
图式 1.
CGC型催化剂的结构通式
Scheme 1.
Schematic structure of CGC-type catalysts

1990年美国麻省理工学院的Bercaw课题组报道了一种桥联单茂-胺基的“限制性几何催化剂” (constrained geometric catalyst,CGC)[1],随后Dow公司结合Insite聚合工艺技术,成功地开发了高1-辛烯含量的乙烯/1-辛烯共聚产品,即聚乙烯弹性体[2]。如图式 1所示,CGC型催化剂物种(代表性化合物[Me2Si(η5-C5Me4)(NtBu)]TiCl2[3])一方面有较小的Cp-M- N键角(a通常小于115°),因而b角具有更开放的配位空间,烯烃底物分子易于接近金属中心,相应地提供了乙烯与α-烯烃单体共聚及α-烯烃单体均聚的反应空间;另一方面胺基有较少的价层电子与M键联合作用(通常是2e~4e,较茂环的6e少),因而M能够与空间位阻大的α-烯烃单体有强的电子作用,有效地促进反应的进行[4]。
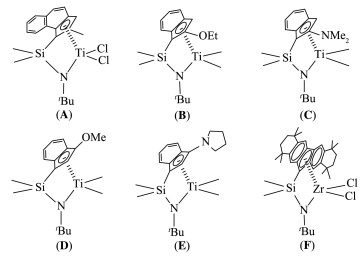
CGC型催化剂呈现高效的催化性能,包括高反应活性、高α-烯烃共聚率和高热稳定性[5]。这些性能受催化剂微观结构的电子和立体空间化学的调控,进而调控目标聚合物的生成。如图式 2所示,Ruckenstein课题组[6]合成了茂基团为苯并茚基的CGC型催化剂(A),催化乙烯/1-辛烯共聚,反应活性高达3.08×107 gproduct·molTi-1·h-1,共聚物中1-辛烯含量达到42.5%,重均分子量为327 000 g·mol-1,且分子量分布小于2。DOW公司的Klosin等[7]合成了烷氧基(B、C)和胺基(D、E)取代的4种CGC型催化剂,并探索了茂环上取代基的改变对乙烯/1-辛烯共聚性能的影响,其中E的催化活性更是达到2.41×109 gproduct·molTi-1·h-1。Miller课题组[8]报道大环芴基(F)催化体系,活性达4.38×108 gproduct·molZr-1·h-1,1-辛烯的共聚含量可以达到30%以上。这些研究表明,CGC型催化剂的反应活性及其对共聚产物微观结构的影响在很大程度上取决于茂环的立体空间和其中的取代基。我们也发现,胺基主要是叔丁胺基。
本文报道一类桥联芴基-芳胺基型受限几何型钛、锆和铪金属催化剂。相比于代表性的CGC型催化剂[Me2Si(η5-C5Me4)(NtBu)]TiCl2,这些化合物中芴基和芳胺基都具有大的立体空间,且芳基与N配位点有共轭效应。本文报道这些化合物的合成和结构表征,也考察这些化合物分别与助剂AliBu3和(Ph3C)+[B(C6F5)4]-组成的催化体系催化乙烯与1-辛烯聚合的反应性能,同时对比研究芴基-芳胺基的电子和空间效应对催化性能的影响。
所有涉及水、氧敏感化合物的实验操作均采用Schlenk技术或在氩气氛手套箱中操作。溶剂四氢呋喃(THF)、甲苯和正己烷经过钠丝预干燥、然后加入钠钾合金/二苯甲酮蒸馏、显示蓝紫色后收集馏分使用。试剂原料二氯二甲基硅烷、叔丁胺、2,6-二异丙基苯胺和2,4,6-三甲基苯胺使用氢化钙除水、重蒸后使用。C6D6经钠钾合金除水后使用,CDCl3则是经氢化钙除水后使用。上述溶剂和试剂以及正丁基锂、甲基锂、三异丁基铝(TIBA)、无水TiCl4、无水ZrCl4、无水HfCl4购买于上海国药和百灵威试剂公司。化合物[Me2Si(C5Me4) (NtBu)]TiCl2[9]、[Me2Si(Ind) (NtBu)]TiCl2[10a]、[Me2Si(Flu)(NtBu)]TiMe2[11]、ZrCl4(THF)2[12]、(Ph3C)+[B(C6F5)4]- [13]按照文献方法制备。
1H(400 MHz)和13C(100 MHz)核磁共振谱在Bruker Avance Ⅱ 400 MHz仪器上测试,测试温度为298 K。用以下符号描述核磁共振信号峰:s(单峰),d (双重峰),t(三重峰),q(四重峰),hept(七重峰),m(多重峰),br(宽峰)。熔点在Büchi B-540型显微熔点仪上测试,红外光谱在Nicolet FT-IR330光谱仪上测试得到,C、H、N元素含量在Vario EL Ⅲ上测试。
准确称取芴(9.97 g,60 mmol)溶于无水乙醚(120 mL)制成溶液。将该溶液冷却至0 ℃,在搅拌下向其中缓慢滴加nBuLi(26.25 mL,2.4 mol·L-1正己烷溶液,63 mmol),溶液变成墨绿色。滴加完毕后自然升至室温,并继续反应6 h。然后再将该反应液冷却至-78 ℃,在搅拌下继续向其中缓慢滴加二氯二甲基硅烷(14.5 mL,0.12 mol),滴加完毕后自然升至室温,并继续反应12 h。反应结束后,过滤除去LiCl和不溶物,滤液在减压下除去溶剂得到无色固体化合物Me2Si[(Flu)H]Cl(11.58 g,产率72.6%)。
准确称取2,6-iPr2C6H3NH2(20 mmol,3.548 g)溶于正己烷(100 mL)制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加nBuLi(8.75 mL,2.4 mol·L-1正己烷溶液,21 mmol)。滴加完毕后自然升至室温,并继续反应6 h。反应结束后,过滤收集白色固体化合物2,6-iPr2C6H3NHLi,用正己烷洗涤、干燥,产量为3.18 g,产率为86.8%。
准确称取上述锂盐(2.748 g,15 mmol)溶于THF (60 mL)中制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加Me2Si[(Flu)H]Cl(3.88 g,15 mmol)的THF溶液。滴加完毕后自然升至室温,并继续反应12 h。反应结束后,过滤除去LiCl和不溶物,滤液在减压下除去溶剂,得到目标配体L1,为淡黄色固体,产量为4.676 g,产率为78%。表征分析结果如下:m.p. 108 ℃;1H NMR(400 MHz,C6D6,298 K):δ 7.78(d,3JHH=7.2 Hz,2H),7.57(d,3JHH=7.6 Hz,2H),7.28(t,3JHH=7.1 Hz,2H),7.23(td,3JHH=7.4,1.3 Hz,2H),7.04(s,3H)(C6H3,C6H4),3.93(s,1H,Flu(H)),3.15(hept,3JHH=6.8 Hz,2H,CHMe2),2.03(s,1H,NH),1.05(d, 3JHH=6.9 Hz, 12H, CH(CH3)2), 0.03(s, 6H, Si(CH3)2);13C{1H} NMR(100 MHz,C6D6,298 K):δ 145.59, 144.94, 141.41, 138.82, 126.66,126.02,124.64,124.46, 123.39, 120.56(C6H3,C6H4), 44.50(Flu(C)),28.30(CHMe2), 23.92 (CH(CH3)2),1.88(Si(CH3)2)。Anal. Calcd. for C27H33NSi (%):C 81.14,H 8.32,N 3.50;Found(%):C 81.39,H 8.56,N 3.63。
合成步骤同配体L1,其中原料由芴替换成2,7-二叔丁基芴。最后得到目标配体L2,为淡黄色固体,产量为4.76 g,产率为62%。表征分析结果如下:m.p. 98 ℃;1H NMR(400 MHz,C6D6,298 K):δ 7.87(d,3JHH=8.0 Hz, 2H), 7.78(s,2H),7.50(d,3JHH=6.4 Hz,2H),7.09(s,3H)(C6H3),4.12(s,1H,Flu(H)),3.16(hept,3JHH= 6.8 Hz, 2H, CHMe2), 1.86(s, 1H, NH), 1.49(s, 18H, C(CH3)3), 1.12(d,3JHH=13.5 Hz,12H,CH(CH3)2),0.20(s,6H,Si (CH3)2);13C{1H} NMR(100 MHz,C6D6,298 K):δ 149.15, 145.23, 145.07, 138.84, 138.49, 123.94, 122.94,122.81, 121.15, 119.48(C6H3), 44.10(Flu(C)), 35.00(CMe3), 31.88(C(CH3)3),28.11(CHMe2), 23.76(CH(CH3)2), 1.62(Si(CH3)2)。Anal. Calcd. for C35H49NSi(%):C 82.13,H 9.65,N 2.74;Found(%):C 82.39,H 9.56,N 2.83。
合成步骤同配体L1,其中原料分别由芴、2,6-二异丙基苯胺替换成2,7-二叔丁基芴、2,4,6-三甲基苯胺。最后得到目标配体L3,为淡黄色固体,产量为5.49 g,产率为76%。表征分析结果如下:m. p. 107 ℃;1H NMR(400 MHz,C6D6,298 K):δ 7.98(d,3JHH =8.1 Hz,2H),7.90(s,2H),7.63(d,3JHH=8.0 Hz,2H),6.99(s, 2H)(C6H2,C6H3), 4.26(s,1H,Flu(H)),2.43(s,3H,p-CH3),2.24(s,6H,o-CH3),2.19(s,1H,NH),1.63(s,18H, C(CH3)3), -0.34(s, Si(CH3)2);13C{1H} NMR(100 MHz, C6D6,298 K):δ 149.08, 145.22, 139.84,138.48,132.50,131.34,129.03,122.82,121.25,119.42(C6H3,C6H3),44.11(Flu(C)),34.98(CMe3),31.88(C(CH3)3),20.70(p-CH3),19.54(o-CH3),-1.28(Si(CH3)2)。Anal. Calcd. for C32H43NSi(%):C 81.81,H 9.23,N 2.98;Found(%):C 81.18,H 9.36,N 2.99。
准确称取配体L1(4 g,10 mmol),溶于正己烷(60 mL)制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加nBuLi(4.5 mL,2.4 mol·L-1正己烷溶液,10.8 mmol),滴加完毕后自然升至室温,并继续反应4 h。反应结束后,过滤收集白色固体锂盐,用正己烷(10 mL×3)洗涤、干燥,产量为3.68 g,产率89%。
将该锂盐(3.29 g,8 mmol)溶于THF(60 mL)制成溶液。在搅拌下将其缓慢滴加至已冷却到-78 ℃的ZrCl4(THF)2(3.02 g,8 mmol)的THF(60 mL)悬浊液中,滴加完毕后自然升至室温,并继续搅拌反应48 h。反应结束后,过滤除去LiCl和不溶物,滤液在减压下除去溶剂。得到的固体用正己烷提取5次,收集正己烷溶液,浓缩,并置于-20 ℃冰箱中。12 h后得到黄色晶体化合物1,产量为3.18 g,产率为48%。表征分析结果如下:m. p. 131.6 ℃;1H NMR(400 MHz,CDCl3,298 K):δ 8.11~8.06(m,4H),7.51(t,3JHH= 7.5 Hz,2H),7.36~7.30(m,2H),7.22(d,3JHH=0.9 Hz,1H),7.20(s,1H) (C6H3,C6H4),3.94(hept,3JHH=6.7 Hz,2H,CHMe2),3.30(s,8H,OC4H8),1.63(s,8H,OC4H8),1.37(d,3JHH=6.7 Hz,12H,CH(CH3)2),0.56(s,6H,Si (CH3)2);13C{1H} NMR(100 MHz,CDCl3,298 K):δ 145.74,143.25,142.55,137.29,126.09,124.89,124.83, 122.20, 119.78(C6H3,C6H4), 73.50(OC4H8),30.22 (Flu(C)), 28.45(CHMe2), 27.12(OC4H8), 25.42(CH(CH3)2),0.33(Si(CH3)2);Anal. Calcd. for C35H47Cl2NO2SiZr(%):C 59.72,H 6.73,N 1.99;Found(%):C 59.80,H 6.64,N 2.03。
准确称取配体L2(5.12 g,10 mmol),溶解于无水乙醚(60 mL)制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加MeLi(26.25 mL,1.6 mol·L-1 Et2O溶液,42 mmol),滴加完毕后自然升至室温,并继续反应4 h。继续将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加TiCl4(1.90 g,10 mmol)的乙醚(20 mL)溶液,滴加完毕后自然升至室温,继续搅拌反应12 h。反应结束后,过滤除去LiCl和不溶物,滤液在减压下除去溶剂,用正己烷溶剂多次洗涤提取,-20 ℃下重结晶,得到红棕色晶体化合物2,然后干燥、称重,产量为3.58 g,产率为61%。表征分析结果如下:m.p. 112.4 ℃;1H NMR(400 MHz,C6D6,298 K):δ 8.00(s,2H),7.86(d,3JHH=8.8 Hz,2H),7.41(dd,3JHH=8.8,1.7 Hz,2H),7.20~6.99(m,5H)(C6H3,C13H6),3.32(hept, 3JHH=6.6 Hz, 2H, CHMe2), 1.33(s, 18H, C(CH3)3), 1.24(d, 3JHH=6.7 Hz, 12H, CH(CH3)2), 0.78(s, 6H, Si(CH3)2),0.12(s,6H,Ti(CH3)2)。13C{1H} NMR(100 MHz,C6D6,298 K):δ 151.12, 144.87, 142.24, 136.81, 126.09, 124.60, 124.41,124.39,123.10(C6H3,C13H6),63.97(Ti(CH3)2),35.30(CHMe3), 31.89(Flu(C)), 31.27(C(CH3)3), 27.59(CHMe2), 26.80(CH(CH3)2), 25.86(CH(CH3)2), 5.02(Si(CH3)2)。Anal. Calcd. for C37H53NSiTi(%):C 75.61,H 9.09,N 2.38;Found(%):C 75.84,H 9.14,N 2.46。
准确称取配体L3(4.69 g,10 mmol),溶解于无水乙醚(60 mL)制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加MeLi(26.25 mL,1.6 mol·L-1 Et2O溶液,42 mmol),滴加完毕后自然升至室温,并继续反应4 h。再将该反应液冷却至-78 ℃,在搅拌下继续向其中缓慢滴加TiCl4(1.90 g,10 mmol)的乙醚(20 mL)溶液,滴加完毕后自然升至室温,并继续反应12 h。反应结束后过滤除去LiCl和不溶物,滤液在减压下除去溶剂,用正己烷溶剂多次洗涤提取,-20 ℃下重结晶,得到红棕色晶体化合物3。然后干燥、称重,产量为3.09 g,产率为57%。表征分析结果如下:m.p. 134 ℃;1H NMR(400 MHz,C6D6,298 K):δ 8.09(d,3JHH=8.8 Hz,2H),7.84(s,2H),7.53 (d,3JHH=8.8,1.7 Hz,2H),6.84(s,2H)(C6H2,C13H6),2.36 (s, 3H,p-CH3), 2.01(s, 6H,o-CH3),1.37(s,18H,C(CH3)3),0.78(s,Si(CH3)2),-0.32(s,Ti(CH3)2);13C{1H} NMR(100 MHz,C6D6,298 K):δ 150.99,145.01,136.97,132.09,130.90,129.18,124.14,123.90,122.63(C6H3,C13H6),62.27(Ti(CH3)2),35.45(Flu(C)),35.34(CHMe3),31.29(C (CH3)3),20.74(p - CH3),19.18(o - CH3),4.77(Si(CH3)2)。Anal. Calcd. for C34H47NSiTi(%):C 74.83,H 8.68,N 2.57;Found(%):C 75.06,H 8.74,N 2.63。
准确称取配体L2(5.12 g,10 mmol),溶解于无水乙醚(60 mL)制成溶液。将该溶液冷却至-78 ℃,在搅拌下向其中缓慢滴加MeLi(26.25 mL,1.6 mol·L-1 Et2O溶液,42 mmol),滴加完毕后自然升至室温,并继续反应4 h。再将该反应液冷却至-78 ℃,在搅拌下继续向其中缓慢滴加HfCl4(3.20 g,10 mmol)的甲苯(40 mL)悬浊液,滴加完毕后自然升至室温,并继续反应12 h。反应结束后,过滤除去LiCl和不溶物,滤液在减压下除去溶剂,得到橙色固体化合物4,用正己烷洗涤,然后干燥、称重,产量为2.94 g,产率为41%。表征分析结果如下:1H NMR(400 MHz,C6D6,298 K):δ 7.79(d,3JHH=8.1 Hz,2H),7.68(s,2H),7.42 (dd,3JHH=8.8,1.7 Hz,2H),7.01(s,3H) (C6H3,C13H6),3.07(hept, 3JHH=6.8 Hz, 2H, CHMe2), 2.00(s, 3H, Hf(CH3)2), 1.88(s, 3H,Hf(CH3)2),1.40(s,18H,C(CH3)3),1.04(d,3JHH =6.9 Hz,12H,CH(CH3)2),0.10(s,6H,Si(CH3)2)。Anal. Calcd. for C37H53NSiHf(%):C 61.86,H 7.44,N 1.95;Found(%):C 61.96,H 7.51,N 1.93。
催化剂的衍射数据在Oxford Gemini S Ultra单晶X射线衍射仪上收集。在100(2) K温度下,用石墨单色器单色化的Cu Kα(λ=0.154 178 nm)射线,采用ω-2θ扫描方式收集衍射数据。分子结构中的非氢原子采用直接法由SHELX-97程序[14-15]解出,氢原子由理论计算模型加上。晶体结构数据和精修参数如表 1所示(其中的化合物5有可能来源于合成4的副反应,可以反映4的部分特征)。
 下载:
导出CSV
下载:
导出CSV
| Compound | 1 | 2 | 3 | 5 |
| Formula | C35H47ZrCl2NSiO2 | C37H53TiNSi | C34H47TiNSi | C44H73HfLiNO2Si2 |
| Formula weight | 703.95 | 587.79 | 545.69 | 889.67 |
| Cryst system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/n | Cc | P21/n |
| a / nm | 1.044 330(10) | 1.699 490(10) | 2.131 93(3) | 1.346 04(2) |
| b / nm | 2.218 65(2) | 1.728 08(2) | 1.517 49(2) | 1.387 84(2) |
| c / nm | 1.520 050(10) | 2.464 15(4) | 3.839 87(4) | 1.423 74(10) |
| α / (°) | 100.319 0(10) | 103.85(10) | ||
| β / (°) | 96.761 0(10) | 104.502 0(10) | 99.232 0(10) | 110.56(10) |
| γ / (°) | 96.318 0(10) | 104.513 0(10) | ||
| V / nm3 | 3.497 47(5) | 6.801 18(10) | 12.26 18(10) | 2.248 06(5) |
| Z | 4 | 8 | 16 | 2 |
| Dc / (g·cm-3) | 1.337 | 1.148 0 | 1.182 | 1.832 |
| μ / mm-1 | 4.541 | 2.635 | 2.888 | 9.504 |
| F(000) | 1 472 | 2 544 | 4 704 | 1 245 |
| Crystal size / mm | 0.20×0.10×0.10 | 0.20×0.10×0.10 | 0.42×0.34×0.28 | 0.18×0.17×0.16 |
| θ range / (°) | 3.54~77.19 | 2.72~77.29 | 3.59~70.00 | 3.528~77.212 |
| Index ranges | -12 ≤ h ≤ 13 | -21 ≤ h ≤ 18 | -25 ≤ h ≤ 25 | -16 ≤ h ≤ 17 |
| -18 ≤ k ≤ 26 | -20 ≤ k ≤ 21 | -7 ≤ k ≤ 18 | -17 ≤ k ≤ 14 | |
| -18 ≤ l ≤ 17 | -29 ≤ l ≤ 31 | -45 ≤ l ≤ 46 | -14 ≤ l ≤ 18 | |
| Collected data | 25 458 | 92 320 | 38 619 | 30 336 |
| Unique data | 6 970 (Rint=0.037 5) | 27 864 (Rint=0.050 5) | 17 938 (Rint=0.025 0) | 9 183 (Rint=0.056 5) |
| Completeness to θ / % | 94.1 | 96.6 | 97.5 | 99.8 |
| Data, restraint, parameter | 6 970, 0, 386 | 27 864, 72, 1 559 | 17 938, 2, 1 381 | 9 183, 0, 480 |
| GOF on F2 | 0.986 | 1.013 | 0.943 | 1.073 |
| Final R indices [I>2σ(I)] | R1=0.032 0 | R1=0.056 5 | R1=0.033 0 | R1=0.038 3 |
| wR2=0.082 1 | wR2=0.153 7 | wR2=0.107 9 | wR2=0.101 1 | |
| R indices* (all data) | R1=0.039 1 | R1=0.068 1 | R1=0.051 2 | R1=0.044 8 |
| wR2=0.085 8 | wR2=0.168 1 | wR2=0.115 6 | wR2=0.107 9 | |
| Largest diff. peak and hole / (e·nm-3) | 746, -752 | 2 148, -543 | 342, -420 | 1 171, -1 586 |
| *R1=∑(||Fo|-|Fc||)/∑|Fo|, wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)]1/2, GOF=[∑w(Fo2-Fc2)2/(No-Np)]1/2. | ||||
CCDC:1975179,1;1975180,2;1986175,3;1986178,5。
将300 mL带有搅拌器的Parr不锈钢间歇釜在100 ℃烘箱中干燥6 h,转移至手套箱中,依次加入甲苯溶剂、TIBA、共聚单体1-辛烯、硼助剂、以及茂金属催化剂,密封后转移出手套箱,与加热搅拌装置以及乙烯钢瓶连接。打开搅拌器,调至指定转速;打开加热器,调至指定反应温度;打开乙烯气体阀,调至指定压力值。控制反应时间,完成聚合反应。聚合反应液用HCl/EtOH终止反应。过滤收集聚合物,用去离子水洗,真空干燥至恒重。
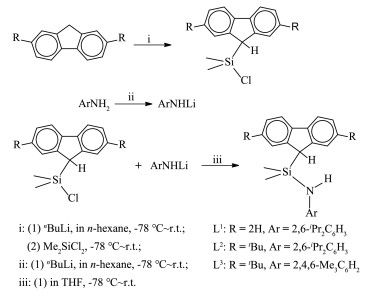
以二甲基硅二氯(Me2SiCl2)为前驱体合成二甲基硅基桥联单茂-胺基配体通常有2条路径,一种是先键联茂基再接上胺基,另一种在键联上胺基后再接茂基。键联胺基通常有2种方法,即以至少过量一倍的胺作为HCl分子的配位剂,同时脱去Me2SiCl2中的一个Cl基和伯胺中的一个H基;或者将伯胺与脱质子金属有机试剂成盐,再与Me2SiCl2发生盐消除反应。键联茂基只能采用金属有机试剂脱质子成盐、再与Me2SiCl2盐消除反应法,因为茂基质子的解离常数很小(pKa≈24)[16],胺配位脱HCl法往往无法进行或进行不完全。
我们选择2,6-二异丙基苯胺和2,4,6-三甲基苯胺为伯胺源,芴基和2,7-二叔丁基芴基为茂源,进行实验合成。我们发现,先键接胺基容易形成混合物,可能是由于Me2SiCl2中的2个Cl发生单个和双个取代,因此我们采用先键接茂基再键联胺基的方法。如图式 3所示,使用等物质的量的正丁基锂与茂环化合物在-78 ℃至室温下发生脱质子反应,生成茂基锂盐;然后再在-78 ℃至室温下与等物质的量的Me2SiCl2中发生脱LiCl的盐消除反应生成Me2Si[(2,7-R2Flu)H]Cl(R=H或tBu)备用。值得注意的是,我们成功地得到了完全单取代的产物,这可能是因为芴基有较大的空间位阻,阻止了另一个芴基的键接。进一步键接胺基时,我们发现采用过量胺脱除HCl的动力学转化不可取,因为得到的产物体系复杂,无法分离目标物。因此我们转而采用LiCl盐消除法。在该法中,当采用“一锅”合成时,即不将等物质的量的正丁基锂与芳胺脱质子反应形成的芳胺基锂从溶液中分离出来,而是直接与Me2Si [(2,7-R2Flu)H]Cl进行LiCl盐消除反应,但是最后得到的配体产率较低(< 20%)。于是我们将得到的芳胺基锂盐分离出来,再与等物质的量的Me2Si[(2,7- R2Flu)H]Cl反应,目标配体的生成产率得到很大提高。
由此,依照图式 3的合成路线,我们分别高产率地得到配体Me2Si[(Flu)H]NH-2,6-iPr2C6H3(L1,淡黄色固体, 78%产率)、Me2Si[2, 7-tBu2Flu(H)]NH-2, 6-iPr2C6H3 (L2,淡黄色固体,62%产率)和Me2Si[2,7-tBu2Flu(H)] NH-2,4,6-Me3C6H2(L3,淡黄色固体,76%产率)。对这3个化合物进行了熔点、1H和13C NMR和元素分析,其中特征基团中Me2Si桥基Me质子的化学位移分别位于δMe=0.03(L1)、δMe=0.20(L2)、δMe=-0.34(L3),与该桥基键联的芴基五元环中CH基团的质子和碳的化学位移分别为δH=3.93和δC=44.50(L1)、δH=4.12和δC=44.10(L2)、δH=4.26和δC=44.1(L3),同时NH基质子的化学位移分别为δH=2.03(L1)、δH=1.86(L2)、δH=2.19 (L3)。此外,L1和L2中的2,6-iPr2C6H3基团可以由异丙基的特征化学位移确认(δCH(CH3)2=3.15(L1)、δCH(CH3)2= 3.16(L2)和δCH(CH3)2=1.05(L1)、δCH(CH3)2=1.12(L2)),且L2中芴基上tBu取代基的化学位移在δH=1.49。在L3中,2,4,6-Me3C6H2基团通过2组Me的共振吸收指认(δp-Me=2.43和δo-Me=2.24),芴基上tBu取代基的化学位移在δH=1.63。
我们采用2种不同的方法分别合成桥联单茂-芳胺基金属氯化物和金属甲基化物。化合物[Me2Si (Flu)(N-2,6-iPr2C6H3)]ZrCl2(THF)2 (1)的合成如图式 4所示,将L1溶于正己烷溶剂中,-78 ℃至室温下用2倍物质的量的正丁基锂分别脱去芴基和芳胺基上的质子,得到双锂盐,以89%的产率分离。称取8 mmol量的双锂盐,溶于THF溶剂,然后逐滴加入到冷却至-78 ℃的等物质的量的ZrCl4(THF)2的THF悬浊液中,经升至室温和48 h的搅拌完成反应,再经过滤除去LiCl、脱除THF、正己烷溶剂多次提取、-20 ℃下重结晶,得到黄色晶体化合物1,产率为48%。
我们对化合物1进行了1H和13C NMR、熔点、元素分析的表征。1H NMR谱显示芴基五元环中CH和芳胺基NH基团的质子化学位移消失,13C NMR谱显示该碳原子的δC=30.22,化学位移向高场移动近14,表明该C原子与金属键联,显现部分C中心的电子云密度向金属中心迁移的结果。同时,THF分子的共振吸收峰在δC4H8O为3.30和1.63处,与游离的THF分子相比(δC4H8O=3.76、1.85),化学位移明显向高场偏移,这表明THF是配位态。
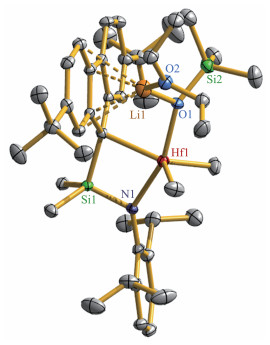
化合物1的结构经X射线单晶衍射表征,分子结构示于图 1。图中可以看到金属Zr与芴基中五元环的独特的键合作用,其中Zr(1) - C(1)的键长为0.235 6(2) nm,明显短于Zr(1)与其它4个碳原子的距离(0.310 0(1)、0.326 8(1)、0.406 5(1)和0.414 4(1) nm)。在报道的化合物[Me2Si(Flu) (NtBu)]ZrR2(R=Cl[17]、CH2SiMe3[18])中,Zr与芴基上环戊二烯环的5个碳原子之间的距离在0.237 9(3)~0.270 8(3) nm范围,可认为是η5配位模式。其它系列η5配位模式化合物[Me2Si(2,7-tBu2Flu) (NtBu)]ZrCl2 (0.237 7(7)~ 0.263 8(8) nm) [19]、{Me2Si[3-Et-6,7-(CMe2CH2)2Flu](NtBu)}ZrCl2 (0.237 0(2)~0.263 5(2) nm)、{Me2Si[3-tBu-6,7-(CMe2CH2)2Flu] (NtBu)}ZrCl2 (0.236 82(18)~ 0.261 93(17) nm)、{Me2Si[6,7-(CMe2CH2)2Flu] (NtBu)} ZrMe2(0.239 97(13)~0.266 687(13) nm)、{Ph2Si[3,4- (CMe2CH2)2-6,7-(CMe2CH2)2Flu](NtBu)}ZrCl2 (0.237 2(6)~ 0.262 7(7) nm)[20]也显示类似的Zr-CFlu键长。由此,我们认为1中的芴基与Zr应该是η1配位模式,与Alt课题组报道的化合物(η5-C5H4Me)2Zr(η1-Flu)Cl[21]相近。基于这样的键合构型,Zr中心被额外的2个THF溶剂分子配位,Zr-O键长分别为0.230 79(15)和0.230 80(14) nm。这说明Zr中心在η1-Flu键合下显现强的Lewis酸性。值得注意的是,1中的Zr-η1-Flu键合方式明显与上述报道的其它CGC型的Zr-η5-Flu方式不同,可以推测是由于芳胺基N-2,6-iPr2C6H3与NtBu基相比有较大的旋转空间,且与N端电子共轭,在电子和立体空间方面对Zr中心与其它基团的键作用方面产生了很大程度的影响。
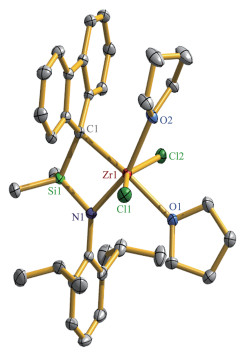
Thermal ellipsoids are at 50% probability and hydrogen atoms are omit‑ ted for clarity; Selected bond lengths (nm) and angles (°): Zr(1) ‑ N(1) 0.204 67(17), Zr(1)‑O(1) 0.230 79(15), Zr(1)‑O(2) 0.230 80(14), Zr(1)‑ C(1) 0.235 6(2), Zr(1)‑Cl(1) 0.243 82(5), Zr(1)‑Cl(2) 0.244 71(5), Si(1)‑ N(1) 0.176 18(18), Si(1)‑C(1) 0.186 6(2); Si(1)‑N(1)‑Zr(1) 99.60(8), N(1)‑ Zr(1)‑C(1) 75.92(7)
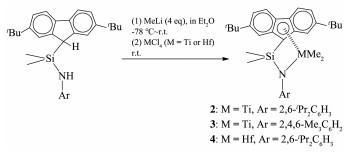
化合物2~4的合成如图式 5所示。配体L2在无水乙醚溶剂中-78 ℃下与4倍物质的量的甲基锂混合,在升至室温的过程中分别脱去芴基和芳胺基质子,形成L2的双锂盐和未反应甲基锂的混合物。在再冷却至-78 ℃时,缓慢滴加一倍物质的量的TiCl4乙醚溶液,升至室温并继续搅拌12 h完成该反应。过滤除去LiCl、减压除去溶剂、正己烷提取以及-20 ℃下重结晶,得到红棕色晶体化合物[Me2Si(2,7-tBu2Flu)(N-2,6-iPr2C6H3)]TiMe2(2,产率为61%)。采用类似的方法使用配体L3得到红棕色晶体化合物[Me2Si(2,7-tBu2Flu)(N-2,4,6-Me3C6H2)]TiMe2(3,产率为57%)。这种方法与化合物1的合成不同,其中甲基锂既是脱质子试剂也是甲基化试剂,甲基化反应是指预生成的[Me2Si(2,7-tBu2Flu)(N-2,6-iPr2C6H3)] TiCl2进一步和未反应的MeLi发生LiCl盐消除反应给出目标物。所有这些反应能够在“一锅”中进行。化合物2和3均通过1H和13C NMR、熔点、元素分析以及X射线单晶衍射进行表征。1H和13C{1H} NMR谱清楚地显示芴基上tBu(对2:δH=1.33、δC(CH3)3= 35.30、δC(CH3)3=31.27;对3:δH=1.37、δC(CH3)3=35.34、δC(CH3)3=31.29)、Si(CH3)2(对2:δH=0.78、δCH3=5.02;对3:δH=0.78、δCH3=4.77)和Ti(CH3)2(对2:δH=0.12、δCH3 = 63.97;对3:δH=-0.32、δCH3 =62.27)基团的共振吸收信号;同时13C{1H} NMR谱中位于δC=31.89(2)和δC= 35.45(3)处的共振吸收可指认为芴基五元环中较短Ti-C键中的C。这些基团的化学位移与类似化合物[Me2Si(2,7-tBu2Flu)(N-1-adamantyl)]TiMe2中的相应基团相当(对tBu:δH=1.37、δC(CH3)3=35.3、δC(CH3)3=31.4;对Si(CH3)2:δH=0.85、δCH3 =6.5;对Ti(CH3)2:δH=-0.47、δCH3 =54.9;对CTi-Flu(the shortest one):δC=59.8)[22]。2中芳胺基N-2,6-iPr2C6H3由异丙基的特征化学位移δCH(CH3)2=3.32和δCH(CH3)2=1.24确认;3中N-2,4,6-Me3C6H2则是基于2组CH3的共振吸收δp-CH3=2.36和δo-CH3=2.01确认。
化合物2和3的单晶结构解析(图 2和3)确认了核磁共振谱学给出的结果。可以明显地看出,Ti与芴基上的五元环呈典型的η5-键联,对于2,Ti-C键长为0.226 7(2)、0.237 5(2)、0.239 1(2)、0.252 0(2)和0.253 8(2) nm;对3则为0.229 0(3)、0.240 4(4)、0.242 3(3)、0.250 4(4)和0.253 1(4) nm。这些键参数与已经报道的化合物[Me2Si(Flu) (NtBu)]TiMe2 (0.225 3(3)~0.257 3(3) nm)[11]、[Me2Si(Flu)(N-1-adamantyl)] TiMe2 (0.224 1(4)~0.264 3(4) nm) [23]和[Me2Si(3, 6- tBu2Flu) (NtBu)]TiMe2(0.224 7(3)~0.261 9(3) nm) [24]相当。化合物2中的Ti-N键长是0.193 16(17) nm,与3中的0.193 6(3) nm相近,但是较[Me2Si(Flu) (NtBu)]TiMe2 (0.192 3(3) nm) [11]稍长。此外,茂环中心与Ti以及N原子形成的Flu(centroid) - Ti - N键角为110.399(1)°(2)和109.575(1)°(3),比[Me2Si(Flu)(NtBu)]TiMe2 (101.0(1)°)中的大。这些数据表明芳胺基的键联与NtBu的键联相比,键参数有一定程度的差异。
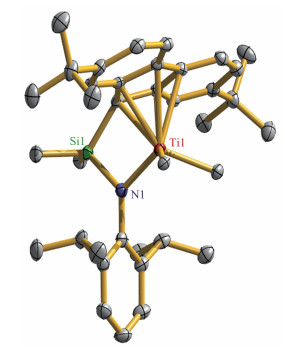
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Ti(1) ‑ N(1) 0.193 16(17), Ti(1)‑C(1) 0.226 7(2), Ti(1)‑C(2) 0.237 5(2), Ti(1)‑ C(3) 0.239 1(2), Ti(1) ‑ C(4) 0.252 0(2), Ti(1) ‑ C(5) 0.253 8(2), Ti(1) ‑ C(24) 0.214 1(2), Ti(1)‑C(25) 0.210 2(2), Si(1)‑N(1) 0.175 17(19), Si(1)‑ C(1) 0.187 0(2); Si(1) ‑ N(1) ‑ Ti(1) 101.96(9), Cp(centroid) ‑ Ti(1) ‑ N(1) 110.399(1)

Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Ti(1)‑N(1) 0.193 6(3), Ti(1)‑C(1) 0.229 0(3), Ti(1)‑C(2) 0.240 4(4), Ti(1)‑C(3) 0.242 3(3), Ti(1)‑C(4) 0.250 4(4), Ti(1)‑C(5) 0.253 1(4), Ti(1)‑C(22) 0.208 9(4), Ti(1)‑C(23) 0.210 1(4), Si(1)‑N(1) 0.174 1(3), Si(1)‑C(1) 0.189 4(3); Si(1)‑N(1)‑Ti(1) 102.59(16), Flu(centroid)‑ Ti(1)‑N(1) 109.575(1)
使用配体L2和HfCl4并按照上述2和3的合成方法,我们得到了橙色固体化合物[Me2Si(2,7-tBu2Flu)- (N-2,6-iPr2C6H3)]HfMe2(4,产率为41%),1H NMR谱显示其特征基团的共振吸收峰分别是δ=3.07(CHMe2)、2.00和1.88(Hf(CH3)2)、1.40(C(CH3)3)、1.04(CH(CH3)2)和0.10(Si(CH3)2),但是13C NMR谱显示其含有少量的杂质。我们试图培养单晶进行X射线衍射分子结构确认,没有成功。但是在重复合成中,我们得到了少量的晶体,经X射线衍射测试和结构解析,是化合物[Me2Si(2,7-tBu2Flu) (N-2,6-iPr2C6H3)]HfMe2·LiO-SiMe3(OEt2)(5,痕量,图 4),推测是副反应所致。尽管化合物5是副反应的产物,但是它在一定程度上显现4的部分结构特征。在此仅将化合物5的结构以及相关键参数给出,不做讨论。
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Hf(1)‑N(1) 0.213 4(3), Hf(1)‑C(1) 0.23 71(3), Hf(1)‑C(24) 0.221 5(4), Hf(1)‑C(25) 0.221 8(4), Si(1)‑N(1) 0.173 5(3), Si(1)‑C(1) 0.186 7(3); Si(1)‑N(1)‑Hf(1) 99.59(13), C(7)‑Hf(1)‑N(1) 73.76(11)
以1/AliBu3/(Ph3C)+[B(C6F5)4]-为催化体系,考察了温度、乙烯压力、1-辛烯浓度、催化剂浓度、三异丁基铝量对催化性能的影响。在催化剂用量5 μmol、nAl/ nZr=300、nB/nZr=1.46、甲苯溶剂150 mL、乙烯压力1.4×103 kPa、1-辛烯浓度0.80 mol·L-1、时间10 min的条件下,当温度分别设定于90、100、110、120 ℃时,催化活性在4.94×105~1.055×106 gproduct·molZr-1·h-1内变化(表 2,Entry 1~4),其中在100 ℃时达到峰值,而120 ℃时显现最低活性。所得聚合物的GPC测试显示,重均分子量Mw在90和100 ℃时分别达到2.599× 105和1.978×105 g·mol-1,分子量分布(PDI)为13.96和16.24;而升温到110和120 ℃时则降低至1.209×105和1.431×105 g·mol-1,PDI为6.20和5.95。温度升高通常会提升聚合速率,提高反应活性,但是温度升高也易于导致聚合增长链的β-H消除反应[25]。由此可以看出,该催化剂体系显示接近中等的催化活性,但是容易发生β-H消除反应,温度升高,消除反应速率提升,并接近于一定的值,而分子量分布随着温度的提升变窄。DSC结果显示聚合物的熔融温度Tm在124.35~126.73 ℃之间,但是最大结晶度Cm在90和100 ℃时为45.85%和37.51%,而升温到110和120 ℃时则变为11.85%和14.88%。我们在相同条件下考察了仅乙烯在110 ℃时的聚合反应(Entry 5),活性只有4.33×105 gproduct·molZr-1·h-1。值得注意的是,Tm为134.14 ℃和Cm是45.63%,这是常规聚乙烯的物性数值[26]。因此,可以推测该催化体系在达到100 ℃时开始显现共聚反应效果,100~120 ℃时较佳;同时在此温度内聚合物呈现较低的PDI,这符合茂金属催化剂体系的单活性中心作用机理[27]。
下载:
导出CSV
| Entry | Catalyst | nAl/nM | V1-octene / mL | mB co-catb / mg | T / ℃ | mpolymer / g | Activity /(g·mol-1·h-1) | Mwc /(g·mol-1) | PDIc | Cmd / % | Tmd / ℃ |
| 1 | 1 | 300 | 18.6 | 4.9 | 90 | 0.756 | 9.07×105 | 2.599×105 | 13.96 | 45.85 | 125.72 |
| 2 | 1 | 300 | 18.6 | 4.9 | 100 | 0.879 | 1.055×106 | 1.978×105 | 16.24 | 37.51 | 126.73 |
| 3 | 1 | 300 | 18.6 | 4.9 | 110 | 0.573 | 6.87×105 | 1.209×105 | 6.20 | 11.85 | 124.35 |
| 4 | 1 | 300 | 18.6 | 4.9 | 120 | 0.412 | 4.94×105 | 1.431×105 | 5.95 | 14.88 | 126.23 |
| 5 | 1 | 300 | 0 | 4.9 | 110 | 0.361 | 4.33×105 | 8.063×105 | 31.02 | 45.63 | 134.14 |
| 6 | 1 | 300 | 3.1 | 4.9 | 110 | 0.449 | 5.39×105 | 2.940×105 | 22.39 | 48.76 | 128.72 |
| 7 | 1 | 300 | 6.2 | 4.9 | 110 | 0.545 | 6.54×105 | 2.886×105 | 17.27 | 26.95 | 129.43 |
| 8 | 1 | 300 | 9.3 | 4.9 | 110 | 0.733 | 8.79×105 | 1.145×105 | 4.55 | 11.37 | 123.02 |
| 9 | 1 | 300 | 27.9 | 4.9 | 110 | 0.449 | 5.39×105 | 1.018×105 | 6.79 | 10.75 | 121.91 |
| 10 | 1 | 200 | 18.6 | 4.9 | 110 | 0.561 | 6.73×105 | 1.425×105 | 6.43 | 11.90 | 123.43 |
| 11 | 1 | 600 | 18.6 | 4.9 | 110 | 1.003 | 1.204×106 | 1.539×105 | 5.02 | 10.56 | 122.57 |
| 12 | 1 | 1 000 | 18.6 | 4.9 | 110 | 0.892 | 1.070×106 | 9.29×104 | 15.74 | 7.57 | 111.44 |
| 13 | 1 | 1 400 | 18.6 | 4.9 | 110 | 0.818 | 9.82×105 | no detected | no detected | 5.22 | 99.43 |
| 14 | 2 | 300 | 18.6 | 4.9 | 110 | 3.477 | 4.172×106 | 1.667×105 | 4.63 | 6.16 | 115.42 |
| 15 | 3 | 300 | 18.6 | 4.9 | 110 | 3.312 | 3.974×106 | 1.432×105 | 4.51 | 5.98 | 114.81 |
| 16 | 4 | 300 | 18.6 | 4.9 | 110 | 0.910 | 1.092×106 | 1.821×105 | 6.74 | 6.78 | 113.75 |
| 17 | Ⅰ | 300 | 18.6 | 4.9 | 110 | 10.900 | 1.3080×107 | 1.482×105 | 3.27 | 3.19 | 72.46 |
| 18 | Ⅱ | 300 | 18.6 | 4.9 | 110 | 8.700 | 1.0440×107 | 1.455×105 | 3.01 | 1.29 | 60.04 |
| 19 | Ⅲ | 300 | 18.6 | 4.9 | 110 | 7.876 | 9.551×106 | 1.843×105 | 7.65 | 7.72 | 115.58 |
| aPolymerization conditions: catalyst 5.0 μmol; AliBu3 1.5 mmol (based on nAl/nM of 300); (CPh3)+[B(C6F5)4]- 7.30 μmol (based on nB/nM of 1.46); ethylene pressure 1.4×103 kPa; toluene 80 mL; time 10 min; b B co-cat: (CPh3)+[B(C6F5)4]-; c Determined by GPC; d Determined by DSC; Ⅰ=[Me2Si(C5Me4)(NtBu)]TiCl2; Ⅱ=[Me2Si(Ind)(NtBu)]TiCl2; Ⅲ=[Me2Si(Flu)(NtBu)]TiMe2. | |||||||||||
我们继续考察1-辛烯浓度的变化对催化性能的影响。在上述条件下并保持温度110 ℃,随着1-辛烯浓度的增加(0.13~1.20 mol·L-1,Entry 3和6~9),催化活性都在变化,并在0.40 mol·L-1时到达最高(8.79×105 gproduct·molZr-1·h-1);在0.40~1.20 mol·L-1间,聚合物的PDI稳定在较低水平(10.18~12.09),Tm在121.91~124.35 ℃之间和Cm在10.75%~11.85%范围内。在一定温度下乙烯和1-辛烯的竞聚率通常保持不变,但是1-辛烯的共聚含量受1-辛烯浓度的影响(一定乙烯压力表明乙烯浓度恒定)。这些数据表明该催化剂体系的反应活性受1-辛烯的浓度影响,这显示了共单体的聚合效应[28]。
改变三异丁基铝的浓度也同样对催化反应有影响(Entry 3和10~13)。同样按照上述条件并保持温度110 ℃和1-辛烯浓度0.80 mol·L-1,当nAl/nZr降低至200时,催化活性略有降低(6.73×105 gproduct·molZr-1· h-1),继续降低至100时活性几乎丧失;而提高至600时,催化活性增至最高(1.204×106 gproduct·molZr-1·h-1),再增加到1 000甚至1 400时,催化活性开始逐渐降低。我们注意到,在nAl/nZr=1 000时,重均分子量变低(Mw=9.29×104 g·mol-1)且分子量分布变得很宽(PDI =15.74);到1 400时,已测不出Mw和PDI。三异丁基铝在体系中兼具除杂和激活产生活性物种的作用,量的增加对反应起促进作用,但是过量时会变成链转移剂,引发聚合链的迁移[29]。上述数据显示了这样的结果。
总体上,化合物1的催化活性显示接近中等的催化水平。文献报道茂金属锆化合物催化剂体系[Me2Si(C5Me4)(NtBu)]ZrCl2/MMAO/(Ph3C)+[B(C6F5)4]- (MMAO为改性甲基铝氧烷)在乙烯压力3×102 kPa、环己烷溶剂和10 min时间的条件下,80 ℃时的催化活性为9.9×107 gproduct·molZr-1·h-1,1 -辛烯的共聚含量仅2.20%;140 ℃时催化活性降为7.7×107 gproduct· molZr-1·h-1。而[Me2Si(3,4-Me2C5H2)(NtBu)]ZrCl2/MMAO/(Ph3C)+[B(C6F5)4]-的催化活性更低,分别是2.6×107 (80 ℃)和8×106 gproduct·molZr-1·h-1 [30]。但是桥联并噻吩茚环并苯基含氮杂环茂金属锆化合物(2,3,5,6- Me4C7S-8-R-C9H5N)ZrCl2/MAO/(Ph3C)+[B(C6F5)4]-(MAO为甲基铝氧烷,R为Me或H)在4×102 kPa乙烯压力、甲苯溶剂和80 ℃下的催化活性显示无活性[31]。尽管如此,{Me2Si[3, 4-(CMe2CH2)2-6,7-(CMe2CH2)2Flu] (NtBu)}ZrCl2/MAO体系显示高活性,在5.46×102 kPa乙烯压力、甲苯溶剂和75 ℃、1-辛烯浓度0.98~6.17 mol·L-1时活性达到3.38×107~4.380×108 gproduct·molZr-1·h-1,不过,反应时间仅1 min[8],因此,该数值仅作为参考,不能反映催化剂体系的真实状态。
我们继续探究茂金属钛化合物2和3的催化活性。参考上述反应条件,即保持催化剂用量5 μmol、nAl/nTi=300、nB/nTi=1.46、温度110 ℃、乙烯压力为1.4×103 kPa、1-辛烯浓度0.80 mol·L-1、甲苯溶剂150 mL、时间10 min下,2和3的活性分别达到4.172×106和3.974×106 gproduct·molTi-1·h-1,比相同条件下1的活性高近5.07和4.78倍。得到聚合物的Mw分别为1.667×105和1.432×105 g·mol-1,PDI值为4.63和4.51,Tm显示115.42和114.81 ℃,Cm为6.16%和5.98%(Entry 14和15)。总体上看性能和物性值均优于1,表明在相同或相似配体的配位环境下金属钛的催化效果要好于金属锆。随后我们合成了常用的叔丁胺基的四甲基环戊二烯基化合物[Me2Si (C5Me4)(NtBu)]TiCl2 (Ⅰ)[9]、茚基化合物[Me2Si(Ind)(NtBu)]TiCl2 (Ⅱ)[10a]和芴基化合物[Me2Si(Flu)(NtBu)]TiMe2 (Ⅲ)[11],在相同条件下的聚合反应考察发现其活性明显优于2和3,分别为1.308×107 gproduct·molTi-1·h-1 (Ⅰ)、1.044×107 gproduct·molTi-1·h-1 (Ⅱ)、9.551×106 gproduct·molTi-1·h-1(Ⅲ)(Entry 17~19)。特别的是,Ⅰ和Ⅱ得到聚合物的PDI值窄至3.27和3.01,Tm低至72.46和60.04 ℃,Cm仅有3.19%和1.29%。这表明这2种催化剂能够给出很高的1 -辛烯共聚含量,接近14%[6, 10b]。Ⅲ的效果与2和3相当。最后,我们考察了茂金属铪化合物4,其催化活性达到1.092×106 gproduct·molHf-1·h-1,低于2和3,但是生成聚合物的物性与2和3的相近。
我们合成了3种桥联芴基-芳胺基配体L1~L3,其中合成路线和方法都经过优化,即以Me2SiCl2为前驱体,采用先键联茂基再键接胺基的路线;并都使用nBuLi脱质子和进一步LiCl盐消除反应的方法。利用这3种配体与第四族金属氯化物MCl4反应制备了4种CGC结构的茂金属化合物[Me2Si(Flu)(N-2,6-iPr2C6H3)]ZrCl2(THF)2 (1)、[Me2Si(2,7-tBu2Flu) (N-2,6-iPr2C6H3)]TiMe2 (2)、[Me2Si(2,7-tBu2Flu) (N-2,4,6-Me3C6H2)]TiMe2 (3)和[Me2Si(2,7-tBu2Flu) (N-2,6-iPr2C6H3)]HfMe2 (4)。化合物1和2~4的合成方法不同,前者采用2倍物质的量的nBuLi脱双质子,再与ZrCl4脱LiCl反应;后者则采用4倍物质的量的MeLi脱双质子,与TiCl4或HfCl4脱LiCl反应,再与MeLi基团交换反应。化合物L1~L3和1~4都经过谱学和元素分析表征,其中1~3还经过X射线单晶衍射结构分析确认。化合物4可由副反应产生的[Me2Si(2,7 -tBu2Flu)(N-2,6-iPr2C6H3)]HfMe2·LiOSiMe3(OEt2) (5)进行结构佐证。
以AliBu3和(Ph3C)+[B(C6F5)4]-为助剂,研究了化合物1~4催化乙烯和1-辛烯的共聚合性能,其中以1为主对聚合反应条件进行优化,最高活性达到1.204×106 gproduct·molZr-1·h-1,显示接近中等的聚合水平;聚合物重均分子量Mw(1.018×105~1.539×105 g· mol-1)和分子量分布PDI(4.55~6.79)值显示茂金属单活性中心作用的结果;熔融温度Tm(121.91~ 126.23 ℃)和最大结晶度Cm(10.56%~14.88%)则表明1-辛烯共聚到聚合物中。化合物2~3以及文献报道的[Me2Si(C5Me4)(NtBu)]TiCl2 (Ⅰ) [9]、[Me2Si(Ind)(NtBu)]TiCl2 (Ⅱ) [10a]和[Me2Si(Flu)(NtBu)]TiMe2 (Ⅲ) [11]在上述优化条件下的聚合反应研究表明,其活性以及聚合物的物性均优于1。化合物4的反应性能则介于1和2~3之间。总体来看,CGC型结构的化合物催化乙烯和1-辛烯共聚的性能,在配体相同时,金属钛优于铪优于锆;在金属相同时,茂环中环戊二烯基优于茚基优于芴基;而胺基中叔丁胺基优于芳胺基。
Supporting information is available at http://www.wjhxxb.cn
Shapiro P J, Bunel E, Schaefer W P, et al. Organometallics, 1990, 9:867-869 doi: 10.1021/om00117a055
Fink G, Mulhaupt R, Brintzinger H H. Ziegler Catalysts:Recent Scientific Innovations and Technological Improvements. New York:Springer Verlag, 1995:511
Stevens J C, Timmers F J, Wilson D R, et al. EP Patent, 0416815, 1991-03-13.
(a) McKnight A L, Waymouth R M. Chem. Rev., 1998, 98: 2587-2598
(b)McKnight A L, Masood M A, Waymouth R M, et al. Organometallics, 1997, 16: 2879-2885
(c)Xu G, Cheng D. Macromolecules, 2001, 34: 2040-2047
(d)Lanza G, Fragalà I L, Marks T J. Organometallics, 2002, 21: 5594-5612
Joe D J, Wu C J, Bok T, et al. Dalton Trans., 2006, 25:4056-4062
Xu G, Ruckenstein E. Macromolecules, 1998, 31:4724-4729 doi: 10.1021/ma980498d
Klosin J, Kruper W J, Nickias P N, et al. Organometallics, 2001, 20:2663-2665 doi: 10.1021/om010016d
Irwin L J, Reibenspies J H, Miller S A. J. Am. Chem. Soc., 2004, 126:16716-16717 doi: 10.1021/ja044678g
Dare E O, Ogunniyi D S, Olatunji G A, et al. Bull. Chem. Soc. Ethiop., 2004, 18:131-141
(a) Grandini C, Camurati I, Guidotti S, et al. Organometallics, 2004, 23: 344-360
(b)Lee S, Park S S, Kim J G, et al. Molecules, 2017, 22: 258-273
Nishii K, Hagihara H, Ikeda T, et al. J. Organomet. Chem., 2006, 691:193-201 doi: 10.1016/j.jorganchem.2005.08.027
Görl C, Betthausen E, Alt H G. Polyhedron, 2016, 118:37-51 doi: 10.1016/j.poly.2016.07.033
Ihara E, Young V G, Jordan R F. J. Am. Chem. Soc., 1998, 120:8277-8278 doi: 10.1021/ja9817444
Sheldrick G M. SHELXS-97, Program for the Solution of Crystal Structure, University of Göttingen, Germany, 1997.
Sheldrick G M. SHELXL-97, Program for the Refinement of Crystal Structure, University of Göttingen, Germany, 1997.
Eppinger J, Spiegler M, Hieringer W, et al. J. Am. Chem. Soc., 2000, 122:3080-3096 doi: 10.1021/ja992786a
Alt H G, Föttinger K, Milius W. J. Organomet. Chem., 1999, 572:21-30 doi: 10.1016/S0022-328X(98)00527-0
Okuda J, Schattenmann F J, Wocadlo S, et al. Organometallics, 1995, 14:789-795 doi: 10.1021/om00002a028
Razavi A, Thewalt U. J. Organomet. Chem., 2001, 621:267-276 doi: 10.1016/S0022-328X(00)00863-9
Chai J F, Abboud K A, Miller S A. Dalton Trans., 2013, 42:9139-9147 doi: 10.1039/c3dt50163a
Schmid M A, Alt H G, Milius W. J. Organomet. Chem., 1997, 541:3-7 doi: 10.1016/S0022-328X(96)06471-6
Sun Y J, Xu B, Shiono T. Organometallics, 2017, 36:3009-3012 doi: 10.1021/acs.organomet.7b00415
Tanaka R, Yanase C, Cai Z G, et al. J. Organomet. Chem., 2016, 804:95-100 doi: 10.1016/j.jorganchem.2015.12.028
Cai Z G, Ikeda T, Akita M, et al. Macromolecules, 2005, 38:8135-8139 doi: 10.1021/ma050898i
(a) Mehdiabadi S, Soares J B P, Bilbao D, et al. Macromolecules, 2013, 46: 1312-1324
(b)CHEN Zhi-Kang(陈志康), MAO Yuan-Hong(毛远洪), CAO Yu-Cai(曹育才), et al. Chin. J. Org. Chem.(有机化学), 2018, 38(11): 2937-2992
王克强, 黄红红.石油化工, 1994, 23(9):603-606WANG Ke-Qiang, HUANG Hong-Hong. Petrochemical Technology, 1994, 23(9):603-606
(a) Kaminsky W, Noll A. Polym. Bull., 1993, 31: 175-182
(b)Kaminsky W, Florian R. Makromol. Chem. Rapid Commun., 1993, 14: 239-243
Kravchenko R, Waymouth R M. Macromolecules, 1998, 31:1-6 doi: 10.1021/ma971037f
Kaminsky W. J. Chem. Soc. Dalton Trans., 1998, 9:1413-1418
Wu C J, Nayab S, Woo H Y, et al. Polyhedron, 2014, 67:199-204 doi: 10.1016/j.poly.2013.08.072
Park J H, Do S H, Cyriac A, et al. Dalton Trans., 2010, 39:9994-10002 doi: 10.1039/c0dt00637h

图式 2 不同桥联茂环-叔丁胺基结构的CGC型催化剂
Scheme 2 CGC-type catalysts with different cyclopentadienyl rings

图式 4 桥联单茂-芳胺基锆氯化物 1的合成
Scheme 4 Synthesis of bridged monocene‑arylamino zirconium chloride 1

图 1 化合物1的晶体结构图
Figure 1 ORTEP drawing of compound 1
Thermal ellipsoids are at 50% probability and hydrogen atoms are omit‑ ted for clarity; Selected bond lengths (nm) and angles (°): Zr(1) ‑ N(1) 0.204 67(17), Zr(1)‑O(1) 0.230 79(15), Zr(1)‑O(2) 0.230 80(14), Zr(1)‑ C(1) 0.235 6(2), Zr(1)‑Cl(1) 0.243 82(5), Zr(1)‑Cl(2) 0.244 71(5), Si(1)‑ N(1) 0.176 18(18), Si(1)‑C(1) 0.186 6(2); Si(1)‑N(1)‑Zr(1) 99.60(8), N(1)‑ Zr(1)‑C(1) 75.92(7)

图 2 化合物2的晶体图
Figure 2 ORTEP drawing of compound 2
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Ti(1) ‑ N(1) 0.193 16(17), Ti(1)‑C(1) 0.226 7(2), Ti(1)‑C(2) 0.237 5(2), Ti(1)‑ C(3) 0.239 1(2), Ti(1) ‑ C(4) 0.252 0(2), Ti(1) ‑ C(5) 0.253 8(2), Ti(1) ‑ C(24) 0.214 1(2), Ti(1)‑C(25) 0.210 2(2), Si(1)‑N(1) 0.175 17(19), Si(1)‑ C(1) 0.187 0(2); Si(1) ‑ N(1) ‑ Ti(1) 101.96(9), Cp(centroid) ‑ Ti(1) ‑ N(1) 110.399(1)

图 3 化合物3的晶体图
Figure 3 ORTEP drawing of compound 3
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Ti(1)‑N(1) 0.193 6(3), Ti(1)‑C(1) 0.229 0(3), Ti(1)‑C(2) 0.240 4(4), Ti(1)‑C(3) 0.242 3(3), Ti(1)‑C(4) 0.250 4(4), Ti(1)‑C(5) 0.253 1(4), Ti(1)‑C(22) 0.208 9(4), Ti(1)‑C(23) 0.210 1(4), Si(1)‑N(1) 0.174 1(3), Si(1)‑C(1) 0.189 4(3); Si(1)‑N(1)‑Ti(1) 102.59(16), Flu(centroid)‑ Ti(1)‑N(1) 109.575(1)

图 4 化合物5的晶体结构图
Figure 4 ORTEP drawing of compound 5
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity; Selected bond lengths (nm) and angles (°): Hf(1)‑N(1) 0.213 4(3), Hf(1)‑C(1) 0.23 71(3), Hf(1)‑C(24) 0.221 5(4), Hf(1)‑C(25) 0.221 8(4), Si(1)‑N(1) 0.173 5(3), Si(1)‑C(1) 0.186 7(3); Si(1)‑N(1)‑Hf(1) 99.59(13), C(7)‑Hf(1)‑N(1) 73.76(11)
表 1 化合物1~3和5的晶体结构数据
Table 1. Crystallographic data for compounds 1~3 and 5
| Compound | 1 | 2 | 3 | 5 |
| Formula | C35H47ZrCl2NSiO2 | C37H53TiNSi | C34H47TiNSi | C44H73HfLiNO2Si2 |
| Formula weight | 703.95 | 587.79 | 545.69 | 889.67 |
| Cryst system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/n | Cc | P21/n |
| a / nm | 1.044 330(10) | 1.699 490(10) | 2.131 93(3) | 1.346 04(2) |
| b / nm | 2.218 65(2) | 1.728 08(2) | 1.517 49(2) | 1.387 84(2) |
| c / nm | 1.520 050(10) | 2.464 15(4) | 3.839 87(4) | 1.423 74(10) |
| α / (°) | 100.319 0(10) | 103.85(10) | ||
| β / (°) | 96.761 0(10) | 104.502 0(10) | 99.232 0(10) | 110.56(10) |
| γ / (°) | 96.318 0(10) | 104.513 0(10) | ||
| V / nm3 | 3.497 47(5) | 6.801 18(10) | 12.26 18(10) | 2.248 06(5) |
| Z | 4 | 8 | 16 | 2 |
| Dc / (g·cm-3) | 1.337 | 1.148 0 | 1.182 | 1.832 |
| μ / mm-1 | 4.541 | 2.635 | 2.888 | 9.504 |
| F(000) | 1 472 | 2 544 | 4 704 | 1 245 |
| Crystal size / mm | 0.20×0.10×0.10 | 0.20×0.10×0.10 | 0.42×0.34×0.28 | 0.18×0.17×0.16 |
| θ range / (°) | 3.54~77.19 | 2.72~77.29 | 3.59~70.00 | 3.528~77.212 |
| Index ranges | -12 ≤ h ≤ 13 | -21 ≤ h ≤ 18 | -25 ≤ h ≤ 25 | -16 ≤ h ≤ 17 |
| -18 ≤ k ≤ 26 | -20 ≤ k ≤ 21 | -7 ≤ k ≤ 18 | -17 ≤ k ≤ 14 | |
| -18 ≤ l ≤ 17 | -29 ≤ l ≤ 31 | -45 ≤ l ≤ 46 | -14 ≤ l ≤ 18 | |
| Collected data | 25 458 | 92 320 | 38 619 | 30 336 |
| Unique data | 6 970 (Rint=0.037 5) | 27 864 (Rint=0.050 5) | 17 938 (Rint=0.025 0) | 9 183 (Rint=0.056 5) |
| Completeness to θ / % | 94.1 | 96.6 | 97.5 | 99.8 |
| Data, restraint, parameter | 6 970, 0, 386 | 27 864, 72, 1 559 | 17 938, 2, 1 381 | 9 183, 0, 480 |
| GOF on F2 | 0.986 | 1.013 | 0.943 | 1.073 |
| Final R indices [I>2σ(I)] | R1=0.032 0 | R1=0.056 5 | R1=0.033 0 | R1=0.038 3 |
| wR2=0.082 1 | wR2=0.153 7 | wR2=0.107 9 | wR2=0.101 1 | |
| R indices* (all data) | R1=0.039 1 | R1=0.068 1 | R1=0.051 2 | R1=0.044 8 |
| wR2=0.085 8 | wR2=0.168 1 | wR2=0.115 6 | wR2=0.107 9 | |
| Largest diff. peak and hole / (e·nm-3) | 746, -752 | 2 148, -543 | 342, -420 | 1 171, -1 586 |
| *R1=∑(||Fo|-|Fc||)/∑|Fo|, wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)]1/2, GOF=[∑w(Fo2-Fc2)2/(No-Np)]1/2. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 乙烯与1-辛烯共聚合的催化结果a
Table 2. Results of catalytic copolymerization of ethylene and 1-octenea
| Entry | Catalyst | nAl/nM | V1-octene / mL | mB co-catb / mg | T / ℃ | mpolymer / g | Activity /(g·mol-1·h-1) | Mwc /(g·mol-1) | PDIc | Cmd / % | Tmd / ℃ |
| 1 | 1 | 300 | 18.6 | 4.9 | 90 | 0.756 | 9.07×105 | 2.599×105 | 13.96 | 45.85 | 125.72 |
| 2 | 1 | 300 | 18.6 | 4.9 | 100 | 0.879 | 1.055×106 | 1.978×105 | 16.24 | 37.51 | 126.73 |
| 3 | 1 | 300 | 18.6 | 4.9 | 110 | 0.573 | 6.87×105 | 1.209×105 | 6.20 | 11.85 | 124.35 |
| 4 | 1 | 300 | 18.6 | 4.9 | 120 | 0.412 | 4.94×105 | 1.431×105 | 5.95 | 14.88 | 126.23 |
| 5 | 1 | 300 | 0 | 4.9 | 110 | 0.361 | 4.33×105 | 8.063×105 | 31.02 | 45.63 | 134.14 |
| 6 | 1 | 300 | 3.1 | 4.9 | 110 | 0.449 | 5.39×105 | 2.940×105 | 22.39 | 48.76 | 128.72 |
| 7 | 1 | 300 | 6.2 | 4.9 | 110 | 0.545 | 6.54×105 | 2.886×105 | 17.27 | 26.95 | 129.43 |
| 8 | 1 | 300 | 9.3 | 4.9 | 110 | 0.733 | 8.79×105 | 1.145×105 | 4.55 | 11.37 | 123.02 |
| 9 | 1 | 300 | 27.9 | 4.9 | 110 | 0.449 | 5.39×105 | 1.018×105 | 6.79 | 10.75 | 121.91 |
| 10 | 1 | 200 | 18.6 | 4.9 | 110 | 0.561 | 6.73×105 | 1.425×105 | 6.43 | 11.90 | 123.43 |
| 11 | 1 | 600 | 18.6 | 4.9 | 110 | 1.003 | 1.204×106 | 1.539×105 | 5.02 | 10.56 | 122.57 |
| 12 | 1 | 1 000 | 18.6 | 4.9 | 110 | 0.892 | 1.070×106 | 9.29×104 | 15.74 | 7.57 | 111.44 |
| 13 | 1 | 1 400 | 18.6 | 4.9 | 110 | 0.818 | 9.82×105 | no detected | no detected | 5.22 | 99.43 |
| 14 | 2 | 300 | 18.6 | 4.9 | 110 | 3.477 | 4.172×106 | 1.667×105 | 4.63 | 6.16 | 115.42 |
| 15 | 3 | 300 | 18.6 | 4.9 | 110 | 3.312 | 3.974×106 | 1.432×105 | 4.51 | 5.98 | 114.81 |
| 16 | 4 | 300 | 18.6 | 4.9 | 110 | 0.910 | 1.092×106 | 1.821×105 | 6.74 | 6.78 | 113.75 |
| 17 | Ⅰ | 300 | 18.6 | 4.9 | 110 | 10.900 | 1.3080×107 | 1.482×105 | 3.27 | 3.19 | 72.46 |
| 18 | Ⅱ | 300 | 18.6 | 4.9 | 110 | 8.700 | 1.0440×107 | 1.455×105 | 3.01 | 1.29 | 60.04 |
| 19 | Ⅲ | 300 | 18.6 | 4.9 | 110 | 7.876 | 9.551×106 | 1.843×105 | 7.65 | 7.72 | 115.58 |
| aPolymerization conditions: catalyst 5.0 μmol; AliBu3 1.5 mmol (based on nAl/nM of 300); (CPh3)+[B(C6F5)4]- 7.30 μmol (based on nB/nM of 1.46); ethylene pressure 1.4×103 kPa; toluene 80 mL; time 10 min; b B co-cat: (CPh3)+[B(C6F5)4]-; c Determined by GPC; d Determined by DSC; Ⅰ=[Me2Si(C5Me4)(NtBu)]TiCl2; Ⅱ=[Me2Si(Ind)(NtBu)]TiCl2; Ⅲ=[Me2Si(Flu)(NtBu)]TiMe2. | |||||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: