Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Institute of Physical Chemistry, Zhejiang Normal University, Jinhua, Zhejiang 321004, China
Corresponding author:
XIE Guanqun, senior engineer; Tel:0579-82291381; E-mail:gqxie@zjnu.cn; Research interests:catalytical chemistry; LUO Mengfei, professor; Tel:0579-82291381; E-mail:mengfeiluo@zjnu.cn; Research interests:catalysis chemistry
Received Date:
17 July 2018 Accepted Date:
27 November 2018 Revised Date:
12 October 2018 Available Online:
01 May 2019
Fund Project:
Supported by the Natural Science Foundation of Zhejiang Province, China(No.LY16B070001)
Abstract:
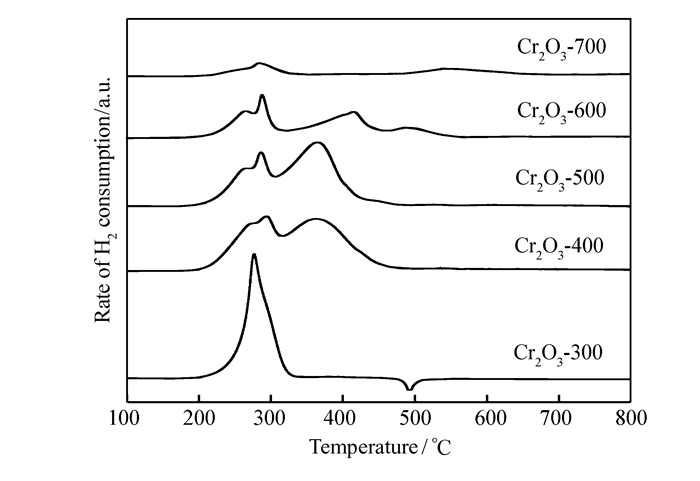
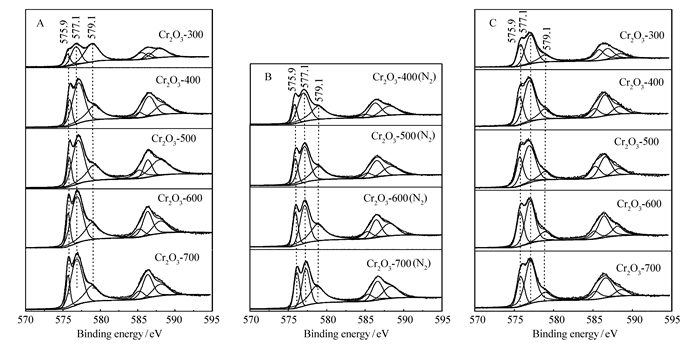
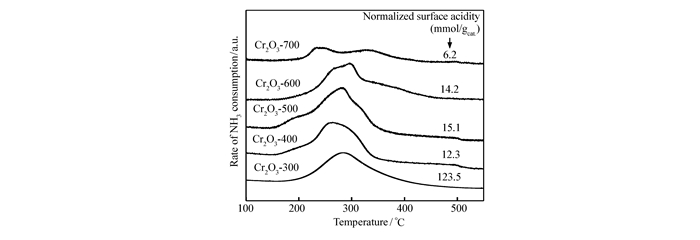
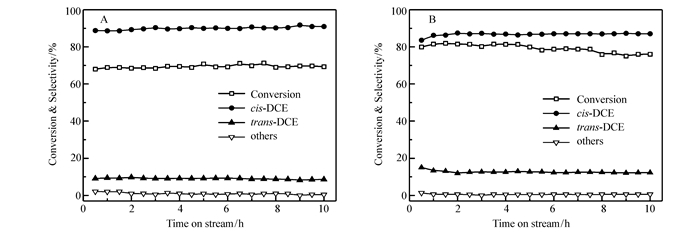
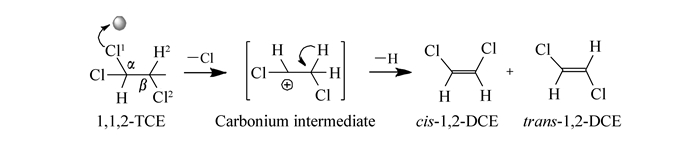
A series of Cr2O3 catalysts was prepared by a precipitation method and tested for the gas phase dehydrochlorination of 1, 1, 2-trichloroethane(TCE) to synthesize cis-1, 2-dichloroethylene(cis-DCE). X-ray diffraction(XRD), hydrogen temperature-programmed reduction(H2-TPR), ammonia temperature-programmed desorption(NH3-TPD) and X-ray photoelectron spectroscopy(XPS) were used to study the dehydrochlorination of TCE on Cr2O3 catalyst and its reaction mechanism. It is found that the conversion ratio of TCE on the catalysts decreases with the increase of the calcination temperature, while the selectivity to cis-DCE first increases and then decreases. The best performance is obtained on the catalyst calcined at 400℃, with a TCE conversion ratio of 70.8% and a cis-DCE selectivity of 90.0%. In addition, the areal specific reaction rate also first increases and then decreases with the increase of the calcination temperature, with the highest value being obtained on the catalysts calcined at 400℃(0.801×10-2 μmol/(s·m2). The catalytic behaviors of the catalysts are well related to their surface Cr2O3 species. Turnover frequencies(TOFs) calculated based on the surface acidity and the highest value being obtained on the catalysts calcined at 400℃(2.82×10-5 s-1) show that the oxidation states of the surface Cr species are important for the reaction and the Cr species with an average valence of 3.2 are appropriate for the reaction.
靳利蕊, 张晓君, 李辉. 三氯乙烷污染地地下水中Dehalobacter属细菌定量与多样性分析[J]. 微生物学通报,
2013,40,(8): 1522-1530.
JIN Lirui, ZHANG Xiaojun, LI Hui. Diversity of 1, 1, 1-Trichloroethane Degrading Dehalobacter spp. in Contaminated Groundwater[J]. Microbiol China,
2013, 40(8):
1522-1530.
[2]
Mukhopadhyay S, Tung H S, Nair H. Method for Producing Fluorinated Organic Compounds.WO2007056149Al[P]. 2007-05-18
[3]
Wang H, Tung H S. Dehydrochlorination of Hydrochlorofluorocarbons Using Pre-treated Activated Carbon Catalysts: WO, 2009021154A2[P]. 2009-02-12
[4]
Li X G, Li T, Zhang T. Nano-TiO2-Catalyzed Dehydrochlorination of 1, 1, 2, 2-Tetrachloroethane:Roles of Crystalline Phase and Exposed Facets[J]. Environ Sci Technol,
2018, 52(7):
4031-4039.
doi: 10.1021/acs.est.7b05479
[5]
Dofling J, Janssen D B. Estimates of Gibbs Free Energies of Formation of Chlorinated Aliphatic Compounds[J]. Biodegradation,
1994, 5(1):
21-28.
[6]
Kokubo K, Kitasaka K, Oshima T. Effects of the Cation Binding Mode on E-Z Isomerization of 1, 2-Dichloroethylene[J]. Org Lett,
2006, 8(8):
1597-1600.
[7]
Turton D A, Martin D F, Wynne K. Optical Kerr-Effect Study of Trans- and Cis-1, 2-Dichloroethene:Liquid Liquid Transition or Super-Arrhenius Relaxation[J]. Phys Chem Chem Phys,
2010, 12(16):
4191-4200.
doi: 10.1039/b918196b
[8]
Van der Heijden A W A M, Mens A J M, Bogerd R. Dehydrochlorination of Intermediates in the Production of Vinyl Chloride over Lanthanum Oxide-based Catalysts[J]. Catal Lett,
2008, 122(3/4):
238-246.
[9]
Tang C, Jin Y, Lu J. Highly Efficient Mg(OH)Cl/SiO2 Catalysts for Selective Dehydrochlorination of 1, 1, 2-Trichloroethane[J]. Appl Catal A,
2015, 508:
10-15.
[10]
靳燕霞, 汤岑, 孟秀清. 气相法合成偏二氯乙烯的高稳定性CsNO3/SiO2催化剂[J]. 物理化学学报,
2016,32,(2): 510-518.
JIN Yanxia, TANG Cen, MENG Xiuqing. Highly Stable CsNO3/SiO2 Catalysts for the Synthesis of Vinylidene Chloride Using a Gaseous Phase Method[J]. Acta Phys-Chim Sin,
2016, 32(2):
510-518.
[11]
胡益浩, 宋通洋, 王月娟. Zn/SiO2气相催化裂解1, 1, 2-三氯乙烷脱HCl:酸性与失活[J]. 物理化学学报,
2017,33,(5): 1017-1026.
HU Yihao, SONG Tongyang, WANG Yuejuan. Gas Phase Dehydrochlorination of 1, 1, 2-Trichloroethane over Zn/SiO2 Catalysts:Acidity and Deactivation[J]. Acta Phys-Chim Sin,
2017, 33(5):
1017-1026.
[12]
Song T Y, Dong Z X, Song J D. Dehydrochlorination of 1, 1, 2-Trichloroethane over SiO2-Supported Alkali and Transition Metal Catalysts:Tunable Selectivity Controlled by the Acid-base Properties of the Catalysts[J]. Appl Catal B,
2018, 236:
368-376.
doi: 10.1016/j.apcatb.2018.04.018
[13]
汪云, 梁艳, 何军. Cr2O3和CrO3/Cr2O3催化剂的2-氯-1, 1, 1-三氟乙烷气相氟化反应的活性物种和失活[J]. 无机化学学报,
2017,33,(1): 123-133.
WANG Yun, LIANG Yan, HE Jun. Catalytic Behaviors of Cr2O3 and CrO3/Cr2O3 Catalysts for Gas Phase Fluorination of 2-Chloro-1, 1, 1-trifluoroethane:Active Spedes and Catalyst Deactivation[J]. Chinese J Inorg Chem,
2017, 33(1):
123-133.
[14]
Han W, Li X, Tang H. Preparation of fluorinated Cr2O3 Hexagonal Prism and Catalytic Performance for the Dehydrofluorination of 1, 1-Difluoroethane to Vinyl Fluoride[J]. J Nanopart Res,
2015, 17(9):
1-12.
[15]
Lim S, Kim M S, Choi J W. Catalytic Dehydrofluorination of 1, 1, 1, 2, 3-Pentafluoropropane(HFC-245eb) to 2, 3, 3, 3-Tetrafluoropropene(HFO-1234yf) Using in-situ Fluorinated Chromium Oxyfluoride Catalyst[J]. Catal Today,
2017, 293:
42-48.
[16]
Luo J W, Song J D, Jia W Z. Catalytic Dehydrofluorination of 1, 1, 1, 3, 3-Pentafluoropropane to 1, 3, 3, 3-Tetrafluoropropene over Fluorinated NiO/Cr2O3 Catalysts[J]. Appl Surf Sci,
2018, 433:
904-913.
[17]
Wang F, Fan J L, Zhao Y. Effects of Yttrium-doping on the Performance of Cr2O3, Catalysts for Vapor Phase Fluorination of 1, 1, 2, 3-Tetrachloropropene[J]. J Fluorine Chem,
2014, 166:
78-83.
doi: 10.1016/j.jfluchem.2014.07.030
[18]
Cheng Y X, Fan J L, Xie Z Y. Effects of M-promoter(M = Y, Co, La, Zn) on Cr2O3 Catalysts for Fluorination of Perchloroethylene[J]. J Fluorine Chem,
2013, 156:
66-72.
doi: 10.1016/j.jfluchem.2013.08.016
[19]
Zhang W X, Liang Y, Luo J W. Morphological Effects of Ordered Cr2O3, Nanorods and Cr2O3, Nanoparticles on Fluorination of 2-Chloro-1, 1, 1-trifluoroethane[J]. J Mater Sci,
2016, 51(13):
6488-6496.
[20]
Grzybowska B, Sloczyn'ski J, Grabowski R. Chromium Oxide/Alumina Catalysts in Oxidative Dehydrogenation of Isobutane[J]. J Catal,
1998, 178(2):
687-700.
[21]
Niedersen K U, Schreier E, Kemnitz E. Isomerization Reaction of 1, 1, 2, 2-Tetrafluoroethane on Chromia—A Study of the Active Sites on the Surface[J]. J Catal,
1997, 167(1):
210-214.
[22]
Wang X, Xie Y C, Wang X. Total Oxidation of CH4 on Sn-Cr Composite Oxide Catalysts[J]. Appl Catal B,
2001, 35(2):
85-94.
[23]
范镜莲.气相氟化四氯丙烯合成四氟丙烯的催化研究[D].浙江师范大学, 2014.FAN Jinglian. Vapor-Phase Fluorination of 1, 1, 2, 3-Tetrachloropropene to Synthesis Tetrafluoropropene[D]. Zhejiang Normal University, 2014(in Chinese).
[24]
Bobet J L, Desmoulins-Krawiec S, Grigorova E. Addition of Nanosized Cr2O3 to Magnesium for Improvement of the Hydrogen Sorption Properties[J]. J Alloys Compd,
2003, 351(1):
217-221.
[25]
Lin Y, Cai W, Tian X. Polyacrylonitrile/Ferrous Chloride Composite Porous Nanofibers and Their Strong Cr-Removal Performance[J]. J Mater Chem,
2011, 21(4):
991-997.
doi: 10.1039/C0JM02334E
[26]
Liu B, Terano M, Liu B. Investigation of the Physico-Chemical State and Aggregation Mechanism of Surface Cr Species on a Phillips CrOx/SiO2 Catalyst by XPS and EPMA[J]. J Mol Catal A,
2001, 172(1/2):
227-240.
[27]
Gaspar A B, Perea C A C, Dieguez L C. Characterization of Cr/SiO2 Catalysts and Ethylene Polymerization by XPS[J]. Appl Surf Sci,
2005, 252(4):
939-949.
doi: 10.1016/j.apsusc.2005.01.031
[28]
Gao S, Dong C, Luo H. Scanning Electrochemical Microscopy Study on the Electrochemical Behavior of CrN Film Formed on 304 Stainless Steel by Magnetron Sputtering[J]. Electrochim Acta,
2013, 114:
233-241.
doi: 10.1016/j.electacta.2013.10.009
[29]
Fu X Z, Luo X X, Luo J L. Ethane Dehydrogenation over Nano-Cr2O3 Anode Catalyst in Proton Ceramic Fuel Cell Reactors to Co-produce Ethylene and Electricity[J]. J Power Sources,
2011, 196(3):
1036-1041.
doi: 10.1016/j.jpowsour.2010.08.043
[30]
Berteau P, Delmon B, Berteau P. Modified Aluminas: Relationship Between Activity in 1-Butanol Dehydration and Acidity Measured by NH3 TPD[J]. Catal Today,
1989, 5(2):
121-137.
doi: 10.1016/0920-5861(89)80020-3
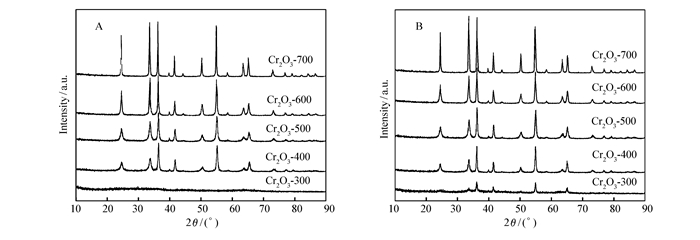
图 1
不同焙烧温度下制备的Cr2O3催化剂催化反应前(A)后(B)XRD图谱
Figure 1
XRD patterns of Cr2O3 catalysts calcined at different temperatures before(A) and after(B) catalytic reaction

 下载:
下载:

 下载:
下载:







 下载:
下载:
