi: 10.1039/C3CC49629E
Freitas, J. C.; Couto, T. R.; Paulino, A. A.; Freitas Filho, J. R.; Malvestiti, I.; Oliveira, R. A.; Menezes, P. H. Stereoselective synthesis of pseudoglycosides catalyzed by TeCl4 under mild conditions. Tetrahedron 2012, 68, 10611−10620. doi: 10.1016/j.tet.2012.09.073
Moons, S. J.; Mensink, R. A.; Bruekers, J. P. J.; Vercammen, M. L. A.; Jansen, L. M.; Boltje, T. J. α-Selective glycosylation with β-glycosyl sulfonium ions prepared via intramolecular alkylation. J. Org. Chem. 2019, 84, 4486−4500. doi: 10.1021/acs.joc.9b00022
Sheldrick, G. M. SHELXS 97, Program for Crystal Structure Determinations. University of Göttingen, Germany 1997.
Sheldrick, G. M. SHELXL 97, Program for the Refinement of Crystal Structure. University of Göttingen, Germany 1997.
Stockert, J. C.; Blázquez-Castro, A.; Cañete, M.; Horobin, R. W.; Villanueva, Á. MTT assay for cell viability: intracellular localization of the formazan product is in lipid droplets. Acta Histochem. 2012, 114, 785–796. doi: 10.1016/j.acthis.2012.01.006
Paolini, J. P. The bond order-bond length relationship. J. Comput. Chem. 1990, 11, 1160–1163. doi: 10.1002/jcc.540111007
Yao, Y.; Xiong, C. P.; Zhong, Y. L.; Bian, G. W.; Huang, N. Y.; Wang, L.; Zou, K. Intramolecular and Ferrier rearrangement strategy for the construction of C1-β-D-xylopyranosides: synthesis, mechanism and biological activity study. Adv. Syn. Cat. 2019, 361, 1012–1017. doi: 10.1002/adsc.201801423
Jian, S. X.; Tian, Y. Y.; Wang, J. Z.; Hu, W. M.; Huang, N. Y. Research progress on the natural anti-peptic ulcer chemical structures. Chin. J. Struct. Chem. 2018, 37, 1703–1710.
Huang, N. Y.; Wang, W. B.; Chen, L.; Luo, H. J.; Wang, J. Z.; Deng, W. Q.; Zou, K. Design, synthesis and biological evaluation of bisabolonalone oxime derivatives as potassium-competitive acid blockers (P-CABs). Bioorg. Med. Chem. Lett. 2016, 26, 2268–2272. doi: 10.1016/j.bmcl.2016.03.051
Cumming, G.; Fidler, F.; Vaux, D. L. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. doi: 10.1083/jcb.200611141
West, B. T. Analyzing longitudinal data with the linear mixed models procedure in SPSS. Eval. Health Prof. 2009, 32, 207–228. doi: 10.1177/0163278709338554

Figure 1 ORTEP drawing of compound 8 showing thermal ellipsoids at the 50% probability level

Figure 2 Packing diagram of compound 8. The O–H···O interactions are shown as dashed lines
Table 1. Screening Suitable Reaction Conditions
| Entrya | Catalyst | Solvent | Yieldb |
| 1 | I2 | CH2Cl2 | N.D. c |
| 2 | AgOTf | CH2Cl2 | N.D. |
| 3 | Au(PPh3)Cl, AgOTf | CH2Cl2 | N.D. |
| 4 | Sc(OTf)3 | CH2Cl2 | 31% |
| 5 | FeCl3 | CH2Cl2 | 9% |
| 6 | ZnCl2 | CH2Cl2 | 18% |
| 7 | AlCl3 | CH2Cl2 | N.D. d |
| 8 | CuCl2 | CH2Cl2 | N.D. |
| 9 | ZnBr2 | CH2Cl2 | 55% |
| 10 | ZnBr2 | Tetrahydrofuran (THF) | 35% |
| 11 | ZnBr2 | Dimethyl sulfoxide (DMSO) | N.D. |
| 12 | ZnBr2 | Diethyl ether (Et2O) | 21% |
| 13 | ZnBr2 | Ethanol (EtOH) | N.D. |
| 14 | ZnBr2 | Toluene | 43% |
| 15 | ZnBr2 | CH2Cl2 | 89% d |
| 16 | ZnBr2 | CHCl3 | 88% d |
| 17 | ZnBr2 | CH2ClCH2Cl | 86% d |
| a All reactions were carried out with 0.1 mmol of 5 and 10 mol% catalyst in 2 mL of solvent at –20 ℃. b Isolated yield. c A sugar ring-opened product (7) was isolated. d The reaction was conducted under –78 ℃ | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected Bond Lengths (Å) and Bond Angles (°) for Compound 3
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| Cl(01)–C(23) | 1.807(4) | N(2)–N(1) | 1.377(4) | C(8)–C(18) | 1.499(5) | ||
| O(2)–C(9) | 1.428(5) | N(2)–C(24) | 1.351(5) | C(19)–C(18) | 1.339(6) | ||
| O(5)–N(3) | 1.232(5) | N(1)–C(22) | 1.277(5) | C(19)–C(20) | 1.442(6) | ||
| O(7)–N(4) | 1.229(5) | C(21)–C(22) | 1.450(5) | C(21)–C(20) | 1.351(5) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| O(2)–C(9)–C(8) | 109.9(3) | C(22)–N(1)–N(2) | 114.3(3) | C(18)–C(8)–C(9)–O(2) | –47.87 | ||
| C(19)–C(18)–C(8) | 126.7(4) | O(4)–N(3)–C(29) | 118.6(3) | C(18)–C(19)–C(20)–C(21) | –178.02 | ||
| C(20)–C(21)–C(22) | 117.9(3) | O(6)–N(4)–O(7) | 123.9(4) | C(20)–C(21)–C(22)–N(1) | –175.49 | ||
| C(20)–C(21)–C(23) | 124.0(4) | C(21)–C(23)–Cl(01) | 109.0(3) | C(21)–C(22)–N(1)–N(2) | 179.82 |
下载: 导出CSV
Table 3. Hydrogen Bonds for Compound 3
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠(DHA) |
| O(2)–H(2)···O(3) | 0.82 | 2.19 | 2.6573(1) | 116 |
| O(2)–H(2)···O(4)(a) | 0.82 | 2.32 | 2.8364(1) | 121 |
| N(2)–H(2A)···O(2)(b) | 0.86 | 2.50 | 3.2817(1) | 152 |
| N(2)–H(2A)···O(4) | 0.86 | 1.99 | 2.6152(1) | 128 |
| C(10)–H(10B)···O(1) | 0.97 | 2.59 | 2.9485(1) | 102 |
| C(13)–H(13)···O(3) | 0.93 | 2.47 | 2.7895(1) | 100 |
| C(16)–H(16)···O(5)(c) | 0.93 | 2.53 | 3.3946(1) | 156 |
| C(22)–H(22)···O(2)(c) | 0.93 | 2.37 | 3.2436(1) | 156 |
| C(25)–H(25)···N(1) | 0.93 | 2.43 | 2.7525(1) | 101 |
| Symmetry codes: (a) 1 + x, y, z; (b) –1 + x, y, z; (c) –1 – x, 1/2 + y, 1/2 – z | ||||
下载: 导出CSV
Table 4. Bioactivity Evaluation of the Compounds
| Compound | Anti-proliferative results by MTT assays (IC50, μmol/L) | |
| Human gastric cancer cell lines (HGC-27) | Human gastric epithelial cell lines (GES-1) | |
| 6 | 45.71 | > 500 |
| 7 | > 100 | > 500 |
| 8 | > 100 | > 500 |
| Paclitaxel a | 4.16 | > 500 |
| a Positive control. | ||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: