College of Chemistry, Nankai University, Tianjin 300071, China
2.
Collaborative Innovation Center of Chemical Science and Engineering, Tianjin 300071, China
3.
Key Laboratory of Advanced Energy Materials Chemistry(Ministry of Education), Nankai University, Tianjin 300071, China
Received Date:
12 June 2018 Revised Date:
04 October 2018 Available Online:
10 December 2018
Abstract:
Novel silicate nanotubes supported amorphous Co-B (Co-B/MgSNTs) were synthesized via a hydrothermal method followed by impregnation-chemical reduction processes. The catalysts were characterized with X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectrometer (XPS), atomic emission spectrometer (ICP) and brunauer-Emmett-Tellerv (BET) surface-area analyzers. The catalytic activity and recyclability of catalysts for hydroformylation of cyclohexene were evaluated. The conversion of cyclohexene over the catalysts is 75.8% and selectivity for aldehyde is 65.8%. The results indicate that Co-B nanoparticles were deposited on the inner and outer surface of MgSNTs, which had large specific surface area and high stability. Furthermore, the tubular structure of nanotubes could prevent nanoparticle from agglomeration. The catalyst used for 3 recycles shows good catalytic activity.
(a) MgSNTs; (b) MgSNTs-300; (c) Co-B/MgSNTs; (d) Co-B/MgSNTs-300; (e) Co-B/MgSNTs-300-4; (f) Co-B/MgSNTs; (g) Co-B/MgSNTs-300; (h) Co-B/MgSNTs-300-4; d is average particle size and σ is standard deviation
Figure 2.
TEM images and particle size distributions graphs of particles of samples
Figure 2
TEM images and particle size distributions graphs of particles of samples
(a) MgSNTs; (b) MgSNTs-300; (c) Co-B/MgSNTs; (d) Co-B/MgSNTs-300; (e) Co-B/MgSNTs-300-4; (f) Co-B/MgSNTs; (g) Co-B/MgSNTs-300; (h) Co-B/MgSNTs-300-4; d is average particle size and σ is standard deviation

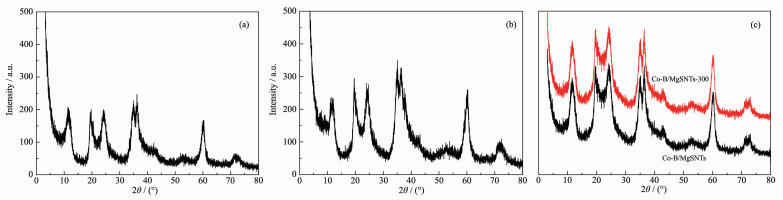
 下载:
下载:


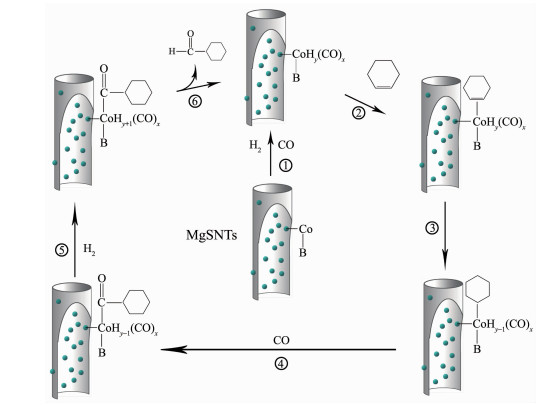

 下载:
下载:





 下载:
下载:
