引用本文:
姚会影, 杨涛, 黄幸, 朱嘉, 李青, 徐伟, 迟力峰. 基于MX4结构的配位化合物析氢反应催化性能[J]. 应用化学,
2018, 35(3): 328-341.
doi:
10.11944/j.issn.1000-0518.2018.03.170453

Citation: YAO Huiying, YANG Tao, HUANG Xing, ZHU Jia, LI Qing, XU Wei, CHI Lifeng. Coordination Complexes Based on MX4 Structure as Catalyst for Hydrogen Evolution Reaction[J]. Chinese Journal of Applied Chemistry, 2018, 35(3): 328-341. doi: 10.11944/j.issn.1000-0518.2018.03.170453
Citation: YAO Huiying, YANG Tao, HUANG Xing, ZHU Jia, LI Qing, XU Wei, CHI Lifeng. Coordination Complexes Based on MX4 Structure as Catalyst for Hydrogen Evolution Reaction[J]. Chinese Journal of Applied Chemistry, 2018, 35(3): 328-341. doi: 10.11944/j.issn.1000-0518.2018.03.170453
基于MX4结构的配位化合物析氢反应催化性能
English
Coordination Complexes Based on MX4 Structure as Catalyst for Hydrogen Evolution Reaction
Abstract:
The coordination complexes based on MX4(M=Fe, Co, Ni, Cu, et al, X=N, S, Se, et al.) structure have attracted a lot of attentions due to their novel structure and good electrocatalytic performance in hydrogen evolution reaction(HER). This review shows the recent research progresses of their catalytic properties for HER. The HER activities of MX4 catalysts are affected by many factors, such as the metal center, coordination atoms, the ligand, the morphology and size of catalysts. Theoretical calculations are very helpful for understanding their influences on the activities of the catalysts and can further help us design more catalysts with higher activities.
-
能源问题是21世纪最重要的问题,人类社会对能源的需求一直呈现迅速而持续的增长。化石能源储量有限,生产和使用过程中会造成环境污染,因此发展可再生能源、清洁能源备受人们的关注,其中,氢能源具有高热值和使用过程中几乎零污染排放的特征,是最有前途的清洁能源之一[1]。利用氢能源的两种最关键的技术是氢储存技术和制氢技术,其中,制氢技术主要包括天然气制氢、甲醇制氢和水解制氢等[2-7]。相比于其它方式,水解制氢具有工艺简单、制得氢气纯度高等优点[4-6, 8],但是需要正视的是目前使用的水解制氢技术能耗巨大,因为水是一种非常稳定的化合物,从水转化成氢气和氧气的过程是一个能量增大的非自发反应,如何实现高效的水分解仍然是巨大的挑战。高效析氢催化剂的存在,可以大大提高水分解效率,降低产氢过程的能耗[8-10]。本文主要关注电解水制氢中的析氢反应(Hydrogen Evolution Reaction,HER,2H++2e-=H2)所使用的催化剂。金属铂因其稳定性,接近于零的析氢过电势和极高的交换电流密度,成为了最受欢迎的HER催化剂之一[11]。然而,铂价格昂贵、储量少,限制了它的大规模应用。因此,研究人员致力于探索催化性能优异且稳定性好、价格低廉的非贵金属HER催化剂。例如,很多金属硫化物、金属磷化物等受到了持续的关注,具有很好的发展前景[8, 12-13]。
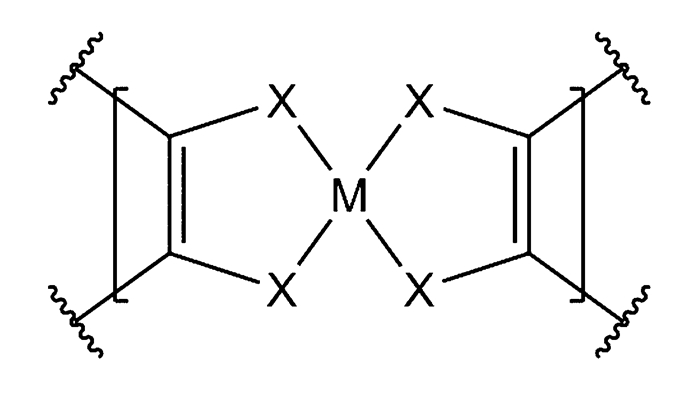
在非贵金属催化剂中,配位化合物因其多样性,为催化性能的探索提供了一个广阔的平台[15-16, 18, 20, 23, 25-26, 31-32]。近年来,具有如图 1所示的MX4结构的(M=Fe, Co, Ni, Cu等,X=N, S, Se等)小分子配位化合物及配位聚合物,它们具有新颖的结构和优异的电催化析氢性能,受到研究人员越来越多的关注。首先受到关注的是具有MX4结构的小分子催化剂,但是小分子HER催化剂大多不能在水溶液中使用,存在过电势高、交换电流密度小等缺陷,且稳定性较差,不利于催化剂活性的保持[31]。解决这一难题的方法之一是将这种MX4单元整合到配位聚合物中,期望得到既具有MX4单元的催化活性,又能利用配位聚合物稳定性的非均相催化剂[17, 19, 22, 33],这种设计思路被证明是成功的。近年来,研究人员制备了一系列含MX4结构的配位聚合物,在催化析氢反应中,具有令人满意的催化活性,而且性质稳定,在长期高强度的使用过程中保持催化活性[16, 18, 27, 31]。理论研究者注意到,基于MX4单元的配位聚合物HER催化剂的成功运用。计算化学相关理论的发展有助于研究者们了解催化剂的反应活性来源和反应机理等,同时为了解这一类催化剂的构效关系提供了有效途径,最终能够帮助研究者们改进、模拟、设计更高催化活性的催化剂。本文将介绍具有MX4单元的小分子配合物和配位聚合物的催化析氢机理以及其发展历程,并讨论如何通过计算化学方法来理解这一类催化剂。
图 1
1. 析氢反应中不同种类电催化剂的催化反应机理
1.1 均相电催化剂的催化反应机理
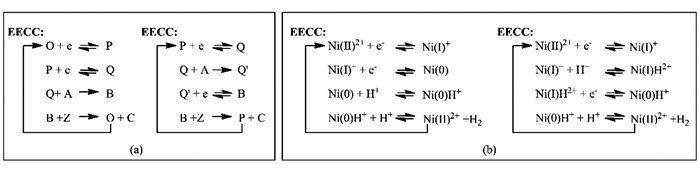
均相催化剂是和反应物同处于一相的催化剂[34]。均相HER催化剂的催化反应过程通常可以划分为两类基本步骤:电子转移(E)和质子化(C)[35],其中E代表电极表面和催化剂之间的电子传递,C代表质子化反应。例如,某一催化反应的第一步是发生电子传递,所得产物继续发生质子化反应,这样的反应历程用EC表示。均相催化剂参与的析氢反应,根据电子转移和质子转移的顺序不同,可能的反应历程有EECC、ECEC、ECCE等[36]。其中,常见的EECC和ECEC反应历程如图 2a所示,例如,Ni(PPh2NPh2)22+作为HER催化剂时的反应历程(如图 2b)[37]。
图 2

HER中涉及到两个电子和质子转移过程,需要通过循环伏安(Cyclic voltammetry,CV)曲线中电流与电势响应确认催化剂的状态,然后,结合理论计算电势大小和吉布斯自由能变化判断倾向于发生质子化还是还原,从而进一步确定反应机理,在后面的计算部分将结合具体反应进行说明[15, 21]。
1.2 非均相电催化剂的催化反应机理
和反应物属于不同相的催化剂是非均相催化剂[34]。非均相催化剂催化HER反应时,普遍认为其在酸性和碱性环境中反应机理如下[38]:
(1) 电化学氢原子吸附(Volmer reaction):
$\begin{array}{c} {\rm H^+ +M+e}^-\rightleftharpoons {\rm M- H}^* \;\;(酸性溶液)\\ {\rm H_2O+ +M+e}^-\rightleftharpoons {\rm M- H}^*+{\rm OH}^- \;\;(碱性溶液) \end{array} $
(2) 电化学解吸附(Heyrovsky reaction):
$ \begin{array}{c} {\rm M- H^* +H^+ + e}^-\rightleftharpoons {\rm M+ H}_2 \;\;(酸性溶液)\\ {\rm M-H^* +H_2O+ e}^-\rightleftharpoons {\rm M+ OH}^-+{\rm H}_2 \;\;(碱性溶液) \end{array} $
(3) 化学解吸附(Tafel reaction):
$ 2{\rm M-H^*=2M+H}_2(酸性和碱性溶液) $
式中,H*代表氢原子与电极表面(M)上的活性位点形成化学吸附。这些反应路径强烈依赖于电极表面固有的(电)化学和电子性质。例如,Conway等[11]通过评估HER极化曲线的塔菲尔(Tafel)斜率值,可以辨别反应中的决速步。
正如反应式(1)、(2)和(3)所示,氢原子在电极表面形成化学吸附和解吸附是两个相互竞争的过程。根据萨巴蒂埃(Sabatier)原理可知,高效的HER催化剂应该与电极表面形成足够强的吸附以促进电子质子转移过程,但是这种吸附也要足够弱以确保两个相近的氢原子结合形成氢分子后能够脱离电极表面。然而,由于不能直接测量吸附质与电极表面的结合能,使得研究人员很难在H*中间体和HER电催化反应速率之间建立定量关系[38]。从物理化学观点出发,H原子在催化剂表面的吸附吉布斯自由能变化(ΔGH*)可以通过HER自由能变图(free energy diagram)来衡量H*吸附和H2解吸附能力[39]。由Sabatier原理可知,ΔGH*等于零时,整个反应的反应速率达到最大值(通过HER中的交换电流密度表示)。接下来主要介绍ΔGH*的计算,探索其在设计HER电催化剂方面的指导意义。通过计算可能的中间体的ΔGH*值,可以衡量它们在电极表面的稳定性,计算方法如下[39]:
$ \Delta {G_{{\rm H^*}}} = \Delta {E_{{\rm H^*}}} + \Delta {E_{\rm ZPE}} - T\Delta S $
吸附能ΔEH*通过下式求得:
$ \Delta {E_{{\rm H^*}}} = {E_{\rm M - {H^*}}} - {E_{\rm M}} - \frac{1}{2}{E_{{\rm H_2}}} $
式中,EH2代表气态氢气分子的能量,EM-H*和EM分别为电极表面质子化之后和之前的能量。ΔEZPE为吸附态的氢原子与孤立的氢气分子之间的零点能差,可以通过振动频率求得。ΔS为熵变,这里的熵包括转动熵,振动熵,平移熵以及电子熵。
2. MX4配合物催化剂的发展历程
MX4配合物由于种类丰富、易合成,在室温和空气中稳定、电子结构多变、化学性质多样化等优点而受到关注[15-16, 27-28, 30-31]。当用作HER催化剂时,MX4配合物可与质子形成稳定的反应中间体,避免了电子或空穴直接向水分子转移生成高能量的反应中间体,降低析氢反应的能垒,获得更高的催化活性[40]。
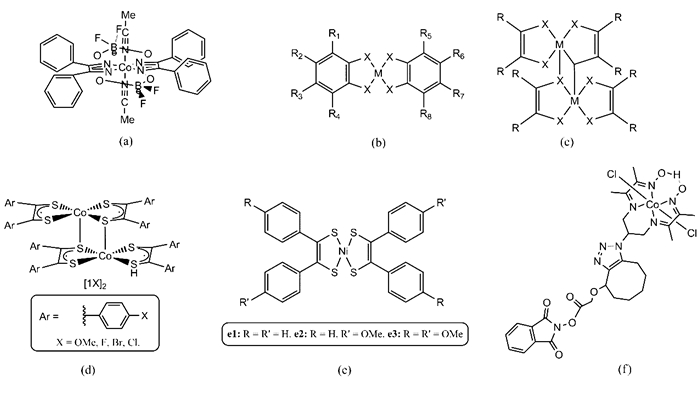
2005年,Hu等[14]研究了具有CoN4结构的钴肟配合物(金属与二乙二肟配体形成的配合物)作为HER均相催化剂的催化活性,其中[Co(dpgBF2)2](如图 3a)体系在乙腈溶液中的起始析氢电势为-280 mV(vs.SCE)。2009年,Dempsey等[15]采用热力学分析方法对类似[Co(dpgBF2)2]的一些钴肟催化剂的析氢反应过程进行了理论研究,认为钴肟氢化物的形成是反应的决速步。
图 3
 图 3 (a)[Co(dpgBF2)2][14];(b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18],[Co(tdt)2]-(M=Co, X=S, R3-6=CH3,其余R=H)[18],[Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, 其余R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20];(c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2、[5F]2、[5Cl]2、[5Br]2[21]; (e)镍芳基二硫烯配合物[27]; (f)钴二胺-二肟催化剂[41]Figure 3. (a)[Co(dpgBF2)2][14]; (b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18], [Co(tdt)2]-(M=Co, X=S, R3-6=CH3, other R=H)[18], [Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, other R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20]; (c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2, [5F]2, [5Cl]2, [5Br]2[21]; (e)nickel dithiolene complexes with p-methoxyphenyl-substituted 1, 2-dithiolene ligands[27]; (f)diimine-dioxime cobalt catalyst[41]
图 3 (a)[Co(dpgBF2)2][14];(b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18],[Co(tdt)2]-(M=Co, X=S, R3-6=CH3,其余R=H)[18],[Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, 其余R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20];(c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2、[5F]2、[5Cl]2、[5Br]2[21]; (e)镍芳基二硫烯配合物[27]; (f)钴二胺-二肟催化剂[41]Figure 3. (a)[Co(dpgBF2)2][14]; (b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18], [Co(tdt)2]-(M=Co, X=S, R3-6=CH3, other R=H)[18], [Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, other R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20]; (c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2, [5F]2, [5Cl]2, [5Br]2[21]; (e)nickel dithiolene complexes with p-methoxyphenyl-substituted 1, 2-dithiolene ligands[27]; (f)diimine-dioxime cobalt catalyst[41]与二乙二肟等配体相比,二硫烯配体更稳定,金属1, 2-二硫烯化合物具有稳定的结构、可逆的电子转移特性和丰富的氧化还原特性[16],使其有望成为高效的HER催化剂[42]。2011年,McNamara等[16]基于二硫乙烯类配体1, 2-二巯基苯合成了钴双-二硫烯配合物[Co(bdt)2]-(bdt=1, 2-二巯基苯,图 3b),探索了其在电催化析氢反应中的活性。实验表明,[Co(bdt)2]-在V(CH3CN):V(H2O)=1:1的弱酸性溶液中具有HER活性,起始电势是-1.01 V(vs.Fc+/Fc)。2012年,McNamara等[18]合成了具有类似的CoS4(图 3b~3c, M=Co, X=S)单元的[Co(bdt)2]-(1,图 3b), [Co(tdt)2]-(2,图 3b), [Co(Cl2bdt)2]-(3,图 3b)和[Co(mnt)2]-(4,图 3c)配合物,探究了配体对这一类催化剂活性的影响。结果表明,4种催化剂的CoⅢ到CoⅡ的还原电势顺序是4>3>1>2,而活性顺序是3>1>2>4。除了化合物4以外,其余3种化合物还原电势的大小与活性顺序一致:当含有苯环的配体上连有吸电子基时,还原电势越正,催化剂活性更好。为什么连有氰基的4号化合物出现了反常?Solis等[43]通过理论计算对这一现象进行了深入的研究,结果表明,化合物4在反应过程中的质子化位点少于化合物1~3,这是造成其活性低的主要原因。
如上文中介绍的[Co(bdt)2]-及其衍生物和[Co(mnt)2]-的对比可知,有机配体的电子结构和性质的改变对催化剂整体性质具有调控作用,这一规律也被来自其它研究组的类似实验结果所证明[21]。2014年,Letko等[21]合成了具有不同对位取代基的芳基的结构[Co(S2C2Ar2)2]2(5),研究了它们作为HER催化剂的活性。其中,被甲氧基、氟、氯、溴取代的结构表示为[5OMe]2、[5F]2、[5Cl]2、[5Br]2(图 3d),它们的催化析氢反应半峰电势(Ep/2)分别为-1.46、-1.37、-0.35和-0.35 V(vs.Fc+/0),即氯和溴取代[Co(S2C2Ar2)2]2的活性最高。2016年,Zarkadoulas等[27]利用镍与对甲氧基苯基取代的1, 2-二硫烯配体合成了3种镍芳基二硫烯配合物,它们的结构中甲氧基的数目不同(图 3e)。在0/-1和-1/-2还原过程中,配合物e3的还原电势最负(最难还原),e1的还原电势最正(最容易还原),而实验结合理论计算结果表明,活性大小顺序为:e2>e3>e1。此研究说明,配体的电子结构对催化反应活性的重要性。
配体对于电催化活性的影响不仅仅体现在配体的取代基上,参与配位的配位原子同样会影响催化性能。Downes等[30]报道了具有平面CoSe4结构的钴与1, 2-二硒醇苯(1, 2-benzenediselenolate,bds)配体形成的配合物Co(bds)2(图 3b, M=Co, X=Se, R=H)作为HER催化剂时,在V(CH3CN):V(H2O)=1:1的酸性溶液中,电流密度达到10 mA/cm2时所需过电势为-1.14 V(vs.Fc+/0)。为了确定催化剂的质子化位点,Downes等[30]通过计算轨道自旋密度分布发现:[Co(bds)2]-的SOMO(singly ocucpied molecular orbital)和SOMO-1轨道只有向上的自旋(α自旋),表明钴和硒原子均发生了自旋局域化。当[Co(bds)2]-还原成[Co(bds)2]2-时,[Co(bds)2]2-的SOMO-1轨道净自旋为零,且硒原子具有很高的电子密度,因此质子化位点为硒原子。[Co(bds)2]-与硫基催化剂[Co(bdt)2]-[16]结构类似,差别在于活性位点分别是配体中的硒原子和硫原子,而Co(bds)2的活性高于[Co(bdt)2]-,说明将巯基配体换成硒醇基配体可以有效地提高催化剂的活性。
在探索和优化MX4催化剂催化活性的过程中,研究人员通过改变金属中心、更换配体或在配体分子上增加供电子或吸电子取代基等方法,调控催化剂分子中活性中心的电子结构,对催化剂结构和性能之间的关系有了进一步了解[15-16, 18, 27, 30]。但如前所述的大多数催化剂只能用于有机相而不能用于水相中,这极大地限制了这一类催化剂的应用前景。2013年,Andreiadis等[41]合成了一种能在水溶液中使用的催化剂:钴二胺-二肟(图 3f),通过与碳纳米管(CNTs)上修饰分子中的酰胺形成共价键连接起来。这是第一次报道修饰在电极表面的钴配合物具有催化活性。钴二胺-二肟在水溶液中催化析氢反应时,起始电势为350 mV(vs.RHE)。电流密度达到1 mA/cm2时所需过电势为590 mV,在此电势下,催化剂在醋酸缓冲液中7 h内转换数(turnover numbers, TONs)达到55000。2014年,Fang等[20]通过使用分子前驱体对电极进行修饰的方法,获得了镍和1, 2-二巯基苯(bdt)配体形成的非均相催化剂[Bu4N][Ni(bdt)2](图 3b, M=Ni, X=S, R=H)。[Bu4N][Ni(bdt)2]在酸性乙腈溶液中沉积到玻碳电极表面形成具有电催化析氢活性的薄膜,在电极冲洗和转移之后仍然保持原来的催化活性,证明是非均相的[Bu4N][Ni(bdt)2]在发挥催化作用,其还原电势相比未经修饰的电极电势正移0.2~0.4 V,说明改变催化剂的状态可以提高其催化活性。
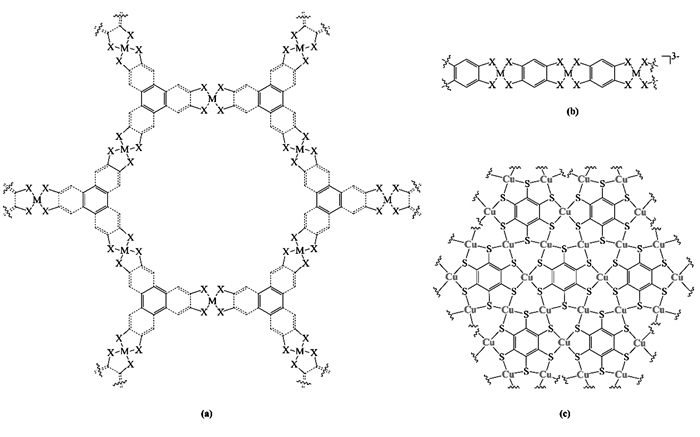
将小分子催化剂固定到电极表面可以在一定程度上解决均相催化剂受溶液扩散影响的局限[20, 41],但是往往负载量较低,最大的催化剂负载量为(1.1±1)×10-8 mol/cm2[44]。设计具有MX4结构单元,同时又具有刚性骨架并可以在水相和有机相中稳定存在的配位聚合物催化剂是解决这个问题的新方法[23]。2015年,Clough等[23]首次研究了这类结构的HER催化活性,采用六巯基苯(benzenehexathiol, BHT)和2, 3, 6, 7, 10, 11-六巯基三亚苯(2, 3, 6, 7, 10, 11-triphenylenehexathiol,THT)配体与Co(Ⅱ)盐合成Co-BHT和Co-THT配位聚合物薄膜(图 4a, M=Co,X=S),并研究了其作为HER催化剂的性质。在pH=1.3的水溶液中,以Co-BHT和Co-THT作为催化剂,法拉第效率均接近100%,相比均相催化剂,法拉第效率高是非均相催化剂的又一大优势。达到10 mA/cm2的催化电流密度分别需要340和530 mV(vs.SHE)的过电势,相比之下,具有类似结构的均相催化剂需要更高的过电势才能达到此电流密度[44]。同时,在Co-BHT中观察到钴负载量为3.7(4)×10-6 mol/cm2,比之前报道的利用小分子配合物直接沉积在电极上的最大负载量高2个数量级[44]。值得一提的是,Clough等[23]将Co-BHT这一类催化剂称为MOS(metal-organic surfaces),即金属有机表面,认为这类材料是以薄膜的形式修饰在电极表面,但没有给出相应的电极形貌的表征,尚不清楚表面修饰层的形貌。2015年,Dong等[24]采用Langmuir-Blodgett(LB)膜方法得到了由Ni(Ⅱ)与2, 3, 6, 7, 10, 11-六巯基三亚苯配体组成的Ni-THT(图 4a,M=Ni,X=S)配位聚合物单原子层薄膜。Ni-THT具有良好的HER电催化活性,在酸性水溶液中(.5 mol/L H2SO4)电流密度达到10 mA/cm2时的过电势仅为333 mV(vs.RHE),塔菲尔斜率也较低,仅为80 mV/dec,这种方法的优势在于利用较低的负载量得到了较高的活性。
图 4
 图 4 (a) Co-BHT(M=Co,X=S)[23], Co-THT(M=Co,X=S)[23],Ni-THT(M=Ni,X=S)[24],含有CoS2N2单元的THTA-Co和具有NiS2N2单元的THTA-Ni(M=Co、Ni, 2S和2N分别位于金属中心两侧的配体中)[28],含有CoS4单元的Co-THT(M=Co, X=S)、含有CoN4单元的Co-THA(M=Co, X=N)[28]; (b)Co-BTT[25]、Fe-BTT、Ni-BTT和Zn-BTT(M=Co、Fe、Ni、Zn,X=S)[26],Co-BTSe和Ni-BTSe(M=Co、Ni,X=S)[29],MS2N2、MS4、MN4(M=Fe、Co、Ni)[32];(c)Cu-BHT[31]Figure 4. (a)Co-BHT(M=Co, X=S)[23], Co-THT(M=Co, X=S)[23], Ni-THT(M= Ni, X=S)[24], CoS2N2 and NiS2N2(M=Co, Ni, where 2S and 2N are on the opposite ligands)[28], CoS4(M=Co, X=S), CoN4(M=Co, X=N)[28]; (b)Co-BTT[25], Fe-BTT, Ni-BTTand Zn-BTT(M=Co, Fe, Ni, Zn, X=S)[26], Co-BTSeandNi-BTSe(M=Co, Ni, X=S)[29], MS2N2, MS4, MN4(M=Fe, Co, Ni)[32]; (c)Cu-BHT[31]
图 4 (a) Co-BHT(M=Co,X=S)[23], Co-THT(M=Co,X=S)[23],Ni-THT(M=Ni,X=S)[24],含有CoS2N2单元的THTA-Co和具有NiS2N2单元的THTA-Ni(M=Co、Ni, 2S和2N分别位于金属中心两侧的配体中)[28],含有CoS4单元的Co-THT(M=Co, X=S)、含有CoN4单元的Co-THA(M=Co, X=N)[28]; (b)Co-BTT[25]、Fe-BTT、Ni-BTT和Zn-BTT(M=Co、Fe、Ni、Zn,X=S)[26],Co-BTSe和Ni-BTSe(M=Co、Ni,X=S)[29],MS2N2、MS4、MN4(M=Fe、Co、Ni)[32];(c)Cu-BHT[31]Figure 4. (a)Co-BHT(M=Co, X=S)[23], Co-THT(M=Co, X=S)[23], Ni-THT(M= Ni, X=S)[24], CoS2N2 and NiS2N2(M=Co, Ni, where 2S and 2N are on the opposite ligands)[28], CoS4(M=Co, X=S), CoN4(M=Co, X=N)[28]; (b)Co-BTT[25], Fe-BTT, Ni-BTTand Zn-BTT(M=Co, Fe, Ni, Zn, X=S)[26], Co-BTSeandNi-BTSe(M=Co, Ni, X=S)[29], MS2N2, MS4, MN4(M=Fe, Co, Ni)[32]; (c)Cu-BHT[31]2015年,Downes等[25]合成了由1, 2, 4, 5-四巯基苯(BTT)配体形成的一维配合物催化剂Co-BTT(图 4b, M=Co,X=S)。虽然Co-BTT用于HER的反应活性较低:在pH=1.3的水溶液中析氢电流密度达到10 mA/cm2时所需过电势为-640 mV vs.SHE(Co-BHT只需要340 mV vs.SHE[23]),但是将其修饰在半导体硅电极上表现出了光电催化析氢活性,在模拟太阳光光照强度和0 V过电势下就能达到3.8 mA/cm2,具有与常用的光阴极材料如InP、GaP相似的活性。2016年,Downes等[26]进一步研究了将Co-BTT的中心金属替换为镍、铁和锌对其催化性能的影响(图 4b, M=Ni、Fe、Zn,X=S),结果表明,Fe-BTT和Zn-BTT配位聚合物几乎无活性;Ni-BTT催化性能良好,在pH=1.3的溶液中电流密度达到10 mA/cm2时所需过电势为470 mV(vs.RHE)。从基于BTT的一维配位聚合物的HER活性,我们可以发现,中心金属的选择对这一类催化剂的活性影响很大,这种差异是如何产生的还需要进一步通过理论计算的方式加以解释。
MX4催化剂的一个重要特点在于,配体的非无辜(Non-innocent)特性,在氧化还原反应中,反应的活性位点不仅可以在金属上,也可能在与金属相连的配体原子上。从前面通过M-BTT类配位聚合物的HER活性的研究中可知,金属的选择对这一类材料的催化活性具有较大的影响,那么配体中配位原子的改变是否会对活性产生影响呢?Downes等[29]合成了基于苯-1, 2, 4, 5-四硒烯醇(BTSe)配体的钴和镍配位聚合物:Co-BTSe、Ni-BTSe(图 4b, M=Co、Ni,X=Se)。结果表明,在钴负载量为9.2×10-7 mol/cm2时,Co-BTSe在pH=1.3的溶液中电流密度达到10 mA/cm2所需过电势仅为343 mV(vs. RHE),而类似的Co-BTT达到相同电流密度时需要更高的过电势:560 mV(vs.RHE)[25]。Downes等[29]对用Se代替S后催化活性的加强提出了可能的解释,认为这一类配位聚合物的反应机理有两种:ECCE和ECEC,而将配位聚合物中的S换成Se之后,反应倾向于以ECCE的机理进行,因此降低了在较高催化剂负载下HER的过电势。同样地,将配体中的S置换为Se前后得到的Ni-BTT和Ni-BTSe,在电流密度达到10 mA/cm2时需要的过电势分别为470 mV、353 mV(vs.RHE)。说明这一类配位聚合物中将配体中S换成Se可以有效提高此类催化剂的活性。
前面讨论的配位聚合物中使用的均是单一的配位原子,使用混合配位原子会对材料的催化活性带来什么样的影响同样是值得探索的。Dong等[28]利用THT配体(2, 3, 6, 7, 10, 11-triphenylenehexathiol)与THA配体(2, 3, 6, 7, 10, 11-triphenylenehexamine)形成的混合配体,分别与金属钴、镍合成了具有CoS2N2单元的THTA-Co和具有NiS2N2单元的THTA-Ni(图 4a, M=Co、Ni, 2S和2N分别位于金属中心两侧的配体中),对比了它们和单一配体合成的具有CoS4、CoN4单元的Co-THT、Co-THA(图 4a,M=Co, X=S、N)二维配位聚合物的HER活性。实验结果表明:当电催化析氢电流密度达到10 mA/cm2时,CoS2N2、NiS2N2、CoS4所需的过电势分别为283、315、323 mV(实验中未得到CoN4),并计算了这4种配合物的活性位点的ΔGH*值,当ΔGH*越接近于零时活性越高,4种配合物的ΔGH*值接近零的程度与活性顺序一致,说明理论计算结果与实验结果相一致。Wang等[32]也报道了类似的工作,利用2, 5-二氨基-1, 4-二巯基苯、1, 2, 4, 5-四巯基苯、1, 2, 4, 5-四苯胺形成的分别含有MS2N2、MS4、MN4(图 4b, M=Fe、Co、Ni)一维配位聚合物,研究了金属和配体原子对析氢催化活性的影响,测试结果表明,对于具有不同配位原子的配位聚合物,其活性性能顺序为MS2N2>MN4>MS4,这与Dong等[28]的文献研究结果一致。Wang等[32]认为金属、硫和氮的配位协同促进了氢在MSxNy催化剂上的吸附和解吸附过程。改变MS2N2中的金属中心发现HER催化性能大小顺序为FeS2N2>CoS2N2>NiS2N2,电流密度达到10 mA/cm2所需要的过电势分别为576、621和696 mV。虽然这些催化剂的过电势仍然较高,但是通过使用不同配体形成杂原子配位结构提升催化剂活性的方法为以后的研究提供了有益的思路。
除了中心金属和配位原子,实际上催化剂的尺寸,形貌及结晶度同样对催化活性具有重要的影响[31]。二维配位聚合物Cu-BHT具有极高的电导率(1580 S/cm)[45],使它在电化学催化剂领域具有应用潜力[31]。Huang等[31]研究了其做为HER催化剂的性能:第一, 实验中通过改变合成条件制备得到3种形态的Cu-BHT(图 4c),分别为薄膜、纳米晶和纳米颗粒,三者具有相似的组成,其中纳米晶和纳米颗粒具有电催化活性,而且纳米颗粒的催化活性远高于纳米晶;第二,在pH值为0的H2SO4溶液中,Cu-BHT纳米晶和纳米颗粒作为催化剂时,催化电流密度达到10 mA/cm2所需过电势分别为760和450 mV;第三, 通过改变Cu-BHT催化剂的形态,实现了催化性能的优化;第四,同时提出Cu-BHT纳米晶和纳米颗粒之间的活性差异部分来源于两者尺度和形貌的不同,纳米颗粒具有更低的结晶度,更小的尺寸,更大的比表面积,但是比表面积的变化并不能完全解释两者催化活性的差异,催化活性位点的改变也有相当程度的贡献。同时,他们通过理论计算确认,位于Cu-BHT(110)晶面上的边界铜原子具有较高的催化活性,Cu-BHT纳米颗粒的催化活性的提升应该与这一位点的相对比例的变化存在联系。这种形貌变化的策略为制备更高效催化剂提供了新的方向,同时也表明在未来研究这一类催化剂时应注意对材料形貌的表征。
对于基于MX4结构的配合物作为HER催化剂的研究,始于小分子配合物均相催化剂,逐渐发展到一维、二维结构的配位聚合物非均相催化剂,将催化剂的使用范围从有机溶剂扩展到了水溶液。从改变配体和金属中心、改变纳米催化剂形貌等方面不断提高其作为HER催化剂的催化活性和应用前景。表 1总结了具有代表性的MX4配合物的催化活性。
表 1
 表 1 MX4配合物作为HER催化剂的活性Table 1. The activities of MX4 complexes as catalyst for HER
表 1 MX4配合物作为HER催化剂的活性Table 1. The activities of MX4 complexes as catalyst for HER 下载:
导出CSV
下载:
导出CSV
Ligand Catalyst 107×Catalyst loadings(calculated by metal)/(mol·cm-2) Overpotentialη/mVa
@10 mA/cm2Tafel Slopea/(mV·dec-1) Exchange current density/(A·cm-2) Ref. 
Co-BHT
Cu-BHT7.0
-
-340b(pH=1.3)
760b, c
450b, d108(pH=2.6)
-
-10-5.3
-
-[23]
[31]
[31]Co-BTT
Ni-BTT
Fe-BTT5.5
-
6.8
-
-560b(pH=1.3)
734e
470b(pH=1.3)
766e
756e70(pH=1.3)
214
76(pH=2.6)
225
21110-9.4
-
10-8.1
-
-[25]
[32]
[26]
[32]
[32]Co-BTSe
Ni-BTSe9.2
9.0343e(pH=1.3)
353e(pH=1.3)97(pH=1.3)
-10-4.4
-[29]
[29]Co-THT
Ni-THT11.0
-
-
-530b(pH=1.3)
323e
415b(pH=1.3)
333b161(pH=2.6)
82
81
80.510-5.4
-
6×10-7
~6×10-4[23]
[28]
[24]
[24]Co-THA - 382e 120 - [28] Co-THTA
Ni-THTA-
-283e
315e71
76-
-[28]
[28]Co-S2N2
Ni-S2N2
Fe-S2N2-
-
-621e
696e
576e193
217
158-
-
-[32]
[32]
[32]Co-BTA
Ni-BTA
Fe-BTA-
-
-679e
705e
631e195
220
173-
-
-[32]
[32]
[32]a.The pH is 0 if there is no special description; b.vs.SHE; c.nanocrystal; d.nanoparticle; e.vs.RHE. 3. MX4催化剂的理论研究
3.1 均相MX4催化剂理论研究
在催化析氢反应时,溶解在有机溶液或水溶液中的MX4配合物往往会使质子在MX4分子平面发生质子电子转移反应,最终形成氢气分子。实验能够测定催化剂的活性,却不能提供具体的反应细节,而理论计算可以帮助研究人员深入地了解反应历程,通过计算吉布斯自由能变化可以确定催化剂的质子化位点,计算各个反应步骤的能垒,进而确定反应路径。对于均相催化剂,研究者们常通过选取可能的质子化位点,计算能量变化,分步确定质子化位点和反应路径。
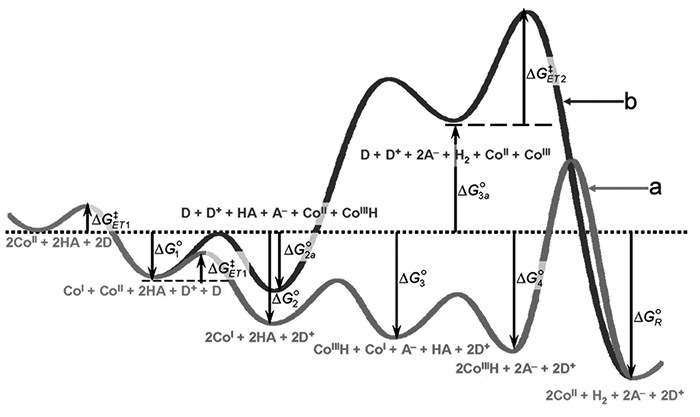
2009年,Dempsey等[15]采用热力学方法分析研究了钴肟配合物(结构如图 3a)的催化析氢历程如图 5所示。反应起始步骤是CoⅡ接受来自电子供体(D)的电子还原为CoⅠ,这一步的反应能垒取决于驱动力和重组参数(λ)。接下来CoⅠ的质子化反应与驱动力(ΔG30-ΔG20)有关,该驱动力体现催化剂质子化的难易程度,其大小与质子供体(HA)和CoⅢH的pKa之差成正比。形成氢化物之后,其稳定程度直接影响后续反应中H2释放的方式:当钴-氢键较弱时,通过具有较低能垒的同源耦合反应路径(Homo-coupling pathway,图 5a)析出氢气,而当氢化物较稳定时,催化剂与第二个质子结合,通过异质耦合反应路径(Hetero-coupling pathway,图 5b)析出氢气。整个析氢反应的驱动力取决于电子供体的还原电势(相对于HA(EHA/H20)析氢的标准电势)。在同源耦合反应路径中,当使用足够强的还原剂和质子供体时,质子还原过程中的所有步骤都是放热的,最大的反应能垒可能与H2的形成有关,这说明质子与催化剂的相互作用过强。在异质耦合路径中,CoⅢH发生质子化形成H2,形成H2的基本步骤的驱动力取决于E0[CoⅢ/Ⅱ]和E0[CoⅡ/Ⅰ](相对于质子还原电势)的平均值。从图 5中可以看出,异质耦合反应路径非常不利于反应发生,大多数情况下更倾向于发生同源耦合反应。值得注意的是,H2的形成方式不仅取决于基元步骤的能垒,还与HA和CoⅢH的相对浓度有关,例如,在酸浓度非常高时,即使在很高的能垒下,异质耦合反应机理也会变得更加有利。通过热力学分析计算不同反应步骤的能垒可确定能量较低的反应路径。
图 5
为了深入地理解镍芳基二硫烯配合物的析氢反应机理及催化剂的活性位点,Zarkadoulas等[27]通过计算镍芳基二硫烯配合物(图 3e)质子化过程中的能量变化以及氧化还原电势提出了反应路径(如图 6所示)。在反应过程中,第1步是催化剂分子还原为一价阴离子,由于一价阴离子在S原子和Ni原子处发生质子化均需要吸收能量,因此一价阴离子更倾向于还原为二价阴离子,接着二价阴离子发生质子化释放能量。由于S原子质子化比Ni原子质子化后的结构更稳定,因此二价阴离子第1次质子化最有利的位点是二硫烯配体上的S原子。在第1次质子化之后,可能发生两个不同的反应过程,即直接进行第2次质子化(路径A)或还原后进行第2次质子化(路径B)。计算表明第2次质子化发生在不同配体单元中相近的两个S原子上(比其它情况稳定)。在此结构中质子化和电子转移主要发生在配体上。3种催化剂的活性顺序为e2>e3>e1,这主要是由于他们的结构中苯环上甲氧基的个数不同,甲氧基的供电子性质导致S原子上的电荷密度增加,使得配合物e2和e3结构不易发生还原反应,却更容易发生质子化。由于在反式的配合物e2结构中,甲氧基的电子更容易聚集在S原子上,所以配合物e2结构更易发生质子化,具有更高的活性。前文介绍了4种催化剂[18]:[Co(bdt)2]-、[Co(tdt)2]-、[Co(Cl2bdt)2]-和[Co(mnt)2]-,前3种结构中与硫原子相连的是苯环,具有相似结构;[Co(mnt)2]-与配合物e1~e3类似,配合物e1~e3结构中与S原子相连的为1, 2-二苯基乙烯,[Co(mnt)2]-中与S原子相连的是1, 2-二氰基乙烯。在这4个结构中,[Co(mnt)2]-的配体中连有吸电子能力强的氰基,S原子电荷密度下降,与质子结合能力减弱;而在配合物e1~e3结构中,甲氧基的存在使S原子电荷密度增大,更易发生质子化,因此含有甲氧基的配合物e2和e3结构的活性更高。通过这两项研究,我们推论活性位点电荷密度减小有利于电子转移,但是活性位点的电荷密度增大有利于质子化。
图 6

一些研究通过计算还原电势和吉布斯自由能变化等确定了反应历程和活性位点,也有其它报道采用比较不同状态下催化剂pKa计算结果的方法进行研究[43]。在McNamara等[18]关于[Co(bdt)2]-、[Co(tdt)2]-、[Co(Cl2bdt)2]-和[Co(mnt)2]-的报道中发现,[Co(mnt)2]-虽然拥有吸电子能力最强的取代基—CN和最正的CoⅢ/Ⅱ还原电势,但是它的电催化活性却最低。Solis等[43]通过计算解释了[Co(mnt)2]-异常电催化活性的原因,通过对比计算得到的相对还原电势与实验测得的还原电势,结合各种状态的[Co(mnt)2]-的pKa计算结果对比,发现[Co(mnt)2]-的活性低于其它催化剂的主要原因是[Co(mnt)2]-中发生质子化的S位点少于其他结构。质子化位点是硫原子,结合更多质子的配合物更容易被还原,这是由于质子引入了更多正电荷。对比催化剂的活性发现,S原子的电荷密度对[Co(bdt)2]-衍生物的催化活性有关键的影响,S的质子化促使还原电势更正[21]。在均相催化剂反应机理中涉及到电子转移步和质子转移步,因此虽然[Co(mnt)2]-在电子转移步表现出最正的还原电势,但是氰基的吸电子性质使得S原子的电荷密度减小,不利于质子化,导致质子化位点少于其他结构,活性低于其它3种催化剂,这也说明了质子化位点数量对于催化剂活性的重要性。
在MX4催化剂结构中,质子化位点的本质活性和数量对催化剂的活性有重要的影响,其中质子化位点的电荷密度是关键因素,电荷密度越高,催化剂越难还原,相应的质子化越容易实现。由于在析氢反应中,两个氢原子结合之后需要脱吸附形成氢气,结合Sabatier原理可知,质子与催化剂活性位点的相互作用不能过强或过弱。对反应机理的研究一般通过对比还原电势和吉布斯自由能变化的大小来确定:每一步反应是更有利于发生还原还是质子化,从而进一步确定整体的反应过程,通过能量变化可以确定反应的决速步,为未来设计具有更小的反应决速步能垒的分子结构选择提供理论依据。同时考虑反应机理受实验环境和反应物浓度改变的影响,应进一步提升理论计算的准确度。
3.2 非均相MX4催化剂理论研究
在溶液中,MX4催化剂通过同源耦合或者异质耦合反应路径释放氢气,但是对于修饰在电极表面的非均相催化剂来说,无法通过同源耦合机理发生还原消除反应生成氢气[41]。近年来,Volmer-Tafel/Heyrovskey析氢反应机理以及吉布斯自由能变化等的计算在非均相催化剂活性的理论研究中得到广泛应用,且与实验结果具有很好地一致性[28, 46]。
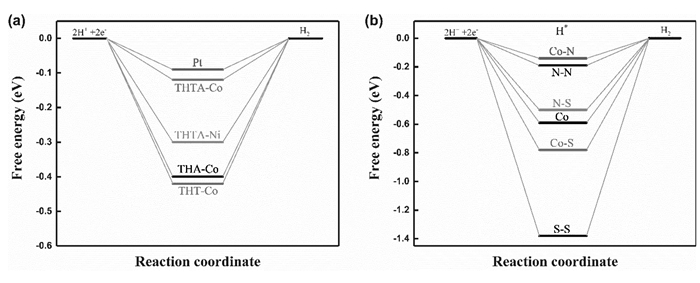
根据HER反应机理结合Sabatier原理可知,平衡电势下吉布斯自由能变化值等于零时,相应催化活性最好[39]。Dong等[28]通过DFT计算研究含有MSxNy(M=Co、Ni; x/y=2/2、4/0、0/4)的2D MOFs作为HER催化剂的催化活性和活性位点。通过对比平衡电势下吉布斯自由能变化,可以判断配合物中金属中心对HER催化活性的影响。如图 7(a)所示,对于CoS2N2配合物,其活性位点具有绝对值更接近于零的吉布斯自由能变化ΔGH*, 这与金属Pt[39]对应的ΔGH*十分接近,而NiS2N2配合物的ΔGH*的绝对值更大, 表明CoS2N2配合物具有比NiS2N2更好的的HER电催化活性。同理对比3种Co中心催化剂的ΔGH*可得,其活性顺序依次为CoS2N2>CoN4>CoS4;对于Ni体系,可以得到相同的结论即NiS2N2>NiN4>NiS4。以上计算结果与实验结果一致,说明上述计算方法能较好的反映此类催化剂活性与组成的关系。为了确定CoS2N2的活性位点,Dong等[28]计算了在可能的位点上的1H原子和2H原子吸附能,分别与H原子和H2的解离能对比,结果表明质子化优先发生在Co原子上,第二次质子化发生在N原子上。对比可能的活性位点的氢吸附吉布斯自由能变化ΔGH*(图 7b)计算结果可知,Co-N(-0.14 eV)具有最高的活性。吸附能与吉布斯自由能变化计算结果表明:与S原子配位增强了H2在Co原子上的吸附能力(ΔGH*),不利于H2脱吸附,而Co-N单元促进了H2脱吸附,因此与CoN4和CoS4相比,综合了两种过程的CoS2N2的催化活性更高。以上研究结果说明, 质子分别与配合物中Co-N单元中的Co原子和N原子结合,电催化析氢机理为ECEC机理,通过异质耦合路径生成H2。通过配体的简单改性以及金属中心的调整实现了电催化剂活性的优化。基于DFT计算能够给出与实验一致的活性预测[28],Wang等[32]通过理论计算进一步研究了MSxNy-基(M=Fe、Co、Ni; x/y=2/2、4/0、0/4)一维结构的催化析氢能力。采用DFT方法计算不同催化剂的吉布斯自由能变化得FeS2N2(-0.1 eV)>CoS2N2(-0.18 eV)>NiS2N2(-0.27 eV),与实验所测催化剂活性顺序一致。同一金属中心不同配体的HER电催化活性满足MS2N2>MN4>MS4。其通过氢原子吸附能和吉布斯自由能变化计算发现,对于MS2N2和MS4催化剂,质子化优先发生在金属原子上,随后H2的生成更倾向于分别在M-N与M-S单元上发生;对于MN4体系,最初的H吸附发生在N原子上。
图 7

Cu-BHT催化剂的活性优化是采用改变催化剂形态的方法实现的[31]。为了深入理解Cu-BHT的催化活性来源和活性提高的原因,Huang等[31]通过DFT计算实验中观测到的晶面上可能的活性位点的ΔGH*值,通过比较各位点的ΔGH*值大小,发现(110)晶面上的边界铜原子的ΔGH*值更接近于零,是活性最高的位点。当改变催化剂形态时,暴露活性位点的数量增多。因此,最小尺寸的Cu-BHT纳米颗粒具有更优的电催化性能。
MX4催化剂中含有金属和非金属原子,活性位点的确定是研究其活性的基础,在理论计算中,通过计算可能的活性位点的氢吸附吉布斯自由能变化,可以帮助确认MX4催化剂的活性位点。一般来说,提高催化剂的活性有2种方法:一是提高活性位点的本质活性;另一种是增加活性位点数量[47]。实验中常通过改变催化剂形态增加比表面积,使得更多活性位点得以发挥作用,此时,催化剂的暴露晶面发生变化,活性位点的性质也可能会发生变化。目前,理论计算中所采用的模型多为理想结构,与实验中催化剂的实际结构有不同程度的差别,并且催化剂的工作环境对其性能的影响往往被忽略。进一步完善模型以及考虑周围环境因素的影响,将有助于理论计算提供更准确和精密的证据以解析实验结果。
4. 总结与展望
配合物因为其多样性和化学反应活性而活跃在催化剂研究领域的方方面面。近年来,具有MX4结构的配合物在析氢反应中的应用吸引了越来越多的关注。本文系统地总结了具有此类结构的小分子配合物催化剂和配位聚合物催化剂的发展和研究现状。总结归纳后可以得出,不管是MX4小分子配合物还是配位聚合物,影响其活性的因素是多样的,既可以是金属中心,也可以是配位原子,也可能是配体的结构,而对于作为非均相催化剂的配位聚合物来讲,材料的形貌、尺寸也会显著影响催化剂活性,基于上述两类化合物的研究已经得到了相当高活性的析氢催化剂。通过已有的实验与理论计算相结合的研究可以发现,MX4催化剂活性中心既可能是金属中心也可能是与金属配位的非金属原子。其中对于与金属配位的非金属原子作为活性位点的催化剂,电子转移和质子化受活性位点电荷密度的影响,活性位点质子化的难易程度对活性非常重要,质子和催化剂之间适中强度的相互作用力是析氢反应催化剂获得高活性的必要条件,然而如何获得具有更高活性的活性位点需要进一步进行研究。对于金属中心作为活性位点的催化剂,在已经研究过的MX4催化剂中,以金属钴和镍为中心的催化剂性能较好,金属中心与催化剂活性之间的关系也需要进一步的理论研究探索。通过反应机理的研究,根据每一步的能量变化确定决速步,比如文中介绍的钴肟配合物更倾向于发生同质耦合反应,反应中决速步与H2的释放有关,这启示我们氢原子与催化剂的吸附作用力较强,据此可以做结构的优化以提高整体的反应速率。
-
-
[1]
Borup R, Meyers J, Pivovar B. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation[J]. Chem Rev, 2007, 107(10): 3904-3951. doi: 10.1021/cr050182l
-
[2]
Amphlett J, Evans M, Jones R. Hydrogen Production by the Catalytic Steam Reforming of Methanol Part 1:The Thermodynamics[J]. Can J Chem Eng, 1981, 59(6): 720-727. doi: 10.1002/cjce.v59:6
-
[3]
Steinberg M, Cheng H C. Modern and Prospective Technologies for Hydrogen Production from Fossil Fuels[J]. Int J Hydrogen Energ, 1989, 14(11): 797-820. doi: 10.1016/0360-3199(89)90018-9
-
[4]
Bard A J, Fox M A. Artificial Photosynthesis:Solar Splitting of Water to Hydrogen and Oxygen[J]. Acc Chem Res, 1995, 28(3): 141-145. doi: 10.1021/ar00051a007
-
[5]
Dresselhaus M S, Thomas I L. Alternative Energy Technologies[J]. Nature, 2001, 414(6861): 332-337. doi: 10.1038/35104599
-
[6]
Turner J A. Sustainable Hydrogen Production[J]. Science, 2004, 305(5686): 972-974. doi: 10.1126/science.1103197
-
[7]
Subbaraman R, Tripkovic D, Strmcnik D. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces[J]. Science, 2011, 334(6060): 1256-1260. doi: 10.1126/science.1211934
-
[8]
Walter M G, Warren E L, McKone J R. Solar Water Splitting Cells[J]. Chem Rev, 2010, 110(11): 6446-6473. doi: 10.1021/cr1002326
-
[9]
Cook T R, Dogutan D K, Reece S Y. Solar Energy Supply and Storage for the Legacy and Nonlegacy Worlds[J]. Chem Rev, 2010, 110(11): 6474-6502. doi: 10.1021/cr100246c
-
[10]
Yin H, Zhao S, Zhao K. Ultrathin Platinum Nanowires Grown on Single-Layered Nickel Hydroxide with High Hydrogen Evolution Activity[J]. Nat Commun, 2015, 6: 6430. doi: 10.1038/ncomms7430
-
[11]
Conway B E, Tilak B V. Interfacial Processes Involving Electrocatalytic Evolution and Oxidation of H2, and the Role of Chemisorbed H[J]. Electrochim Acta, 2002, 47(22/23): 3571-3594.
-
[12]
McKone J R, Marinescu S C, Brunschwig B S. Earth-Abundant Hydrogen Evolution Electrocatalysts[J]. Chem Sci, 2014, 5(3): 865-878. doi: 10.1039/C3SC51711J
-
[13]
Zheng Y, Jiao Y, Zhu Y. Hydrogen Evolution by a Metal-Free Electrocatalyst[J]. Nat Commun, 2014, 5: 3783.
-
[14]
Hu X, Cossairt B M, Brunschwig B S. Electrocatalytic Hydrogen Evolution by Cobalt Difluoroboryl-Diglyoximate Complexes[J]. Chem Commun, 2005, 0(37): 4723-4725.
-
[15]
Dempsey J L, Brunschwig B S, Winkler J R. Hydrogen Evolution Catalyzed by Cobaloximes[J]. Acc Chem Res, 2009, 42(12): 1995-2004. doi: 10.1021/ar900253e
-
[16]
McNamara W R, Han Z, Alperin P J. A Cobalt-Dithiolene Complex for the Photocatalytic and Electrocatalytic Reduction of Protons[J]. J Am Chem Soc, 2011, 133(39): 15368-15371. doi: 10.1021/ja207842r
-
[17]
Barnett S M, Goldberg K I, Mayer J M. A Soluble Copper-Bipyridine Water-Oxidation Electrocatalyst[J]. Nat Chem, 2012, 4(6): 498-502. doi: 10.1038/nchem.1350
-
[18]
McNamara W R, Han Z, Yin J C. Cobalt-Dithiolene Complexes for the Photocatalytic and Electrocatalytic Reduction of Protons in Aqueous Solutions[J]. Proc Natl Acad Sci USA, 2012, 109(39): 15594-15599. doi: 10.1073/pnas.1120757109
-
[19]
Jahan M, Liu Z, Loh K P. A Graphene Oxide and Copper-Centered Metal Organic Framework Composite as a Tri-Functional Catalyst for HER, OER, and ORR[J]. Adv Funct Mater, 2013, 23(43): 5363-5372. doi: 10.1002/adfm.v23.43
-
[20]
Fang M, Engelhard M H, Zhu Z. Electrodeposition from Acidic Solutions of Nickel Bis(Benzenedithiolate) Produces a Hydrogen-Evolving Ni-S Film on Glassy Carbon[J]. ACS Catal, 2014, 4(1): 90-98. doi: 10.1021/cs400675u
-
[21]
Letko C S, Panetier J A, Head-Gordon M. Mechanism of the Electrocatalytic Reduction of Protons with Diaryldithiolene Cobalt Complexes[J]. J Am Chem Soc, 2014, 136(26): 9364-9376. doi: 10.1021/ja5019755
-
[22]
Zhang P, Wang M, Yang Y. A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution[J]. Angew Chem Int Edit, 2014, 53(50): 13803-13807. doi: 10.1002/anie.201408266
-
[23]
Clough A J, Yoo J W, Mecklenburg M H. Two-Dimensional Metal-Organic Surfaces for Efficient Hydrogen Evolution from Water[J]. J Am Chem Soc, 2015, 137(1): 118-121. doi: 10.1021/ja5116937
-
[24]
Dong R, Pfeffermann M, Liang H. Large-Area, Free-Standing, Two-Dimensional Supramolecular Polymer Single-Layer Sheets for Highly Efficient Electrocatalytic Hydrogen Evolution[J]. Angew Chem Int Edit, 2015, 54(41): 12058-12063. doi: 10.1002/anie.201506048
-
[25]
Downes C A, Marinescu S C. Efficient Electrochemical and Photoelectrochemical H2 Production from Water by a Cobalt Dithiolene One-Dimensional Metal-Organic Surface[J]. J Am Chem Soc, 2015, 137(43): 13740-13743. doi: 10.1021/jacs.5b07020
-
[26]
Downes C A, Marinescu S C. One Dimensional Metal Dithiolene(M=Ni, Fe, Zn) Coordination Polymers for the Hydrogen Evolution Reaction[J]. Dalton Trans, 2016, 45(48): 19311-19321. doi: 10.1039/C6DT03257E
-
[27]
Zarkadoulas A, Field M J, Papatriantafyllopoulou C. Experimental and Theoretical Insight into Electrocatalytic Hydrogen Evolution with Nickel Bis(aryldithiolene) Complexes as Catalysts[J]. Inorg Chem, 2016, 55(2): 432-444.
-
[28]
Dong R, Zheng Z, Tranca D C. Immobilizing Molecular Metal Dithiolene-Diamine Complexes on 2D Metal-Organic Frameworks for Electrocatalytic H2 Production[J]. Chem Eur J, 2017, 23(10): 2255-2260. doi: 10.1002/chem.201605337
-
[29]
Downes C A, Marinescu S C. Bioinspired Metal Selenolate Polymers with Tunable Mechanistic Pathways for Efficient H2 Evolution[J]. ACS Catal, 2017, 7(1): 848-854. doi: 10.1021/acscatal.6b03161
-
[30]
Downes C A, Yoo J W, Orchanian N M. H2 Evolution by a Cobalt Selenolate Electrocatalyst and Related Mechanistic Studies[J]. Chem Commun, 2017, 53(53): 7306-7309. doi: 10.1039/C7CC02473H
-
[31]
Huang X, Yao H, Cui Y. Conductive Copper Benzenehexathiol Coordination Polymer as a Hydrogen Evolution Catalyst[J]. ACS Appl Mater Interfaces, 2017, 9(46): 40752-40759. doi: 10.1021/acsami.7b14523
-
[32]
Wang L, Tranca D C, Zhang J. Toward Activity Origin of Electrocatalytic Hydrogen Evolution Reaction on Carbon-Rich Crystalline Coordination Polymers[J]. Small, 2017, 13(37): 1700783-1700790. doi: 10.1002/smll.v13.37
-
[33]
Coggins M K, Zhang M T, Chen Z. Single-Site Copper(Ⅱ) Water Oxidation Electrocatalysis:Rate Enhancements with HPO42- as a Proton Acceptor at pH 8[J]. Angew Chem Int Edit, 2014, 53(45): 12226-12230. doi: 10.1002/anie.201407131
-
[34]
Nam N T S, Sluys M V D, Jones C W. On the Nature of the Active Species in Palladium Catalyzed Mizoroki-Heck and Suzuki-Miyaura Couplings-Homogeneous or Heterogeneous Catalysis, a Critical Review[J]. Adv Synth Catal, 2006, 348(6): 609-679. doi: 10.1002/(ISSN)1615-4169
-
[35]
Song L C, Yang Z Y, Bian H Z. Diiron Oxadithiolate Type Models for the Active Site of Iron-Only Hydrogenases and Biomimetic Hydrogen Evolution Catalyzed by Fe2(SCH2OCH2S)(CO)6[J]. Organometallics, 2005, 24(25): 6126-6135. doi: 10.1021/om0507373
-
[36]
Costentin C, Sav ant J M. Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions:Benchmarking of Homogeneous Catalysts[J]. ChemElectroChem, 2014, 1(7): 1226-1236. doi: 10.1002/celc.201300263
-
[37]
Wiedner E S, Brown H J, Helm M L. Kinetic Analysis of Competitive Electrocatalytic Pathways:New Insights into Hydrogen Production with Nickel Electrocatalysts[J]. J Am Chem Soc, 2016, 138(2): 604-616. doi: 10.1021/jacs.5b10853
-
[38]
Zheng Y, Jiao Y, Jaroniec M. Advancing the Electrochemistry of the Hydrogen-Evolution Reaction Through Combining Experiment and Theory[J]. Angew Chem Int Edit, 2015, 54(1): 52-65. doi: 10.1002/anie.201407031
-
[39]
Nørskov J K, Bligaard T, Logadottir A. Trends in the Exchange Current for Hydrogen Evolution[J]. J Electrochem Soc, 2005, 152(3): J23-J26. doi: 10.1149/1.1856988
-
[40]
Artero V, Fontecave M. Some General Principles for Designing Electrocatalysts with Hydrogenase Activity[J]. Coord Chem Rev, 2005, 249(15/16): 1518-1535.
-
[41]
Andreiadis E S, Jacques P A, Tran P D. Molecular Engineering of a Cobalt-Based Electrocatalytic Nanomaterial for H2 Evolution under Fully Aqueous Conditions[J]. Nat Chem, 2013, 5(1): 48-53. doi: 10.1038/nchem.1481
-
[42]
Baker-Hawkes M J, Billig E, Gray H B. Characterization and Electronic Structures of Metal Complexes Containing Benzene-1, 2-dithiolate and Related Ligands[J]. J Am Chem Soc, 1966, 88(21): 4870-4875. doi: 10.1021/ja00973a021
-
[43]
Solis B H, Hammes-Schiffer S. Computational Study of Anomalous Reduction Potentials for Hydrogen Evolution Catalyzed by Cobalt Dithiolene Complexes[J]. J Am Chem Soc, 2012, 134(37): 15253-15256. doi: 10.1021/ja306857q
-
[44]
Tran P D, Le Goff A, Heidkamp J. Noncovalent Modification of Carbon Nanotubes with Pyrene-Functionalized Nickel Complexes:Carbon Monoxide Tolerant Catalysts for Hydrogen Evolution and Uptake[J]. Angew Chem Int Edit, 2011, 50(6): 1371-1374. doi: 10.1002/anie.v50.6
-
[45]
Huang X, Sheng P, Tu Z. A Two-Dimensional π-d Conjugated Coordination Polymer with Extremely High Electrical Conductivity and Ambipolar Transport Behaviour[J]. Nat Commun, 2015, 6: 7408. doi: 10.1038/ncomms8408
-
[46]
Jaramillo T F, Jørgensen K P, Bonde J. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2Nanocatalysts[J]. Science, 2007, 317(5834): 100-102. doi: 10.1126/science.1141483
-
[47]
Seh Z W, Kibsgaard J, Dickens C F. Combining Theory and Experiment in Electrocatalysis:Insights into Materials Design[J]. Science, 2017, 355(6321): 146-146.
-
[1]
-

图 3 (a)[Co(dpgBF2)2][14];(b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18],[Co(tdt)2]-(M=Co, X=S, R3-6=CH3,其余R=H)[18],[Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, 其余R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20];(c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2、[5F]2、[5Cl]2、[5Br]2[21]; (e)镍芳基二硫烯配合物[27]; (f)钴二胺-二肟催化剂[41]
Figure 3 (a)[Co(dpgBF2)2][14]; (b)[Co(bdt)2]-(M=Co, X=S, R=H)[16, 18], [Co(tdt)2]-(M=Co, X=S, R3-6=CH3, other R=H)[18], [Co(Cl2bdt)2]-(M=Co, X=S, R1=R4=R5=R8=Cl, other R=H)[18], Co(bds)2(M=Co, X=Se, R=H)[30], [Ni(bdt)2](M=Ni, X=S, R=H)[20]; (c)[Co(mnt)2]-(M=Co, X=S, R=CN)[18]; (d)[Co(S2C2Ar2)2]2(5), [5OMe]2, [5F]2, [5Cl]2, [5Br]2[21]; (e)nickel dithiolene complexes with p-methoxyphenyl-substituted 1, 2-dithiolene ligands[27]; (f)diimine-dioxime cobalt catalyst[41]
图 4 (a) Co-BHT(M=Co,X=S)[23], Co-THT(M=Co,X=S)[23],Ni-THT(M=Ni,X=S)[24],含有CoS2N2单元的THTA-Co和具有NiS2N2单元的THTA-Ni(M=Co、Ni, 2S和2N分别位于金属中心两侧的配体中)[28],含有CoS4单元的Co-THT(M=Co, X=S)、含有CoN4单元的Co-THA(M=Co, X=N)[28]; (b)Co-BTT[25]、Fe-BTT、Ni-BTT和Zn-BTT(M=Co、Fe、Ni、Zn,X=S)[26],Co-BTSe和Ni-BTSe(M=Co、Ni,X=S)[29],MS2N2、MS4、MN4(M=Fe、Co、Ni)[32];(c)Cu-BHT[31]
Figure 4 (a)Co-BHT(M=Co, X=S)[23], Co-THT(M=Co, X=S)[23], Ni-THT(M= Ni, X=S)[24], CoS2N2 and NiS2N2(M=Co, Ni, where 2S and 2N are on the opposite ligands)[28], CoS4(M=Co, X=S), CoN4(M=Co, X=N)[28]; (b)Co-BTT[25], Fe-BTT, Ni-BTTand Zn-BTT(M=Co, Fe, Ni, Zn, X=S)[26], Co-BTSeandNi-BTSe(M=Co, Ni, X=S)[29], MS2N2, MS4, MN4(M=Fe, Co, Ni)[32]; (c)Cu-BHT[31]

表 1 MX4配合物作为HER催化剂的活性
Table 1. The activities of MX4 complexes as catalyst for HER
Ligand Catalyst 107×Catalyst loadings(calculated by metal)/(mol·cm-2) Overpotentialη/mVa
@10 mA/cm2Tafel Slopea/(mV·dec-1) Exchange current density/(A·cm-2) Ref. Co-BHT
Cu-BHT7.0
-
-340b(pH=1.3)
760b, c
450b, d108(pH=2.6)
-
-10-5.3
-
-[23]
[31]
[31]Co-BTT
Ni-BTT
Fe-BTT5.5
-
6.8
-
-560b(pH=1.3)
734e
470b(pH=1.3)
766e
756e70(pH=1.3)
214
76(pH=2.6)
225
21110-9.4
-
10-8.1
-
-[25]
[32]
[26]
[32]
[32]Co-BTSe
Ni-BTSe9.2
9.0343e(pH=1.3)
353e(pH=1.3)97(pH=1.3)
-10-4.4
-[29]
[29]Co-THT
Ni-THT11.0
-
-
-530b(pH=1.3)
323e
415b(pH=1.3)
333b161(pH=2.6)
82
81
80.510-5.4
-
6×10-7
~6×10-4[23]
[28]
[24]
[24]Co-THA - 382e 120 - [28] Co-THTA
Ni-THTA-
-283e
315e71
76-
-[28]
[28]Co-S2N2
Ni-S2N2
Fe-S2N2-
-
-621e
696e
576e193
217
158-
-
-[32]
[32]
[32]Co-BTA
Ni-BTA
Fe-BTA-
-
-679e
705e
631e195
220
173-
-
-[32]
[32]
[32]a.The pH is 0 if there is no special description; b.vs.SHE; c.nanocrystal; d.nanoparticle; e.vs.RHE.  下载: 导出CSV
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 11
- 文章访问数: 1962
- HTML全文浏览量: 596
 下载:
下载:
