Received Date:
29 August 2019 Revised Date:
31 December 2019 Available Online:
10 April 2020
Abstract:
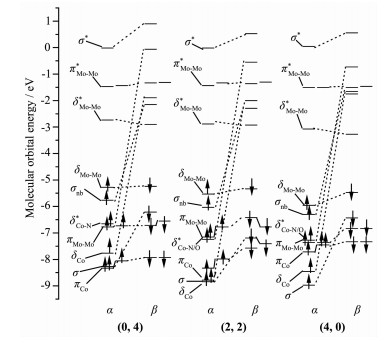
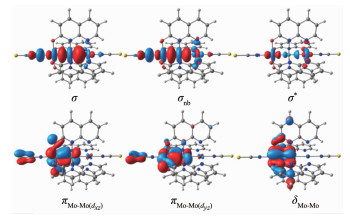
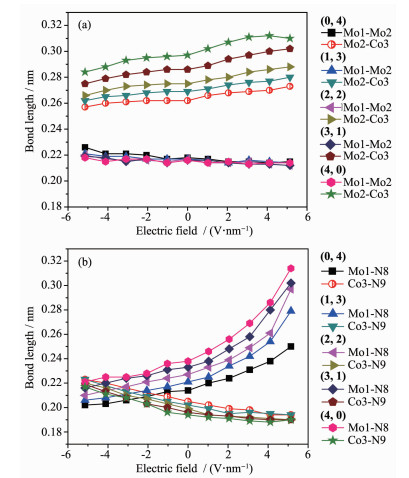
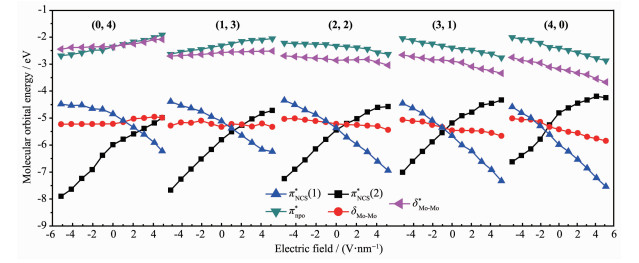
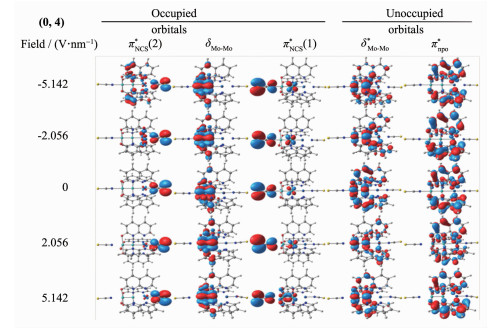
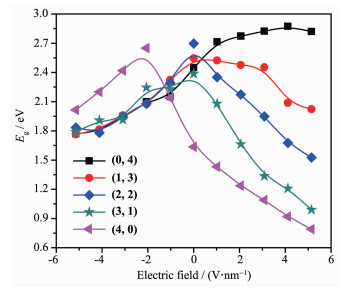
Metal string complexes, with the structure of linear metal chain helically wrapped by four equatorial ligands, have attracted extensively attention due to their unique electronic, magnetic, and potential applications in molecular electronics. Some factors such as difference of metal atoms, axial ligands and equatorial ligands would affect the physical properties of conductance and magnetic properties of metal string complexes. The diversity of equatorial ligands provides more possibilities for such changes. The coordination structures of metal string complexes[MoMoCo(npo)4(NCS)2] (npo=1, 8-naphthyl-2-ketone) with potential applications as molecular wires have been investigated using the density functional theory B3LYP method by considering the effects of an external electric field (EF). The coordination mode is denoted as (n, m), where n and m represent the number of oxygen atoms coordinated with the Co3 and Mo1, respectively, and n=0, 1, 2, 3, 4; m=4, 3, 2, 1, 0. The energies and polarities of these molecules increase gradually as the coordination modes of four npo- ligands become more and more consistent, but all of them can exist stably and compete with each other. The Mo-Mo quadruple bond exists in all molecules, and the bond length decreases with the decrease of the Z-direction dipole moment μ(Z). In addition, as the value of μ(Z) decreases, the orbital energy of πNCS*(1) decreases but that of πNCS*(2) increases. The geometric and electronic structures of the five coordination modes change regularly under the action of electric field. Under the electric field effect of Z direction, the Mo1-N8 bond lengths of all coordination modes except (0, 4) increase obviously, leading to structural instability. Moreover, the phenomenons of energy level interlacing in the frontier orbitals, and the reduction of LUMO-HOMO energy gap are related to the value of μ(Z). When μ(Z) is positive, the energy gaps of (0, 4) and (1, 3) decrease more significantly under the electric field effect of -Z direction. However, when μ(Z) is negative, the energy gaps of (2, 2), (3, 1) and (4, 0) decrease more obviously under the electric field effect of Z direction. Therefore, the complexes of (0, 4), (3, 1) and (4, 0) may have the rectification effect, but (3, 1) and (4, 0) are less stable.


 下载:
下载:


 下载:
下载:









 下载:
下载:
