-
[1]
Dong G C, Duan K F, Zhang Q, Liu Z Z. A New Colorimetric and Fluorescent Chemosensor Based on Schiff Base-Phenyl-Crown Ether for Selective Detection of Al3+ and Fe3+[J]. Inorg. Chim. Acta,
2019, 487:
322-330.
doi: 10.1016/j.ica.2018.12.036
-
[2]
Sun Y, Zhu M Y, Yao Y L, Wang H W, Tong B H, Zhao Z. A Novel Approach for the Selective Extraction of Li+ from the Leaching Solution of Spent Lithium-Ion Batteries Using Benzo-15-Crown-5 Ether as Extractant[J]. Sep. Purif. Technol.,
2020, 237:
116325-116332.
doi: 10.1016/j.seppur.2019.116325
-
[3]
李茹霞, 钟文彬, 谢林华, 谢亚勃, 李建荣. 金属有机框架材料对Cr(Ⅵ)离子的吸附去除研究进展[J]. 无机化学学报,
2021,37,(3): 385-400.
LI R X, ZHONG W B, XIE L H, XIE Y B, LI J R. Recent Advances in Adsorptive Removal of Cr(Ⅵ) Ions by Metal-Organic Frameworks[J]. Chinese J. Inorg. Chem.,
2021, 37(3):
385-400.
-
[4]
田欢, 张梦龙, 王莉莎, 童碧海, 赵卓. 4, 13-二硫杂苯并-18-冠-6的合成及对Ag+的选择性萃取[J]. 高等学校化学学报,
2018,39,(6): 1191-1196.
TIAN H, ZHANG M L, WANG L S, TONG B H, ZHAO Z. Synthesis of 4, 13-Dithio Benzene and-18-Crown-6 and Its Selective Extractability on Ag+[J]. Chem. J. Chinese Universities,
2018, 39(6):
1191-1196.
-
[5]
缪谦, 黄辉, 黄小波, 徐颖, 成义祥. 基于2, 3-萘-18-冠-6单元的共轭高分子对金属离子的荧光传感器[J]. 无机化学学报,
2009,25,(12): 2182-2188.
doi: 10.3321/j.issn:1001-4861.2009.12.018MIAO Q, HUANG H, HUANG X B, XU Y, CHENG Y X. A Fluorescent Chemosensor for Metal Ions Based on the Conjugated Polymer Containing 2, 3-Naphtho-18-Crown-6[J]. Chinese J. Inorg. Chem.,
2009, 25(12):
2182-2188.
doi: 10.3321/j.issn:1001-4861.2009.12.018
-
[6]
Ratch J S, Chivers T. Silicon Analogues of Crown Ethers and Cryptands: A New Chapter in Host-Guest Chemistry? Angew[J]. Chem. Int. Ed.,
2007, 46(25):
4610-4613.
doi: 10.1002/anie.200701822
-
[7]
Ouchi M, Inoue Y, Kanzaki T, Hakushi T. Ring-Contracted Crown Ethers: 14-Crown-5, 17-Crown-6, and Their Sila-Analogues[J]. Drastic Decrease in Cation-Binding Ability. Bull. Chem. Soc. Jpn.,
1984, 57(3):
887-888.
-
[8]
Reuter K, Thiele G, Hafner T, Uhlig F, von Hänisch C. Synthesis and Coordination Ability of a Partially Silicon Based Crown Ether[J]. Chem. Commun.,
2016, 52(90):
13265-13268.
doi: 10.1039/C6CC07520G
-
[9]
Reuter K, Buchner M R, Thiele G, von Hänisch C. Stable Alkali-Metal Complexes of Hybrid Disila-Crown Ethers[J]. Inorg. Chem.,
2016, 55(9):
4441-4447.
doi: 10.1021/acs.inorgchem.6b00267
-
[10]
Dankert F, von Hänisch C. Insights into the Coordination Ability of Siloxanes Employing Partially Silicon Based Crown Ethers: A Comparative Analysis of s-Block Metal Complexes[J]. Inorg. Chem.,
2019, 58(5):
3518-3526.
doi: 10.1021/acs.inorgchem.9b00086
-
[11]
Dankert F, Donsbach C, Rienmüller J, Richter R M, von Hänisch C. Alkaline Earth Metal Template (Cross-) Coupling Reactions with Hybrid Disila-Crown Ether Analogues[J]. Chem. Eur. J.,
2019, 25(69):
15934-15943.
doi: 10.1002/chem.201904209
-
[12]
赵岩, 敖银勇, 陈建, 宋宏涛, 黄玮, 彭静. 基于密度泛函理论研究锂-冠醚配合物的结构和热力学参数[J]. 物理化学学报,
2016,32,(7): 1681-1690.
ZHAO Y, AO Y Y, CHEN J, SONG H T, HUANG W, PENG J. Density Functional Theory Studies of the Structures and Thermodynamic Parameters of Li+-Crown Ether Complexes[J]. Acta Phys.-Chem. Sin.,
2016, 32(7):
1681-1690.
-
[13]
Nametkin N S, Islamov T K, Gusel'nikov L E, Sobtsov A A, Vdovin V M. Gas-Phase Thermal Conversion in a Series of Cyclocarbosiloxanes[J]. Russ. Chem. Bull.,
1971, 20(1):
76-79.
doi: 10.1007/BF00849323
-
[14]
Nametkin N S, Islamov T Kh, Gusel'nikov L E, Vdovin V M. Cyclocarbosiloxanes[J]. Russ. Chem. Rev.,
1972, 41(2):
111-130.
doi: 10.1070/RC1972v041n02ABEH002056
-
[15]
Stephens P J, Devlin F J, Chabalowski C F, Frisch M J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields[J]. J. Phys. Chem.,
1994, 98(45):
11623-1627.
doi: 10.1021/j100096a001
-
[16]
Grimme S, Ehrlich S, Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory[J]. J. Comput. Chem.,
2011, 32(7):
1456-1465.
doi: 10.1002/jcc.21759
-
[17]
Weigend F, Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy[J]. Phys. Chem. Chem. Phys.,
2005, 7(18):
3297-3305.
doi: 10.1039/b508541a
-
[18]
Weigend F. Accurate Coulomb-Fitting Basis Sets for H to Rn[J]. Phys. Chem. Chem. Phys.,
2006, 8(9):
1057-1065.
doi: 10.1039/b515623h
-
[19]
Reed A E, Weinstock R B, Weinhold F. Natural Population Analysis[J]. J. Chem. Phys.,
1985, 83(2):
735-746.
doi: 10.1063/1.449486
-
[20]
Lu T, Chen F W. Multiwfn: A Multifunctional Wavefunction Analyzer[J]. J. Comput. Chem.,
2012, 33(5):
580-592.
doi: 10.1002/jcc.22885
-
[21]
Bader R W F. Atoms in Molecules: A Quantum Theory[J]. New York: Oxford University Press,
1990, :
16-22.
-
[22]
Boys S F, Bernardi F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies-Some Procedures with Reduced Errors[J]. Mol. Phys.,
1970, 19(4):
553-556.
doi: 10.1080/00268977000101561
-
[23]
Marenich A V, Cramer C J, Truhlar D G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions[J]. J. Phys. Chem. B,
2009, 113(18):
6378-6396.
doi: 10.1021/jp810292n
-
[24]
Zhao Y, Schultz N E, Truhlar D G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions[J]. J. Chem. Theory Comput.,
2006, 2(2):
364-382.
doi: 10.1021/ct0502763
-
[25]
Petersson G A, Bennett A, Tensfeldt T G, Al-Laham M A, Shirley W A, Mantzaris J. A Complete Basis Set Model Chemistry. Ⅰ. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Elements[J]. J. Chem. Phys.,
1988, 89(4):
2193-2218.
-
[26]
Petersson G A, Al-Laham M A. A Complete Basis Set Model Chemistry. Ⅱ. Open-Shell Systems and the Total Energies of the First-Row Atoms[J]. J. Chem. Phys.,
1991, 94(9):
6081-6090.
-
[27]
Ho J, Klamt A, Coote M L. Comment on the Correct Use of Continuum Solvent Models[J]. J. Phys. Chem. A,
2010, 114(51):
13442-13444.
doi: 10.1021/jp107136j
-
[28]
Parrish R M, Burns L A, Smith D G A, Simmonett A C, DePrince A E, Hohenstein E G, Bozkaya U, Sokolov A Y, Remigio R D, Richard R M, Gonthier J F, James A M, McAlexander H R, Kumar A, Saitow M, Wang X, Pritchard B P, Verma P, Schaefer H F, Patkowski K, King R A, Valeev E F, Evangelista F A, Turney J M, Crawford T D, Sherrill C D. Psi41. 1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability[J]. J. Chem. Theory Comput.,
2017, 13(7):
3185-3197.
-
[29]
Jeziorski B, Moszynski R, Szalewicz K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes[J]. Chem. Rev.,
1994, 94(7):
1887-1930.
doi: 10.1021/cr00031a008
-
[30]
Dunning T H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. Ⅰ. The Atoms Boron through Neon and Hydrogen[J]. J. Chem. Phys.,
1989, 90(2):
1007-1023.
-
[31]
Dunning T H, Peterson K A, Wilson A K. Gaussian Basis Sets for Use in Correlated Molecular Calculations. Ⅹ. The Atoms Aluminum through Argon Revisited[J]. J. Chem. Phys.,
2001, 114(21):
9244-9253.
-
[32]
Feller D. The Role of Databases in Support of Computational Chemistry Calculations[J]. J. Comput. Chem.,
1996, 17(13):
1571-1586.
doi: 10.1002/(SICI)1096-987X(199610)17:13<1571::AID-JCC9>3.0.CO;2-P
-
[33]
Papajak E, Truhlar D G. Convergent Partially Augmented Basis Sets for Post-Hartree-Fock Calculations of Molecular Properties and Reaction Barrier Heights[J]. J. Chem. Theory Comput.,
2011, 7(1):
10-18.
doi: 10.1021/ct1005533
-
[34]
Schuchardt K L, Didier B T, Elsethagen T, Sun L S, Gurumoorthi V, Chase J, Li J, Windus T L. Basis Set Exchange: A Community Database for Computational Sciences[J]. J. Chem. Inf. Model.,
2007, 47(3):
1045-1052.
doi: 10.1021/ci600510j
-
[35]
Woon D E, Dunning T H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. Ⅲ. The Atoms Aluminum through Argon[J]. J. Chem. Phys.,
1993, 98(2):
1358-1371.
-
[36]
Parker T M, Burns L A, Parrish R M, Ryno A G, Sherrill C D. Levels of Symmetry Adapted Perturbation Theory (SAPT). Ⅰ. Efficiency and Performance for Interaction Energies[J]. J. Chem. Phys.,
2014, 140(9):
094106-1--094106-16.
-
[37]
Boulatov R, Du B, Meyers E A, Shore S G. Two Novel Lithium-15-Crown-5 Complexes: An Extended LiCl Chain Stabilized by Crown Ether and a Dimeric Complex Stabilized by Hydrogen Bonding with Water[J]. Inorg. Chem.,
1999, 38(20):
4554-4558.
doi: 10.1021/ic9904790
-
[38]
Cordero B, Gómez V, Platero-Prats A E, Revés M, Echeverría J, Cremades E, Barragán F, Alvarez S. Covalent Radii Revisited[J]. Dalton Trans.,
2008, (21):
2832-2838.
doi: 10.1039/b801115j
-
[39]
Shannon R D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides[J]. Acta Crystallogr. Sect. A,
1976, A32(5):
751-767.
-
[40]
Guo X J, Zhu Y D, Wei M J, Wu X M, Lü L H, Lu X H. Theoretical Study of Hydration Effects on the Selectivity of 18-Crown-6 Between K+ and Na+[J]. Chinese J. Chem. Eng.,
2011, 19(2):
212-216.
doi: 10.1016/S1004-9541(11)60156-0
-
[41]
Cremer D, Kraka E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy[J]. Croat. Chem. Acta,
1985, 57(6):
1259-1281.
-
[42]
Vener M V, Manaev A V, Egorova A N, Tsirelson V G. QTAIM Study of Strong H-Bonds with the O-H…A Fragment (A=O, N) in Three-Dimensional Periodical Crystals[J]. J. Phys. Chem. A,
2007, 111(6):
1155-1162.
doi: 10.1021/jp067057d
-
[43]
Espinosa E, Alkorta I, Elguero J, Molins E. From Weak to Strong interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X-H…F-Y Systems[J]. J. Chem. Phys.,
2002, 117(12):
5529-5542.
doi: 10.1063/1.1501133
-
[44]
Cameron T S, Decken A, Krossing I, Passmore J, Rautiainen J M, Wang X P, Zeng X Q. Reactions of a CyclodimethyLSiloxane (Me2SiO)6 with Silver Salts of Weakly Coordinating Anions; Crystal Structures of[Ag(Me2SiO)6] [Al] ([Al]=[FAl{OC(CF3)3}3], [Al{OC(CF3)3}4]) and Their Comparison with[Ag(18-Crown-6)]2[SbF6]2[J]. Inorg. Chem.,
2013, 52(6):
3113-3126.
doi: 10.1021/ic3025793

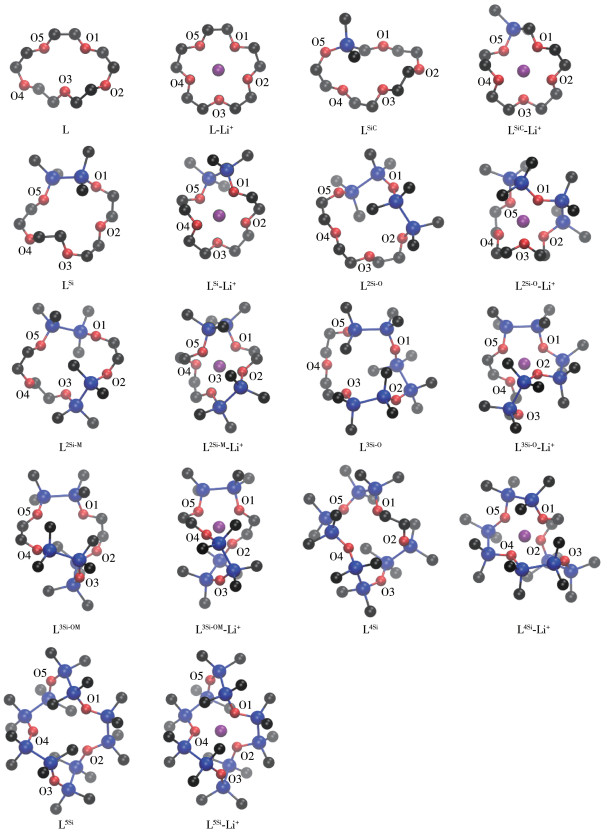
 下载:
下载:
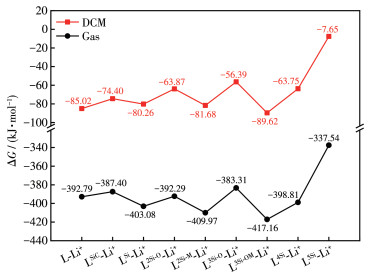

 下载:
下载:



 下载:
下载:
