-
[1]
Moerner W E, Kador L. Phys. Rev. Lett., 1989, 62(21): 2535-2538
-
[2]
Orrit M, Bernard J. Phys. Rev. Lett., 1990, 65(21): 2716-2719
-
[3]
Betzig E, Chichester R J. Science, 1993, 262(5138): 1422-1425
-
[4]
Xie X S, Duun R C. Science, 1994, 265(5170): 361-364
-
[5]
Ambrose W P, Goodwin P M, Keller R A, Martin J C. Science, 1994, 265(5170): 364-367
-
[6]
Macklin J J, Trautman J K, Harris T D, Brus L E. Science, 1996, 272(5159): 255-258
-
[7]
Xie X S, Trautman J K. Ann. Rev. Phys. Chem., 1998, 49: 441-480
-
[8]
Claessen V I, Engelkamp H, Christianen P C M, Maan J C, Nolte R J M, Blank K, Rowan A E. Ann. Rev. Anal. Chem., 2010, 3: 319-340
-
[9]
Lu H P, Xun L Y, Xie X S. Science, 1998, 282(5395): 1877-1882
-
[10]
Xue Q F, Yeung E S. Nature, 1995, 373(6516): 681-683
-
[11]
Yang H, Luo G B, Karnchanaphanurach P, Louie T, Rech I, Cova S, Xun L Y, Xie X S. Science, 2003, 302(3): 262-266
-
[12]
Edman L, Földes-Papp Z, Wennmalm S, Rigler R. Chem. Phys., 1999, 247(1): 11-22
-
[13]
Zheng D, Lu H P. J. Phys. Chem. B, 2014, 118(31): 9128-9140
-
[14]
Guo Q, He Y F, Lu H P. Proc. Natl. Acad. Sci. USA, 2015, 112(45): 13904-13909
-
[15]
Comellas-Aragones M, Engelkamp H, Claessen V I, Sommerdijk N A, Rowan A E, Christianen P C, Maan J C, Verduin B J, Cornelissen J J, Nolte R J. Nat. Nanotechnol., 2007, 2(10): 635-639
-
[16]
Gorris H H, Walt D R. J. Am. Chem. Soc., 2009, 131(17): 6277-6282
-
[17]
Piwonski H M, Goomanovsky M, Bensimon D, Horovitz A, Haran G. Proc. Natl. Acad. Sci. USA, 2012, 109(22): 1437-1443
-
[18]
Tan W H, Yeung E S. Anal. Chem., 1997, 69(20): 4242-4248
-
[19]
Craig D B, Arriaga E A, Wong J C Y, Lu H, Dovichi N J. J. Am. Chem. Soc., 1996, 118(22): 5245-5253
-
[20]
Chiu D T, Wilson C F, Karlsson A, Danielsson A, Lundqvist A, Stromberg A, Ryttsen F, Davidson M, Nordholm S, Orwar O, Zare R N. Chem. Phys., 1999, 247(1): 133-139
-
[21]
Craig D B, Dovichi N J. Can. J. Chem., 1998, 76: 623-626
-
[22]
Iversen L, Tu H L, Lin W C , Christensen S M, Abel S M, Iwig J, Wu H J, Gureasko J, Rhodes C, Petit R S, Hansen S D, Thill P, Yu C H, Stamou Di, Chakraborty A K, Kuriyan J, Groves J T. Science, 2014, 345(6192): 50-54
-
[23]
Lu Y, Wang W P, Kirschner M W. Science, 2015, 348(6231): 10.1126/science.1248737
-
[24]
Lu Y, Lee B H, King R W, Finley D, Kirschner M W. Science, 2015, 348(6231): 10.1126/science.1250834
-
[25]
Zhuang X, Bartley L E, Babcock H P, Russell R, Ha T, Herschlag D, Chu S. Science, 2000, 288(5473): 2048-2051
-
[26]
Pyle A M, Murphy F L, Cech T R. Nature, 1992, 358(6382): 123-128
-
[27]
Narlikar G J, Herschlag D. Nat. Struct. Biol., 1996, 3(8): 701-710
-
[28]
Zhuang X, Kim H, Pereira M J, Babcock H P, Walter N G, Chu S. Science, 2002, 296(5572): 1473-1476
-
[29]
Rueda D, Bokinsky G, Rhodes M M, Rust M J, Zhuang X, Walter N G. Proc. Natl. Acad. Sci. USA, 2004, 101(27): 10066-10071
-
[30]
Paudel B P, Rueda D. J. Am. Chem. Soc, 2014, 136(48): 16700-16703

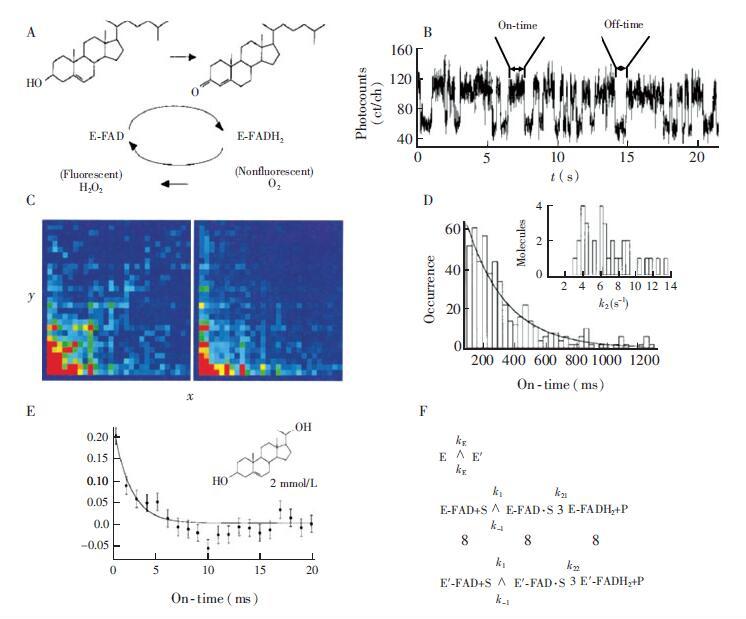
 下载:
下载:
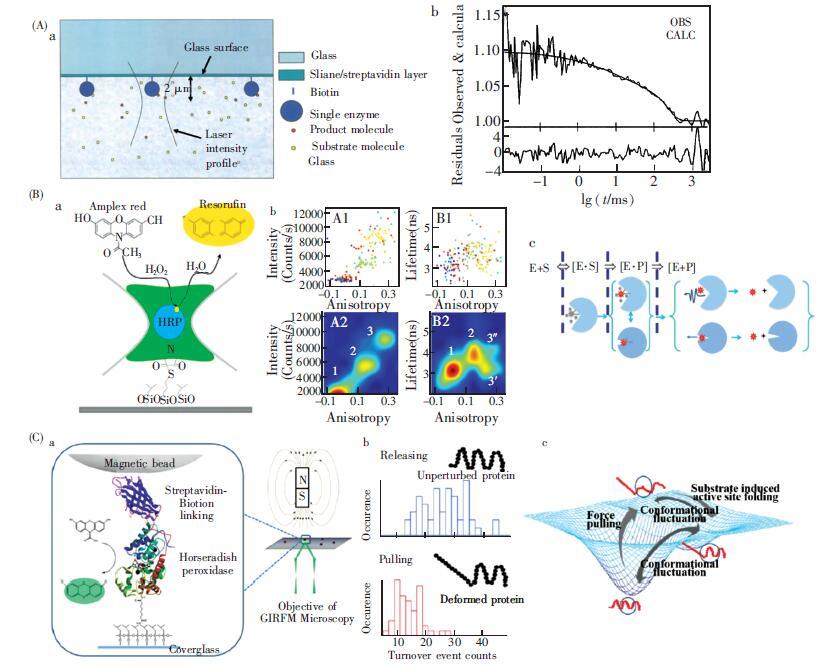
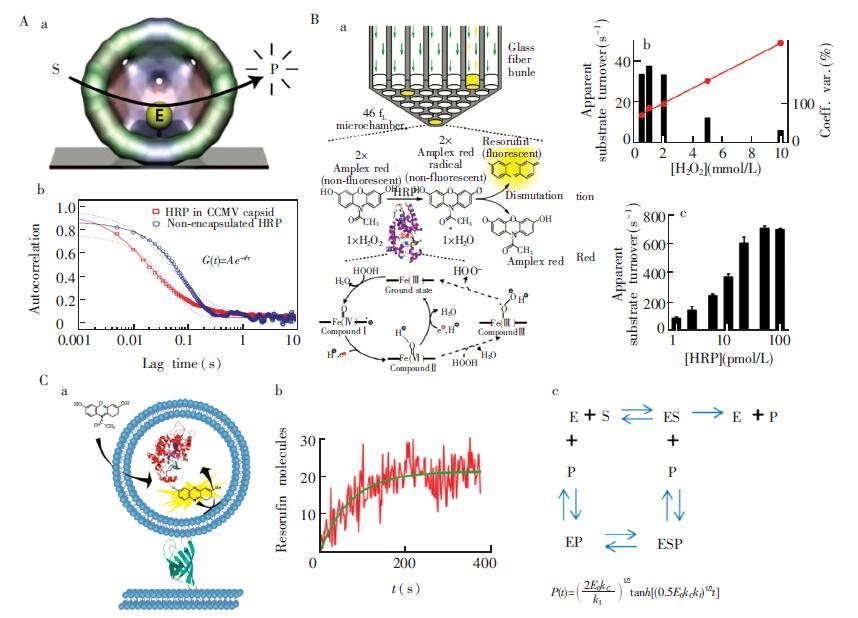
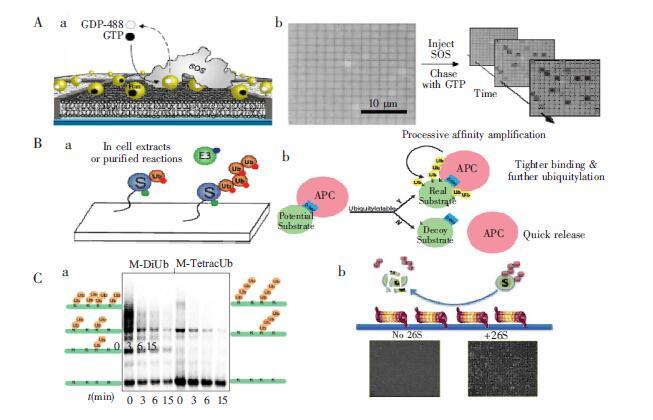

 下载:
下载:






 下载:
下载:
