
图 1.
光催化基本原理图
Figure 1.
Schematic diagram of photocatalysis

氢气作为一种重要的化工原料,其生产主要依赖烃类重整和煤气化。然而,化石能源的过度消耗及带来的能源与环境问题严重影响了人类社会的可持续发展。1972年,Honda等首次报道了单晶TiO 2半导体电极的分解水现象,此后,半导体光催化技术作为一种绿色制氢技术得到了广泛关注和飞速发展[1]。目前开发高效、稳定的分解水光催化剂仍然是光解水制氢领域的热点与核心问题。
有机聚合物半导体(g-C3N4等)、金属氧化物(TiO2、钛酸盐)、金属硫化物等一直是光解水制氢领域的热点材料,但单一光催化剂存在电荷复合率高、光吸收能力弱等缺点,调控形貌、构建异质结、负载助催化剂、掺杂等手段是克服上述缺点的有效方法。在诸多材料中,ZnS与CdS具有相同的晶体结构、配位模式以及近似的原子半径,两者可以形成无限互溶的CdxZn1-xS固溶体,其能带位置可通过改变Zn/Cd比连续调控,使光生电荷可以在连续的能带中移动,而不是在离散的施主或受主能级中迁移。因此,其活性通常优于掺杂的CdS和ZnS。近年来,以Cd1-xZnxS固溶体为代表的双金属硫化物因优异的催化活性而受到越来越多的关注。
我们将首先介绍光催化产氢的基本原理及其热力学和动力学特征。其次,详细阐述近年来Cd1-xZnxS固溶体在光催化产氢方面的研究现状。最后,对其面临的挑战和问题进行了分析,并对近期研究进行了展望,以期为合理设计和制备高性能产氢光催化剂提供借鉴。
如图 1所示,在适当的光照条件下,半导体价带(VB)中的电子(e-)跃迁到导带(CB)上,并在原位留下空穴(h+),跃迁的电子具有还原性,空穴则具有氧化性。所产生的电子-空穴一部分迁移至材料表面后引发化学反应,另一部分在材料表面或者体相重新复合。光催化剂通常以粉末或者薄膜的形式参与反应,因此,光催化反应通常也是一种非均相催化过程。
水分解是一个吉布斯自由能变大于0的非自发反应。标准状态下,1 mol水分解为H2和O2需要237 kJ的能量(式1),即当电场强度大于1.23 eV时,电子可使H+还原产生H2(式2),而空穴可将H2O氧化释放出O2(式3)[2]。
|
$ \begin{array}{l} {{\rm{H}}_{\rm{2}}}{\rm{O}}\left( {\rm{l}} \right){\rm{ }} \to {\rm{ }}{{\rm{H}}_{\rm{2}}}\left( {\rm{g}} \right){\rm{ + 1/2}}{{\rm{O}}_{\rm{2}}}\left( {\rm{g}} \right)\\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;(\Delta {G^{⊖}} = 237.2{\rm{ kJ \cdot mo}}{{\rm{l}}^{ - 1}}, \Delta E = 1.23{\rm{ V}}) \end{array} $ |
(1) |
|
$ {\rm{2}}{{\rm{H}}^{\rm{ + }}}{\rm{ + 2}}{{\rm{e}}^ - } \to {\rm{ }}{{\rm{H}}_{\rm{2}}}\left( {\rm{g}} \right)\;\;\;\;\;\;\;\;\;\;{\rm{ }}(\Delta {E^{⊖}}{\rm{ = 0 V}}, {\rm{pH = 0)}} $ |
(2) |
|
$ \begin{array}{l} {\rm{2}}{{\rm{H}}_{\rm{2}}}{\rm{O}}\left( {\rm{l}} \right){\rm{ }} \to {\rm{ }}{{\rm{O}}_{\rm{2}}}\left( {\rm{g}} \right){\rm{ + 4}}{{\rm{H}}^{\rm{ + }}}{\rm{ + 4}}{{\rm{e}}^ - }\\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;(\Delta {E^{⊖}}{\rm{ = + }}1.23{\rm{ V}}, {\rm{pH = 0)}} \end{array} $ |
(3) |
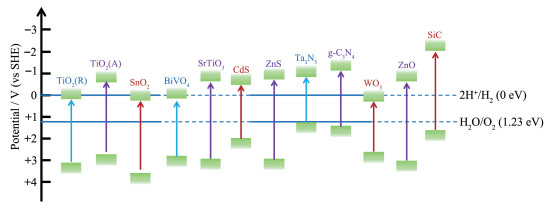
光催化分解水体系通常可分为2类[3]:(1)有牺牲剂的半反应体系。通过向体系加入更易氧化的电子给体,不仅能降低水分解的热力学能垒,也能不可逆地加快光生空穴或羟基自由基(·OH)的消耗速率,从而使更多的电子参与产氢半反应,常用电子给体包括生物质(如蔗糖、纤维素、葡萄糖、乳酸等)、还原性有机物(三乙醇胺、甲醇、乙醇等)及无机物(Na2S、Na2SO3、Na2S2O4、NaI等)。(2)全解水体系。该体系对催化剂能带结构要求较为苛刻:其导带和价带电势必须与水的还原和氧化电位相匹配,即导带底须比H+的还原电位(0 V vs SHE)更负,而价带顶须比H2O的氧化电位(1.23 V vs SHE)更正,考虑到非均相过程过电势的存在,其禁带宽度应不小于1.8 eV。
图 2为一些常见光催化剂的能带结构,图中导带/价带电势与水分解还原/氧化电势间的差异决定了反应驱动力的大小,也就是说CB电势更低有利于还原反应,而VB电势更高则有利于氧化反应[2-3]。然而,能够实现全解水的光催化剂往往太阳光利用率不足,而窄带隙光催化剂尽管具有良好的光吸收能力,其光生电荷氧化还原电势却相对较低,导致光生电荷氧化还原能力较弱。因此,平衡和优化光生电荷氧化还原电势(CB/VB位置)和光吸收能力(带隙)之间的矛盾显得十分关键。固溶体材料的带隙等性质随其组成变化而连续变化,因此,形成固溶体是解决以上矛盾的有效方法[4-6]。
除热力学因素外,光催化产氢效率还受到材料微-纳米结构、光子捕获能力、表面/界面形态、结晶度、助催化剂等的影响[2-3]。决定光催化反应效率的过程包括4个串联步骤:(1)光子的捕获;(2)电荷载流子的产生与分离;(3)电荷载流子迁移;(4)表面氧化还原反应;总太阳能转化效率由上述步骤共同构成,由如下公式表示[2]:
|
$ {\eta _{\rm{c}}} = {\eta _{{\rm{abs}}}}{\eta _{{\rm{cs}}}}{\eta _{{\rm{cmt}}}}{\eta _{{\rm{cu}}}} $ |
(4) |
其中,ηc为太阳能总转化效率;ηabs为光吸收效率;ηcs为电荷激发与分离效率;ηcmt为电荷迁移效率;ηcu表示表面氧化还原反应效率。通常复杂的载流子动力学和缓慢的表面反应动力学是导致太阳能转化效率偏低的关键因素[7-8]。
构建具有丰富微孔道结构和大比表面积的光催化剂可使光子在其孔道内多次反射和散射,使光吸收增强,从而产生更多的光生电荷[9]。其次,构建异质结、负载助催化剂均能有效抑制光生电荷在催化剂表面的随机复合。另外,迟缓的表面反应动力学导致光催化剂表面光生电荷累积,甚至引发光催化剂的自氧化或自还原(光腐蚀过程),导致催化剂活性降低[2]。因此,提高反应物在反应介质界面的吸附扩散动力学,增强表面反应过程也能显著提高光催化效率[10]。与热力学因素相比,上述动力学因素如载流子动力学、表面反应动力学等对光催化体系的整体效率具有重要影响[11]。
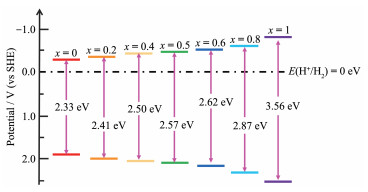
Cd1-xZnxS固溶体具有立方闪锌矿和六方纤锌矿2种晶体结构[13-14],其导带由Zn4s4p与Cd5s5p轨道杂化而成,导带底电势相比于CdS更低,而禁带宽度相比于ZnS更小,同时兼顾了光吸收性能与光生电荷的氧化还原能力(图 3),但其光生电荷较高的复合率和空穴诱导的自腐蚀仍是其应用的主要障碍。为此,研究者们试图通过微观结构调控、负载助催化剂、构建半导体异质结等方法对其进行改性,以进一步抑制光生电荷的复合并增强其稳定性。
通过结构调控既能改变催化剂微观形态,也可以影响其表界面物理化学性质,从而促进光生电荷的分离传递或增强光能利用率。由于Cd1-xZnxS复杂的元素组成和固有的密堆积结构,其通常表现为纳米颗粒或微球,通过简单一锅水热法对其微观形貌的调控较为困难,一般采用模板法或直接通过优化合成条件与元素组成而达到对其活性优化的目的。
2017年,李映伟课题组以ZIF-8为硬模板合成了中空ZnS纳米笼,后又通过阳离子交换法获得了中空Cd1-xZnxS光催化剂。其中,Cd0.4Zn0.6S的比表面积达到156 m2·g-1,为光催化反应提供了丰富的活性位点,中空结构使光子在其内部多次散射,显著提升了材料光吸收效率[15]。钱海生等通过前驱体硫化-阳离子交换,得到了双壳层中空Cd1-xZnxS微球。研究发现镉源和硫源的加入量影响其双壳层的间距,当以乙二醇代替去离子水为溶剂时,通过相同方法可得到单壳层Cd1-xZnxS。理论模拟进一步表明,双壳层相比于单壳层有更高的光子利用率[16]。上述2个工作分别利用模板法和离子交换法首次构建了单分散的中空Cd1-xZnxS微球,借助中空结构的“聚光效应”使Cd1-xZnxS活性相比常规块体纳米颗粒或纳米微球有了较大提高,不但丰富了Cd1-xZnxS材料的微结构,也为Cd1-xZnxS在其他领域的研究提供了借鉴。
除中空结构外,利用胺类多齿配体与Zn2+和Cd2+的鳌合配位作用,可制备出具有棒状或片状结构的Cd1-xZnxS。靳治良等研究了当乙二胺(en)为溶剂时,硫源添加量对Cd1-xZnxS形貌的影响。研究发现硫源不足时纳米棒和纳米颗粒共存于催化剂结构中,当增加硫源用量时,其结构转变为由纳米棒自组装的纳米花,当(nZn+nCd):nS=1:3.5时,催化剂表现出最优活性和稳定性[17]。该工作表明Cd1-xZnxS固溶体的微结构及光催化活性可能与制备条件密切相关,通过优化制备方案,即可得到具有良好活性的光催化剂,但作者并未对催化剂与其活性间的构效关系进行深入探讨。我们课题组以乙二胺/水(en/ H2O)混合溶液为溶剂,通过阳离子交换得到了2D介孔超薄Cd0.5Zn0.5S纳米片。得益于载流子迁移距离的缩短、表面不饱合原子及比表面积的增大,Cd0.5Zn0.5S纳米片的析氢活性达到相应纳米颗粒的2倍多,即使在纯水中,Cd0.5Zn0.5S纳米片产氢速率仍可达到1 395 μmol·h-1·g-1,超过了目前所报道的未加修饰的光催化剂的活性,但其在纯水中的光腐蚀却更为严重[18]。代凯课题组以二乙烯三胺/水(DETA/H2O)混合溶液为溶剂,分别制备了ZnS-DE- TA、CdS-DETA、Cd1-xZnxS-DETA无机-有机杂化光催化剂。其中,Zn0.2Cd0.8S-DETA具有最优异的产氢活性,作者认为这与其恰到好处的能带位置及增强的光吸收密切相关[19]。但实际上除ZnS-DETA外,DE- TA是以物理吸附的形式存在于Cd1-xZnxS- DETA或CdS-DETA表面,并未形成真正的无机-有机杂化结构。桑换新等则成功制备了Zn0.2Cd0.8S-en无机-有机杂化纳米片,并对纳米片微观性质如厚度、大小等做了简单研究,发现适量的硫源有利于减少材料表面缺陷,也能对样品晶面暴露比例产生影响[20]。然而,以上关于无机-有机杂化Cd1-xZnxS的研究中,均忽略了有机分子在光催化反应中所起到的作用及其含量等关键参数对光催化反应的影响。另外,Cd1-xZnxS基无机-有机杂化材料在光催化产氢领域的研究还相对较少,其相比于全无机Cd1-xZnxS的优势或劣势尚不明确。
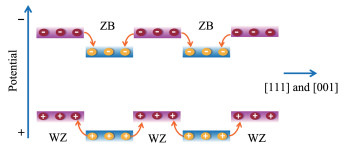
如前所述,Cd1-xZnxS固溶体存在闪锌矿(ZB)和纤锌矿(WZ)两种晶体结构,且具有近似的晶格常数。其中,闪锌矿沿[111]晶面簇方向以A-B-C-A-B-C方式密堆积排列,而纤锌矿以A-B-A-B的方式沿[001]晶面簇密堆积排列[21]。因此,ZB和WZ可以沿[111]和[001]方向交替排列,形成一种无晶格失配和缺陷的ZB/WZ共格孪晶结构[14, 22-23, 26]。WZ结构的[001]晶面簇方向存在自发极化,而ZB结构高度对称无极化,因而在ZB/WZ超晶格中形成了Ⅱ型能带结构的锯齿状电位分布(图 4),光生电子和空穴分别于孪晶结构的ZB段和WZ段富集,这可显著提高Cd1-xZnxS体相光生电荷的分离效率。
曹荣等通过水热法制备了Cd1-xZnxS(x=0.2、0.4、0.6、0.8)纳米固溶体。在可见光照射下(Na2S/Na2SO3体系中),Cd0.6Zn0.4S的产氢速率是相同条件下CdS产氢速率的690余倍,是ZnS的2 000余倍。由于纳米固溶体兼具S空位、相异质结和孪晶同质结,有效促进了载流子分离,强化了S2-在催化剂表面的吸附。作者还发现Cd0.6Zn0.4S在Na2S/Na2SO3中活性最好,其次为乳酸,并认为这种活性差异源于各种电子给体不同的氧化电势和介电常数[24]。对于析氢半反应来讲,多电子转移的氧化半反应是整个反应的速控步骤,这种活性的差异更有可能是不同牺牲剂分子在催化表面的氧化速率的差异导致的。最近,乔振安等借助配体辅助首次制备了单分散的孪晶Cd1-xZnxS介孔纳米球,相比于纳米颗粒其具有更大的比表面积和丰富的孔道结构。此外,通过控制Zn/Cd元素比,可调节WZ相与ZB相的比例,同时还能实现对纳米球半径的有效控制(图 5)[25]。但是,元素物质的量之比调变的同时引起了WZ相与ZB相的比例及纳米球半径的变化,因此,Cd0.2Zn0.8S高活性究竟源于适当的异相结还是其他因素仍无法下结论。尽管目前研究者普遍认为孪晶Cd1-xZnxS相比于纯相ZB相或WZ相Cd1-xZnxS具有更高的产氢活性,但从原子水平揭示其高活性起源的研究仍然较少。宫建茹及郭烈锦等通过对Cd1-xZnxS纳米晶进行二次溶剂热处理,不但在Cd1-xZnxS纳米晶中成功引入了ZB/WZ超晶格孪晶结构,且随热处理时间的延长,Cd1-xZnxS纳米晶中ZB/WZ孪晶结构逐渐增多,实现了对ZB/WZ孪晶结构相对含量的调控,作者还首次阐明了ZB/WZ孪晶间锯齿状的电位分布是由于不同相态Cd1-xZnxS的分子极化所引起的[26]。

通过溶剂调控、控制制备过程中S2-释放速率等方法也能实现可控、高效地制备Cd1-xZnxS纳米颗粒。例如,李建荣等以离子液体(氯化1-丁基-2,3-二甲基咪唑鎓)为溶剂,利用元素直接反应得到了Cd1-xZnxS固溶体,发现Cd0.2Zn0.8S的产氢活性是CdS的163倍[27]。由于离子液体具有低蒸汽压的特征,该方法可能适用于规模化制备Cd1-xZnxS光催化剂,但元素直接合成却增加了原料成本。Kaur等以4,4′-dipyr- idyldisulfide(DPDS)为硫源,利用其分解速率随温度变化的特点控制S2-的释放速率,成功合成了Cd1-xZnxS纳米微球,发现Zn0.7Cd0.3S具有最佳的产氢活性,而Zn0.9Cd0.1S具有最佳的还原硝基苯制苯胺活性[28]。由此可见,将Cd1-xZnxS用于不同反应过程时应对其元素组成进行优化,从而调整光生电荷的氧化还原能力使之与相应反应过程匹配。
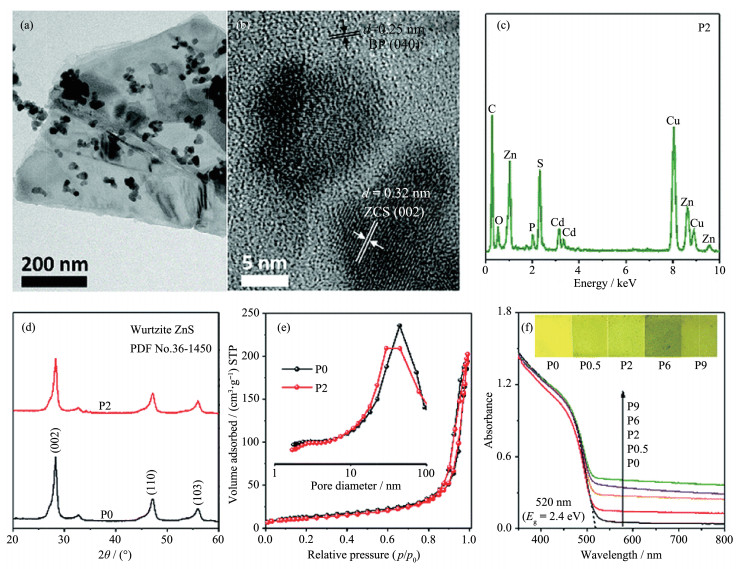
尽管Cd1-xZnxS相比于g-C3N4、TiO2具有超高的产氢活性,但其低的光化学稳定性对于实际应用是一个很大的挑战[29]。Abideen等借助锌独特的两性性质,通过NaOH溶液热处理制备了富含锌空位的Cd0.7Zn0.3S(VZn-CZS)。(VZn可以作为空穴陷阱抑制光生电荷的复合,此外,由于(VZn导致样品VB氧化电位降低,VZn-CZS还具有更好的抗光腐蚀性[30]。该工作首次提出可通过对Cd1-xZnxS进行适当的后处理同时实现活性的提高和稳定性的增强,具有操作简单且成本低廉的特点,有望扩展到其他光催化剂。但是,作者并未对VZn-ZCS抗光腐蚀性的增强进行深入研究,这可能与浓碱处理过程中样品表面化学组成的变化密切相关,这一点在未来研究中应更值得关注。针对纳米Zn0.5Cd0.5S在水溶液中回收利用难的问题,余家国课题组在聚丙烯腈纳米纤维上包覆了一层Zn0.5Cd0.5S纳米颗粒,成功制备了Zn0.5Cd0.5S薄膜光催化剂(图 6),其在420 nm处的量子效率达到了27.4%[31]。该光催化薄膜不但具有良好的柔韧性和机械强度,还克服了粉末光催化体系难以回收的缺陷,为光催化产氢技术的实用化提供了一定借鉴。
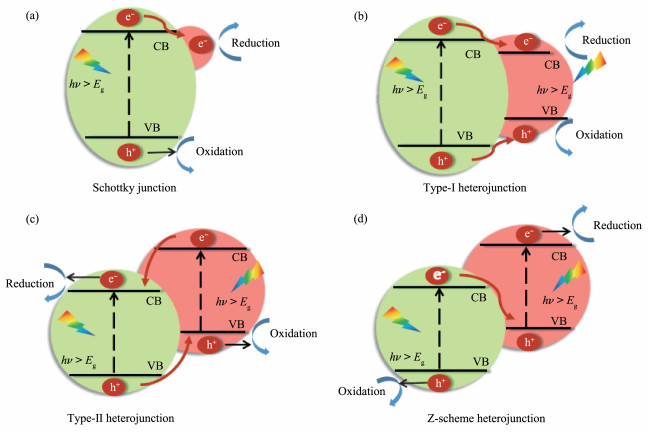
在半导体表面构建异质结是另外一种能有效强化光催化产氢效率的方法,表面异质结能够有效促进载流子的界面分离。构建高效界面、研究其光生电荷迁移机制仍是光催化领域的重要课题。典型的异质结可分为两大类,具体如图 7所示:(1)金属/类金属-半导体异质结(肖特基结);(2)半导体-半导体异质结,包括Ⅰ型异质结、Ⅱ型异质结及Z型异质结等。
以Pt、Au、Ag为代表的贵金属是一类高效的析氢助剂,不仅能有效加快表面反应速率,而且借助等离子体效应和肖特基结能有效提高光能利用率并抑制光生电荷表面复合。Ng等制备了具有高分散性的超小Pt颗粒(~2 nm)修饰的孪晶Zn0.5Cd0.5S,同质结(ZB/WZ)和异质结(Pt/Cd0.5Zn0.5S)的同时存在有效促进了光生电荷的空间分离,并为H2析出提供了更多的活性位点。在Na2S/Na2SO3和抗坏血酸中,最优样品的可见光产氢活性分别比原始Zn0.5Cd0.5S提高了4.9倍和27.9倍[32]。这一结果再次说明了在不同牺牲剂中,光催化剂表面反应速率的差异是引起其表观活性差异的重要原因。Chen等在Cd1-xZnxS纳米线表面先沉积了一层Au薄膜,通过进一步煅烧获得了Au纳米颗粒修饰的Cd1-xZnxS。研究发现不同厚度Au薄膜前驱体可产生不同分布状态的Au颗粒,当Au薄膜厚度为3.5 nm时(3.5-Au/Cd0.5Zn0.5S),由于Au的局域表面等离子体效应可使体系产生更多的光生电荷,复合物活性亦达到了最高[33]。该工作表明,等离子体金属对光催化活性的促进与其粒径及分布有密切关系。值得注意的是,表面等离子体态包含大量不同态密度的等离激元,其与等离子体材料的光吸收范围及强度有密切关系,而材料的能带结构,即半导体催化剂导带与等离子体材料能级间的差异是决定等离子体“热电子”注入过程的关键因素,而上述研究并未涉及Cd1-xZnxS能带结构的调控。Pu等则通过热注入法得到了Au@Cd1-xZnxS (x=0~0.37)核-壳催化剂,发现随着壳层Cd1-xZnxS中Zn含量的逐渐增大,界面载流子迁移驱动力逐渐增大,使载流子迁移动力学速率加快,同时,材料载流子浓度也随Zn含量的增大呈现出与活性变化规律相一致的特点[34]。这一工作首次通过调控Cd1-xZnxS导价带位置,细致研究了其导带位置与界面热电子传输动力学间的联系。
然而,贵金属价格昂贵、储量有限,不利于大规模使用,且单金属所提供的活性位点有限,因此开发高效非贵金属助催化剂显得很有必要。Yue等制备了NiCo合金修饰的Cd1-xZnxS,通过优化Ni/Co元素比与牺牲剂组成得到的Ni0.8Co0.2/Zn0.75Cd0.25S在模拟太阳光照射下获得了211.4 mmol·h-1·g-1的超高产氢速率。功函数测试发现Ni0.8Co0.2相比于单金属Ni或金属Co有更低的费米能级和更小的产氢过电势,使光生电子向合金纳米颗粒迁移时的驱动力相比于单金属明显增大[35]。该研究表明,通过优化元素结构与组成,过渡金属合金纳米粒子有潜力作为一类高效的非贵金属析氢助剂,但目前关于过渡金属合金纳米粒子与Cd1-xZnxS相结合的报道仍较为少见。
近年来,研究者们发现过渡金属磷化物是一类性能优异的非贵金属光催化析氢助剂,并对此做了大量工作。戈磊课题组首次通过热磷化法制备了CoP/Cd0.5Zn0.5S纳米棒复合光催化剂。其中,CoP/ Cd0.5Zn0.5S(CoP的质量分数为5%)在乳酸溶液中的活性约为纯Cd0.5Zn0.5S的20倍[36],X射线光电子能谱、SPV(表面光电压谱)等证明CoP与Cd0.5Zn0.5S产生了紧密的界面接触,这提高了光生电荷的分离效率,作者认为CoP与Cd0.5Zn0.5S间形成了I型异质结接触。随后该课题组又以红磷为磷源,制备了Ni2P/ Zn0.5Cd0.5S,其在Na2S/Na2SO3溶液中的活性为Zn0.5Cd0.5S的13倍,理论模拟发现Ni2P的费米能级高于Zn0.5Cd0.5S,两者紧密接触后Zn0.5Cd0.5S能带将向下弯曲,光生电荷很快地迁移至Ni2P表面,Ni2P作为活性位起着收集电子的作用,使电子-空穴对的分离效率得到提高[37]。以上研究表明,Ni2P、CoP可有效提高Cd0.5Zn0.5S的产氢活性,但是由于作者所选择的电子给体不同,上述2个研究工作并不具有横向对比性。Dhingra等在同一时期发现Ni2P/Zn0.5Cd0.5S在Na2S/Na2SO3中的析氢活性不但优于Ni2P/ZnS和Ni2P/CdS而且优于Pt/Zn0.5Cd0.5S[38]。2018年,Wang等则首次将1D CoP纳米线(NWs)与Cd0.5Zn0.5S相结合,借助CoP纳米线良好的导电性与其特殊的1D纳米结构间的协同作用,CoP NWs/Cd0.5Zn0.5S的活性是原始Cd0.5Zn0.5S的22倍,且达到同等条件下CoP纳米颗粒修饰Zn0.5Cd0.5S的2倍多[39],但Wang等认为CoP与Cd0.5Zn0.5S间形成了肖特基结,这与戈磊等的观点相左。FeP是另外一种重要的过渡金属磷化物,Zhu等通过原位磷化成功获得了FeP/Cd1-xZnxS光催化剂,发现FeP不仅促进了光生电荷载流子的分离还降低了析氢过电势,FeP/Cd0.5Zn0.5S(FeP质量分数为2%)的析氢活性为纯Cd0.5Zn0.5S的130倍[40]。向全军等进一步制备了负载多种过渡金属磷化物(Cu3P、Ni2P、CoP、Fe2P)的Cd0.5Zn0.5S,并首次横向对比了不同磷化物助剂对Cd0.5Zn0.5S产氢活性的影响,当控制磷化物质量分数为0.3%时,样品在Na2S/Na2SO3体系的活性强弱遵循Cu3P>Ni2P>Fe2P>CoP的顺序[41]。但由于电子给体对光催化表面反应过程有较大影响,同一催化剂往往在不同牺牲剂溶液中表现出巨大的活性差异,该活性规律是否适用于其他牺牲剂体系仍有待探索。除上述研究较多的Ni、Co、Fe磷化物外,靳治良等还报道了一种磷化钨(WP)与Cd0.5Zn0.5S结合的Ⅰ型异质结光催化剂,瞬态光致发光光谱(PL)证明Cd0.5Zn0.5S的光生电子快速地注入了WP的导带,提高了光生电荷的分离效率。WP的引入还使体系的光吸收能力增强,同时使产氢过电势、界面载流子传输阻力均明显降低[42]。过渡金属磷化物虽可作为一类高效的非贵金属析氢助剂,但其制备工艺较为复杂,对设备要求较高,多采用高温磷化或毒性磷源,尽管以红磷为磷源制备过渡金属磷化物复合光催化剂已取得较大进展,但以红磷为磷源时,磷化物微观形貌难以有效控制。因此,开发更加简便易操作的低廉制备方法仍有很大意义。
除过渡金属磷化物外,研究者们围绕NiS、MoS2等的廉价析氢助剂也做了大量工作。安长华等制备了同时负载NiS和MoS2的Cd1-xZnxS纳米棒,发现MoS2和NiS都能明显提高Cd1-xZnxS的光催化活性。NiS和MoS2的共负载则进一步提高了光生电荷分离效率,在可见光照射下NiS/MoS2/Cd0.8Zn0.2S的产氢活性达到Pt/Zn0.2Cd0.8S的5倍多[43]。王辉等分别在孪晶Cd1-xZnxS(T-Cd1-xZnxS)和非孪晶Cd1-xZnxS颗粒表面修饰了富有缺陷的MoS2。T-Cd1-xZnxS相比于非孪晶Cd1-xZnxS纳米颗粒能有效地促进光生电荷的体相分离,而这种富含S22-与Mo5+的MoS2不仅拓宽了催化剂光吸收范围,同时也促进了光生电子在MoS2缺陷位的富集和水分子在其表面的吸附[44]。Zhao等通过DFT计算发现,NiS的引入促进了H2O分子在NiS/ Cd1-xZnxS表面的解离,加快了表面反应动力学,使得产氢活性得到大幅度提高[45]。然而,通过简单负载对Cd1-xZnxS的活性提高仍然有限,针对催化剂形貌、电子能带结构和表面活性位点的综合调控仍鲜见报道。林海峰课题组借助调控Zn/Cd比与生长富含O缺陷的MoS2,并进一步光沉积NiOx纳米颗粒,使催化剂能带结构、活性位点和界面电荷分离实现了同步调节[46]。值得注意的是,上述研究仍主要致力于牺牲剂体系产氢半反应的研究,而牺牲剂的使用导致反应体系的经济成本大大提高。上官文峰课题组最近将MoS2@Cd1-xZnxS用于同时降解阿莫西林废水和分解水产氢,在实现降解阿莫西林的同时实现了分解水产氢,尽管其活性较低,但却极大地降低了析氢反应成本,为Cd1-xZnxS的实际应用提供了一定参考[47]。
诸如石墨烯、碳点等碳材料因其优异的光热稳定性、良好的导电性及尺寸依赖光学性质有潜力作为另外一类廉价光催化析氢助剂。Zhou等先通过两步水热制备了洋葱状碳(OLC)与Cd0.5Zn0.5S QDs(量子点)核-壳结构,实验发现,这种OLC同时具备上转换和下转换发光特性,复合材料在550~900 nm光照射下仍然具有产氢活性,充分证明了OLC的上转换特性有利于复合材料活性的提高。基于OLC的光学特点,在530 nm下,复合材料遵循Ⅱ型异质结电荷转移机理,530 nm以上则由Cd0.5Zn0.5S QDs吸收上转换光子产生光生电荷引发光催化反应[48]。该工作利用碳材料的上转换发光特性首次将Cd0.5Zn0.5S基复合光催化剂的光响应范围拓展到了近红外区域。由于Zn的引入使Cd1-xZnxS的吸收边相比CdS有所蓝移,光谱响应范围有所变窄,因此,进一步开发具有紫外-可见-近红外光响应的全光谱Cd1-xZnxS基复合光催化剂显得很有必要。另外,关于石墨烯基光催化剂的研究已发表了较多综述,在此不再赘述[49-52]。
如前所述,构建具有相匹配能带结构的半导体-半导体异质结能同样有效地抑制光生电荷的表面复合,从而提高催化活性。刘平课题组合成了1D/2D Cd1-xZnxS/ZnS(en)0.5复合物,其在440 nm处的表观量子产率高达49.95%。低维材料中独特的电子行为、匹配的晶格及能带结构,使光生电荷体相及表面分离效率得到同步增强[53]。但Zn0.41Cd0.59S/ ZnS(en)0.5的催化稳定性欠佳,这可能与2种材料界面接触的减弱、部分en配体分子解离或被H2O、OH-和S2-取代以及氨基官能团的光氧化有关。Yang等制备了具有相匹配能带结构的Cd0.7Zn0.3S/NiTiO3纳米纤维复合光催化剂,实验表明NiTiO3前驱体的制备温度对最终样品的活性有很大影响,Cd0.7Zn0.3S/ NiTiO3-600 ℃表现出更为优异的产氢活性[54]。代凯课题组采用溶剂热法制备了g-C3N4/Cd0.8Zn0.2S-DETA光催化剂[55]。通过XPS峰位移,作者推测认为g-C3N4/Cd0.8Zn0.2S-DETA间形成了阶梯形异质结(S - scheme),但却并未对这一结论做进一步论证。此外,作者发现该复合物在Na2S/NaSO3牺牲剂溶液中连续使用21 h后活性没有明显的衰减,具有良好的稳定性,这与刘平课题组针对Cd1-xZnxS/ZnS(en)0.5类似无机-有机复合物稳定性研究结果并不一致。
金属氢氧化物(LDHs)是一类具有层状结构半导体材料,因其丰富的表面活性位点和可调变的元素组成吸引了很多研究者的兴趣。例如,Shi等通过LDHs的原位转化得到了2D/2D ZnO/Cd1-xZnxS单晶纳米片异质结,通过控制煅烧温度和时间,可实现对ZnO含量的精确控制,其独特的制备方法不仅使两者形成了紧密的界面接触,而且还形成了直接Z-型异质结[56]。随后,该课题组又调节前驱体组成,直接得到了2D/2D Cu2S/Zn0.67Cd0.33S面内异质结。由于它们的面内共生结构源自于前驱体的拓扑转化,其具有一致的晶格取向,这种平面内的共生结构极大地促进了光生电荷的分离[57]。Li等通过静电自组装将Cd0.5Zn0.5S颗粒和CoAl-LDH相复合,当CoAl-LDH的质量分数为20%时,复合样品活性是纯Cd0.5Zn0.5S的6.9倍,且在使用20 h后活性没有明显衰减[58]。以上研究表明,无论是通过对LDHs的化学转化或直接将LDH与Cd1-xZnxS复合都能有效增强其析氢活性。
近年来,黑磷(BP)由于其可调节的带隙、快的载流子迁移率、独特的二维结构等优势迅速成为了一种明星材料,被广泛用于太阳能电池、光热催化及光催化产氢[59-60]。2017年,乔世璋课题组将单层磷烯与Cd1-xZnxS机械混合制备了复合材料,分析均表明二者形成了强的界面电子耦合,促进了光生电荷的迁移与分离,复合样品的最大产氢活性达到纯Cd1-xZnxS的20余倍(图 8)[61]。这项工作不仅证明了单层黑磷可以作为优异的2D基底来制备0D/2D异质结光催化剂,而且也为这种特殊的异质结构在不同领域的应用提供了一定参考。随后,李希友课题组通过类似机械复合成功制备了BP/Cd0.5Zn0.5S光催化剂,当使用510 nm以上波长的光照射时,BP/ Cd0.5Zn0.5S仍然具有高产氢活性,表明BP与Cd0.5Zn0.5S的异质界面不仅有效促进了光生电荷的界面分离,更拓宽了复合样品的有效光吸收范围[62],该工作进一步证明BP可作为高效的析氢助剂,也证明BP可作为光敏剂吸收低能光子,进而注入Cd0.5Zn0.5S导带,使长波段的低能光子得到有效利用,但是BP的制备条件较为苛刻,且在空气中的稳定性存在很大问题。
我们课题组通过溶剂热法成功构建了三元Cd0.5Zn0.5S/RGO/g-C3N4 Z型异质结,其最优活性相比于纯g-C3N4和Cd0.5Zn0.5S分别提高了48.4倍和8.1倍,这归因于RGO存在时Cd0.5Zn0.5S纳米颗粒和g-C3N4纳米片之间的协同效应,RGO则充当了两者之间的载流子传输媒介[63]。我们还将Cd0.5Zn0.5S量子点负载于碳量子点薄膜和TiO2纳米薄膜表面,虽然碳膜作为桥梁可使TiO2导带电子与Cd0.5Zn0.5S价带空穴快速复合形成Z型电荷转移机制,但薄膜形态的催化剂总体产氢效率不高,约38.74 mmol·h-1·m-2,仅为TiO2薄膜制氢速率的3.4倍[64]。王传义课题组将Cd0.8Zn0.2S/Au/g-C3N4三元材料用于光解水产氢,并以生物质葡萄糖作为电子给体代替Na2S/ Na2SO3或三乙醇胺,葡萄糖的引入提高了该体系的经济性,而利用Au作为电子传输媒介形成的Z型异质结显著抑制了载流子的表面复合。相比于Au/g-C3N4,引入硫化物后体系产生的羟基自由基与葡萄糖反应形成的葡萄糖酸对抑制气体副产物的生成起着至关重要的作用[65]。以上工作均有力地证明了选择适当的材料合理构建半导体异质结能显著抑制光生电荷的表面复合,从而增强光催化活性。但三元体系的制备过程较为繁琐,涉及到的界面电荷传输、表面催化反应过程也更为复杂。另外,上述研究主要致力于构建Cd1-xZnxS基复合光催化剂以抑制光生电荷表面复合,对其表面反应动力学过程机理探究仍十分欠缺。
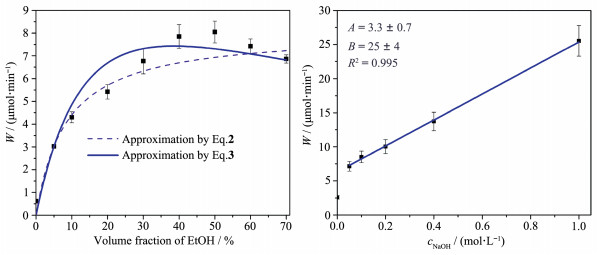
王国荣等将1D Cd0.3Zn0.7S与2D NiWO4相结合,两者由于匹配的能带结构形成了p-n结,形成的内建电场促进了光生电荷的界面分离。此外,作者发现牺牲剂中Na2S的浓度对溶液pH有较大影响,进而影响催化剂平带电位,致使不同浓度牺牲剂下产生活性差异[66]。不同浓度牺牲剂溶液除影响催化剂平带电位外,还直接影响光催化反应的表面反应速率,这给我们提供了一种新的可能,即借助对溶剂环境的调控来提高表面反应速率,从而对催化剂应用于某一反应时的活性进行进一步优化。Markovskaya等在Ni(OH)2/Cd0.3Zn0.7S上负载了质量分数约1%的Pt,在NaOH溶液中,以乙醇为电子给体,借助Langmuir-Hinshelwood吸附模型分别研究了乙醇及NaOH浓度与H2生成速率的关系,发现乙醇与H2生成速率的关系更符合双分子反应,而NaOH浓度与H2生成速率成正比,最终获得了以乙醇浓度和NaOH浓度为参数的总反应速率表达式(图 9)[67]。基于此,作者仅通过反应条件的调控,使催化剂在450 nm处的表观量子效率达到了90%以上。该工作证明,通过反应条件的优化来实现光催化产氢效率的大幅度提升是可行的,并可为进一步相关研究提供重要借鉴,同时,通过动力学研究,可为光催化反应器的设计提供重要理论依据。Mar- kovskaya等还研究了Zn(OH)2/Cd0.3Zn0.7S在不同牺牲剂溶液下的产氢行为与其结构的联系。在Na2S/ Na2SO3牺牲剂中,Zn(OH)2/Cd0.3Zn0.7S在反应完成后其表面Zn(OH)2转变为了ZnS,最终形成ZnS/ Cd0.3Zn0.7S。而在乙醇牺牲剂中,Zn(OH)2/Cd0.3Zn0.7S表面的Zn(OH)2由β相转变为ε相,是真正的活性组分[68]。可见,在光照下,牺牲剂对光催化剂活性及组成有较大影响,后续研究应更应关注这一点,从而更加科学、深入地认识光催化剂活性与其组成及溶液环境间的关系。吕功煊课题组在Cd1-xZnxS表面包覆了一层超薄NiO层,该NiO层不仅能够抑制光腐蚀,同时还能促进光生电荷的分离。但NiO/ Cd1-xZnxS分解纯水产氢的活性仍然十分低下,同时还在液相中检测到了微量H2O2。当向反应液中加入少量全氟萘烷时,其产氢活性明显增大,这是由于体系抑制了H2/O2逆反应的缘故[69]。该工作首次将Cd1-xZnxS基光催化剂用于分解纯水,但是,Cd1-xZnxS的光腐蚀源于光生空穴对晶格硫离子的氧化,即水氧化半反应与光腐蚀反应具有竞争性。此项工作中未说明光生空穴在体系中起何作用,即未阐明水分解的氧化半反应与所制备催化剂良好稳定性间的联系。
Eq.5:
离子掺杂通常能在半导体光催化剂内引入新的施主或受主能级,产生新的能级结构,能够显著影响光催化的光吸收特性。济南大学刘福田课题组以ZIF-8为硬模板,通过离子交换法分别制备了Cd1-xZnxS和具有多孔结构的Co掺杂的Cd1-xZnxS固溶体光催化剂。在可见光照射下,这种Cd1-xZnxS产氢速率比水热法制备的相同样品高2倍多,作者认为这主要是由于材料孔结构的增多,Co的掺杂进一步提高了Cd1-xZnxS在可见光区的光吸收能力[70]。然而,文中缺乏直观的证据以证明多孔结构的存在,另外,应在单色光下进行实验以直接说明Co掺杂引起的光吸收增强是否对产氢过程有直接贡献。Hao等采用一步水热法制备了Cu/Ni共掺杂的Cd0.5Zn0.5S纳米颗粒,当提高Ni/Cu的总掺杂量为1%(质量分数)时则形成了NiS与Cu2S负载于Cd0.5Zn0.5S表面的情况,其活性相比于原始Cd0.5Zn0.5S提高了3倍多,分析表明复合样品在可见光区光吸收能力大幅提升,PL强度则明显降低,有效提高了光生电荷的分离效率[71],但同样地,文中缺乏直接证据证明光吸收能力的提高对析氢过程有直接贡献。谢广文课题组发现当Ni掺杂的质量分数为0.5%时,Cd1-xZnxS在可见光下的产氢活性达到25.4 mmol·h-1·g-1,甚至高于Pt/Cd0.5Zn0.5S(Pt的质量分数为3%)。引入的Ni原子与S原子相键合,不仅促进了光吸收能力的增强还有效降低了表面反应势垒[72]。总的来说,借助掺杂以进一步提高Cd1-xZnxS光催化产氢活性的研究相对较少,导致这一现象的原因可能有2个:(1)原子掺杂对于Cd1-xZnxS的活性提高有限;(2)相比于助催化剂负载和异质结构建,缺乏直接有效的手段对掺杂所引起的如材料能带与结构变化进行表征分析。
由以上可知,目前关于究竟Zn/Cd比为多少时,单体Cd1-xZnxS具有最佳的光催化活性仍然没有统一的认识,Cd0.2Zn0.8S、Cd0.3Zn0.7S、Cd0.5Zn0.5S等均有报道,因此,亟需进一步从分子水平阐明不同Zn/Cd比的Cd1-xZnxS析氢活性与其晶体结构、光吸收、牺牲剂种类等的内在联系。
其次,牺牲剂(Na2S/Na2SO3、乳酸、抗坏血酸等)存在时的析氢半反应的基本反应途径和机理已经被充分认识,并且牺牲剂价格昂贵,增加了反应体系经济成本,但大多数研究者仍在牺牲剂体系对其活性进行评价,因此,进一步拓展Cd1-xZnxS的研究范围,降低体系经济成本显得十分有必要。考虑到Cd1-xZnxS相比于其他催化剂具有十分优异的析氢活性,深入研究Cd1-xZnxS基材料在无牺牲剂时的分解水性能显然更有意义也更具挑战性。CdS与Cd1-xZnxS具有相似的物理化学性质。2019年,卢小泉等采用锌-4-酰苯卟啉修饰的CdS分解纯水,其产氢活性为344.74 μmol·h-1·g-1,但并未检测到O2[73]。陈勇等将P掺杂CdS用于分解纯水,除H2外,在液相产物中检测到少量H2O2,随后在反应液中加入全氟萘烷和氯代血红素时发现其产氢活性大大提高,证明反应产物中存在难以检测到的微量O2[74]。以上工作证明,将硫化物用于分解纯水是可行的。此外,吕功煊课题组的工作及我们课题组的前期探索均表明Cd1-xZnxS用于纯水分解是可行的,但是其在纯水中的光腐蚀却更为严重[18, 69]。
有研究表明,Cd1-xZnxS在碱性Na2S/Na2SO3牺牲剂体系中具有良好的稳定性,而在酸性体系,由于H+与Cd1-xZnxS作用而释放H2S,导致稳定性欠佳[75]。目前对其在光催化反应过程中的光腐蚀机理研究仍非常有限,通常认为,这主要是由于催化剂表面空穴富集而导致的。在无电子给体时,水氧化半反应与晶格S2-离子光腐蚀氧化之间存在竞争性过程,这意味着表面光腐蚀与催化剂表面反应速率密切相关,同时意味着表面反应的增强(空穴的快速利用)将大大提高硫化物光催化剂的稳定性。2010年,Zyoud等将CdS/TiO2用于光降解苯并吡啶时偶然发现随着溶液pH值升高,反应溶液中游离Cd2+浓度降低,表明反应液pH与CdS光腐蚀速率存在联系[76]。我国学者李越湘等以葡萄糖为牺牲剂,发现部分无机盐离子对Pt/Cd0.5Zn0.5S产氢速率有促进作用[77]。Fuku等则发现BiVO4光阳极的水氧化速率及产物与电解质种类密切相关[78-79]。近期,浙江理工大学刘诗咏课题组发现以抗坏血酸为牺牲剂时,共轭有机聚合物半导体在不同溶剂环境下显现出迥异的光解水制氢活性[80]。我们课题组在研究中也发现类似现象,表明存在“电解质-催化剂效应”,也表明光催化剂表面反应速率与其所处溶液环境密切相关,包括溶质种类、pH等均对光催化剂的产氢活性有巨大影响,但关于这些因素的深入研究仍鲜见报道。
因此,十分有必要在充分认识光催化反应过程中光腐蚀反应的基础上,进一步结合结构调控与溶液环境调控,同步强化Cd0.5Zn0.5S的光生电荷表面分离及表面催化过程以达到克服光腐蚀的目的;在通过负载助催化剂、构建异质结等方法对光催化剂进行改性时可以结合反应条件的调控,以期构建具有高活性的Cd1-xZnxS光催化体系,从而推动Cd1-xZnxS基光催化剂走向实际应用。
Fujishima A, Honda K. Nature, 1972, 238:37-38 doi: 10.1038/238037a0
Li X, Yu J G, Low J X, et al. J. Mater. Chem. A, 2015, 3:2485-2534 doi: 10.1039/C4TA04461D
Sun S C, Zhang X Y, Liu X L, et al. Acta Phys.-Chim. Sin., 2020, 36:1905007 doi: 10.3866/PKU.WHXB201905007
Maeda K, Teramura K, Lu D L, et al. Nature, 2006, 440:295-295 doi: 10.1038/440295a
Asahi R, Morikawa T, Ohwaki T, et al. Science, 2001, 293:269-271 doi: 10.1126/science.1061051
Pelaez M, Nolan N T, Pillai S C, et al. Appl. Catal. B, 2012, 125:331-349 doi: 10.1016/j.apcatb.2012.05.036
Tada H, Jin Q, Iwaszuk A, et al. J. Phys. Chem. C, 2014, 118:12077-12086 doi: 10.1021/jp412312m
Pasternak S, Paz Y. ChemPhysChem, 2013, 14:2059-2070 doi: 10.1002/cphc.201300247
Jia H P, Zhang X, Du A J, et al. J. Am. Chem. Soc., 2008, 130:11256-11257 doi: 10.1021/ja803582m
Bai S, Jiang J, Zhang Q, et al. Chem. Soc. Rev., 2015, 44:2893-2939 doi: 10.1039/C5CS00064E
Yang J H, Wang D G, Han H X, et al. Acc. Chem. Res., 2013, 46:1900-1909 doi: 10.1021/ar300227e
Reber J F, Rusek M. J. Phys. Chem., 1986, 90:824-836 doi: 10.1021/j100277a024
Song J G, Zhao H T, Sun R R, et al. Energy Environ. Sci., 2016, 10:225-235
Liu M C, Jing D W, Zhou Z H, et al. Nat. Commun., 2013, 4:1-8
Chen J M, Chen J Y, Li Y W. J. Mater. Chem. A, 2017, 46:24116-24125
Zhang C Y, Liu H H, Wang W N, et al. Appl. Catal. B, 2018, 239:309-316 doi: 10.1016/j.apcatb.2018.08.027
Jin Z L, Liu Y, Hao X Q. J. Colloid Interface Sci., 2020, 567:357-368 doi: 10.1016/j.jcis.2020.02.024
Xue W H, Chang W X, Hu X Y, et al. Chinese J. Catal., 2021, 42:152-163 doi: 10.1016/S1872-2067(20)63593-8
Lv J L, Zhang J F, Dai K, et al. Dalton Trans., 2017, 46:11335-11343 doi: 10.1039/C7DT01892D
桑换新, 葛永慧, 刘兴静,等.化学通报, 2018, 81(4):349-354SANG Huan-Xin, GE Yong-Hui, LIU Xing-Jing, et al. Chemistry Bulletin, 2018, 81(4):349-354
Pemasiri K, Montazeri M, Gass R, et al. Nano Lett., 2009, 9:648-654 doi: 10.1021/nl802997p
Liu M C, Wang L Z, Lu G Q, et al. Energy Environ. Sci., 2011, 4:1372-1378 doi: 10.1039/c0ee00604a
Thelander C, Caroff P, Plissard S, et al. Nano Lett., 2011, 11:2424-2429 doi: 10.1021/nl2008339
Huang H B, Fang Z B, Yu K, et al. J. Mater. Chem. A, 2020, 8:3882-3891 doi: 10.1039/C9TA13836F
Li K Q, Xiong H L, Wang X, et al. Inorg. Chem., 2020, 59:5063-5071 doi: 10.1021/acs.inorgchem.0c00290
Zhang K, Dai Y W, Zhou Z H, et al. Nano Energy, 2017, 41:101-108 doi: 10.1016/j.nanoen.2017.09.021
Hao M T, Hua Q Q, Lia J R, et al. Inorg. Chem. Commun., 2018, 93:20-24 doi: 10.1016/j.inoche.2018.04.027
Kaur M, Nagaraja C M. ACS Sustainable Chem. Eng., 2017, 5:4293-4303 doi: 10.1021/acssuschemeng.7b00325
Yuan Y J, Chen D Q, Yu Z T, et al. J. Mater. Chem. A, 2018, 6:11606-11630 doi: 10.1039/C8TA00671G
Abideen Z U, Teng F. Appl. Surf. Sci., 2019, 465:459-469 doi: 10.1016/j.apsusc.2018.09.195
Fu J W, Zhu B C, You W, et al. Appl. Catal. B, 2018, 220:148-160 doi: 10.1016/j.apcatb.2017.08.034
Ng B J, Putri L K, Kong X Y, et al. Appl. Catal. B, 2018, 224:360-367 doi: 10.1016/j.apcatb.2017.10.005
Chen Y C, Huang Y S, Huang H, et al. Nano Energy, 2020, 67:104225 doi: 10.1016/j.nanoen.2019.104225
Pu Y C, Chen W T, Fang M J, et al. J. Mater. Chem. A, 2018, 6:17503-17513 doi: 10.1039/C8TA05539D
Yue X Z, Yi S S, Wang R W, et al. Appl. Catal. B, 2018, 224:17-27 doi: 10.1016/j.apcatb.2017.10.010
Dai D S, Xu H, Ge L, et al. Appl. Catal. B, 2017, 217:429-436 doi: 10.1016/j.apcatb.2017.06.014
Dai D S, Wang L, Xiao N, et al. Appl. Catal. B, 2018, 233:194-210 doi: 10.1016/j.apcatb.2018.04.013
Dhingra S, Nagaraja C M. Int. J. Hydrogen Energy, 2018, 43:22917-22928 doi: 10.1016/j.ijhydene.2018.10.145
Wang P F, Zhan S H, Wang H T, et al. Appl. Catal. B, 2018, 230:210-219 doi: 10.1016/j.apcatb.2018.02.043
Zhu X L, Yu S J, Gong X Z, et al. ChemPlusChem, 2018, 83:825-830 doi: 10.1002/cplu.201800316
Cheng L, Zhang D N, Fan J J, et al. Appl. Catal. A, 2020, 590:117336 doi: 10.1016/j.apcata.2019.117336
Yan X, Jin Z L, Zhang Y P, et al. Dalton Trans., 2019, 48:11122-11135 doi: 10.1039/C9DT01421G
An C H, Feng J, Liu J X, et al. Inorg. Chem. Front., 2017, 4:1042-1047
Li Y Y, Sun B W, Lin H F, et al. Appl. Catal. B, 2020, 267:118702 doi: 10.1016/j.apcatb.2020.118702
Zhao X X, Feng J R, Liu J. et al. Angew. Chem. Int. Ed., 2018, 57:9790-9794 doi: 10.1002/anie.201805425
Lin H F, Sun B W, Wang H, et al. Small, 2019, 15:1804115 doi: 10.1002/smll.201804115
Wei Z D, Xu M Q, Liu J Y, et al. Chinese J. Catal., 2020, 41:103-113 doi: 10.1016/S1872-2067(19)63479-0
Zhou X L, Wang X N, Feng X, et al. ACS Appl. Mater. Interfaces, 2017, 9:22560-22567 doi: 10.1021/acsami.7b05592
Zhang S, Li B F, Wang X X, et al. Chem. Eng. J., 2020, 390:124642 doi: 10.1016/j.cej.2020.124642
Hou H L, Zeng X K, Zhang X W. Sci. China Mater., 2020, doi: 10.1007/s40843-019-1256-0
Li X, Shen R C, Ma S, et al. Appl. Surf. Sci., 2018, 430:53-107 doi: 10.1016/j.apsusc.2017.08.194
Kuang P Y, Sayed M, Fan J J, et al. Adv. Energy Mater., 2020, 14:1903802
Feng W H, Wang Y Z, Huang X Y, et al. Appl. Catal. B, 2018, 220:324-336 doi: 10.1016/j.apcatb.2017.08.002
Wang L, Yang G R, Wang S L, et al. Int. J. Hydrogen Energy, 2019, 44:30974-30985 doi: 10.1016/j.ijhydene.2019.09.209
Mei F F, Li Z, Dai K, et al. Chinese J. Catal., 2020, 41:41-49 doi: 10.1016/S1872-2067(19)63389-9
Shi J J, Li S D, Wang F M, et al. Catal. Sci. Technol., 2018, 8:6458-6467 doi: 10.1039/C8CY01884G
Shi J J, Li S D, Wang F M, et al. Dalton Trans., 2019, 48:3327-3337 doi: 10.1039/C8DT04154G
Li H Y, Hao X Q, Liu Y, et al. J. Colloid Interface Sci., 2020, 572:62-73 doi: 10.1016/j.jcis.2020.03.052
Zheng Y, Chen Y L, Gao B F, et al. Adv. Funct. Mater., 2020, 30:2002021 doi: 10.1002/adfm.202002021
Pang J B, Bachmatiuk A, Yin Y, et al. Adv. Eng. Mater., 2018, 8:1702093 doi: 10.1002/aenm.201702093
Ran J R, Wang X L, Zhu B C, et al. Chem. Commun., 2017, 53:9882-9885 doi: 10.1039/C7CC05466A
Zhao H T, Liu H Y, Sun R R, et al. ChemCatChem, 2018, 10:4395-4405 doi: 10.1002/cctc.201800827
Xue W H, Hu X Y, Liu E Z, et al. Appl. Surf. Sci., 2018, 447:783-794 doi: 10.1016/j.apsusc.2018.04.048
Liu E Z, Xu C H, Jin C Y, et al. J. Taiwan Inst. Chem. E, 2019, 97:316-325 doi: 10.1016/j.jtice.2019.02.027
Zhao H, Ding X L, Zhang B, et al. Sci. Bull., 2017, 9:602-609
Liu Y, Wang G R, Li Y B, et al. J. Colloid Interface. Sci., 2019, 554:113-124 doi: 10.1016/j.jcis.2019.06.080
Markovskaya D V, Kozlova E A, Gerasimov E Y, et al. Appl. Catal. A, 2018, 563:170-176 doi: 10.1016/j.apcata.2018.07.002
Markovskaya D V, Cherepanova S V, Gerasimov E Y, et al. RSC Adv., 2020, 10:1341-1350 doi: 10.1039/C9RA08833D
Ning X F, Zhen W L, Zhang X Q, et al. ChemSumChem, 2019, 12:1410-1420
Tang X, Zhao J H, Li Y H, et al. Dalton Trans., 2017, 46:10553-10557 doi: 10.1039/C7DT01970J
Yu T P, Sun K L, Wang K H, et al. Mater. Lett., 2019, 239:159-162 doi: 10.1016/j.matlet.2018.12.091
Hao Y J, Kang S Z, Liu X, et al. ACS Sustainable Chem. Eng., 2017, 5:1165-1172 doi: 10.1021/acssuschemeng.6b02499
Ning X M, Wu Y L, Ma X F, et al. Adv. Funct. Mater., 2019, 29:1902992 doi: 10.1002/adfm.201902992
Shi R, Ye H F, Liang F, et al. Adv. Mater., 2018, 30:1705941 doi: 10.1002/adma.201705941
Li S B, Yesodharan S, Griitzel M. Phys. Chem., 1985, 89:121-124
Zyoud A H, Zaatar N, Saadeddin I, et al. J. Hazard. Mater., 2010, 173:318-325 doi: 10.1016/j.jhazmat.2009.08.093
Li Y X, Gao D, Peng S Q, et al. Int. J. Hydrogen Energy, 2011, 36:4291-4297 doi: 10.1016/j.ijhydene.2011.01.038
Fuku K, Miyase Y, Miseki Y, et al. ChemistrySelect, 2016, 1:5721-5726 doi: 10.1002/slct.201601469
Fuku K, Sayama K. Chem. Commun., 2016, 52:5406-5409 doi: 10.1039/C6CC01605G
Cheng J Z, Liu L L, Liao G F, et al. J. Mater. Chem. A, 2020, 8:5890-5899 doi: 10.1039/C9TA13514F

图 4 ZB/WZ超晶格锯齿状的电位分布示意图
Figure 4 Schematic diagram of the sawtooth potential in ZB/WZ superlattice

图 5 ZnS、Zn0.82Cd0.18S、CdS纳米球的扫描电镜图(a~c)和透射电镜图(d~f); Zn0.82Cd0.18S的(g)高分辨透射电镜图; (h、i) (g)中对应于WZ相及ZB相的选区电子衍射图; (j) Zn0.82Cd0.18S纳米球元素mapping图[25]
Figure 5 (a~c) Scanning electron microscope images and (d~f) transmission electron microscope images of ZnS, Zn0.82Cd0.18S, and CdS; (g) High resolution transmission electron microscope image of Zn0.82Cd0.18S; (h, i) Selected area electron diffraction patterns of WZ and ZB region in (g); (j) Element mapping images of Zn0.82Cd0.18S[25]

图 7 不同类型异质结示意图
Figure 7 Schematic diagram of different heterojunctions in photocatalysis

图 8 (a~c) 2%-BP/Cd1-xZnxS(2%为BP质量分数)的透射电镜图、高分辨透射电镜图及能谱图; (d~e) BP和2%-BP/Cd1-xZnxS的XRD图及N2吸附等温线; (f)样品的UV-Vis漫反射光谱[61]
Figure 8 (a~c) Transmission electron microscope, high resolution transmission electron microscope and energy dispersive spectroscope of 2%-BP/Cd1-xZnxS (2% is the mass fraction of BP); (d~e) XRD patterns and N2 absorption isotherms of phosphorene and 2%-BP/Cd1-xZnxS; (f) UV-Vis diffuse reflectance spectra of the samples[61]

图 9 产氢速率的动力学拟合曲线:产氢速率与乙醇浓度的关系(左图, cNaOH=0.1 mol·L-1, 方程式5和6分别对应采用单分子反应和双分子反应模型时的拟合曲线); 产氢速率与NaOH浓度的关系(右图, EtOH体积分数为50%, W=A+BcNaOH, A与B分别为表观速率常数, R2为相关系数)[67]
Figure 9 Dynamics fitting curve of H2 evolution: relationship between H2 production rate and ethanol concentration(left, cNaOH= 0.1 mol·L-1, Eq.5 and 6 correspond to the fitting curves using single molecular and bimolecular model, respectively); Relationship between H2 production rate and NaOH concentration (right, volume fraction of EtOH is 50%, W=A+BcNaOH, A and B are apparent rate constant, and R2 is correlation coefficient)[67]
Eq.5:

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:



 下载:
下载: