Table 1.
Crystal data and structure refinement for 1 and 2
Citation:
LI Xiu-Mei, PAN Ya-Ru, LIU Bo, ZHOU Shi, CHANG Ying-Fei. Syntheses, Crystal Structures and Theoretical Calculations of Cadmium/Zinc Supramolecular Coordination Compounds[J]. Chinese Journal of Inorganic Chemistry,
2018, 34(10): 1923-1928.
doi:
10.11862/CJIC.2018.225

两个镉/锌超分子配合物的合成、晶体结构及理论计算
English
Syntheses, Crystal Structures and Theoretical Calculations of Cadmium/Zinc Supramolecular Coordination Compounds
Abstract:
Two new metal-organic supramolecular coordination compounds[Cd(cba)2(bix)]2 (1) and[Zn(cba)2(bix)]2 (2) (Hcba=2-chlorinebenzoic acid, bix=1, 4-bis(imidazol-1-ylmethyl) benzene) have been hydrothermally synthesized and structurally characterized by elemental analysis, IR spectrum, TG, fluorescence spectrum, single-crystal and powder X-ray diffraction. Complexes 1 and 2 are isomorphism, and all exhibit zero-dimensional framework and display three-dimensional supramolecular network via π-π stacking interactions. In addition, natural bond orbital (NBO) of 1 were analyzed by using the PBE0/LANL2DZ method built in Gaussian09 program. The calculation results showed the obvious covalent interaction between the coordinated atoms and Cd(Ⅱ) ion.
-
Key words:
- hydrothermal synthesis
- / crystal structure
- / cadmium complex
- / zinc complex
- / natural bond orbital
-
Coordination polymers with bridged transition metals have received intense interest and attention for their fascinating architectures and potential applications as materials in optical, electronic, magnetic fields, gas storage, catalysis and so on[1-5]. Consequently, numerous new complexes can be specially designed by the careful selection of metal centers with preferred coordination geometries[6-9]. It has been observed that organic ligands play crucial roles in the preparation of some interesting coordination networks, such as flexibility, donating type, and the geometry of organic ligands[10-11]. Among various organic ligands, aromatic carboxylates have been extensively used because of their extension ability in both covalent bonding and supramolecular interactions (H-bonding and aromatic stacking). For example, benzoic acid, 1, 3 -benzenedicarboxylate, 1, 4-benzenedicarboxylate, 1, 2-benzenedicarboxylate, and 1, 2, 4, 5-benzene tetracarboxylate[12-14] are well used in the construction of CPs due to their structural rigidity, chemical stability and appropriate connectivity. Besides the carboxylate linkers, bis(imidazole) bridging ligands with different length and flexibly, for example, 1, 3-bis(imidazol-1-ylmethyl)-benzene, 1, 4-bis(imidazol-1-ylmethyl)-benzene, 1, 4-bis(imidazol-1-yl)-butane are frequently used in the assembly process of CPs as bridging linker[13-15].
Based on all the aspects stated above, herein we report the synthesis and crystal structure of two coordination supramolecules, [Cd(cba)2(bix)]2 (1) and [Zn(cba)2(bix)]2 (2). In the solid state, complexes 1 and 2 form a three-dimensional (3D) network resulted from π-π stacking interactions.
1. Experimental
1.1 General procedures
All reagents were purchased commercially and used without further purification. Elemental analyses (C, H and N) were measured on a Vario EL Ⅲ Elemental Analyzer. IR spectrum was recorded in the range of 4 000~400 cm-1 on a Nicolet 6700 spectrometer using a KBr pellet. TG studies were carried on a STA7300 analyzer under nitrogen at a heating rate of 10 ℃·min-1. The fluorescent spectra were obtained on a computer-controlled JY Fluoro-Max-3 spectrometer at room temperature. The power X-ray diffraction (PXRD) studies were performed with a Bruker D8 Discover instrument (Cu Kα radiation, λ=0.154 184 nm, U=40 kV, I=40 mA) over the 2θ range of 5°~50° at room temperature.
1.2 Synthesis
The pH value of the mixture of 0.2 mmol M(OAc)2·2H2O (M=Cd, 1; Zn, 2), Hcba (0.3 mmol, 0.047 g), bix (0.3 mmol, 0.071 5 g), 1 mL C2H5OH and 9 mL H2O was adjusted to 7 with 40% NaOH, sealed in a Teflon -lined stainless steel vessel, heated to 120 ℃ for 5 days, and followed by slow cooling (a descent rate of 5 ℃·h-1) to room temperature. Colorless block crystals were obtained.
[Cd(cba)2(bix)]2 (1): Yield: 25% (based on Cd). Anal. Calcd. for C56H44Cd2Cl4N8O8(%): C, 50.81; H, 3.35; N, 8.47. Found(%): C, 50.05; H, 2.96; N, 8.02. IR (cm-1): 3 118w, 1 581s, 1 555s, 1 517s, 1 432w, 1 388s, 1 279w, 1 234w, 1 107m, 1 086m, 1 051w, 934w, 850w, 749m, 723w, 695s, 652s, 620w.
[Zn(cba)2(bix)]2 (2): Yield: 32% (based on Zn). Anal. Calcd. for C56H44Cl4N8O8Zn2(%): C, 54.70; H, 3.61; N, 9.11. Found(%): C, 54.05; H, 3.14; N, 8.62. IR (cm-1): 3 431w, 3 134w, 1 623s, 1 521w, 1 432w, 1 358s, 1 101 m, 1 051w, 955w, 849w, 750m, 651w.
1.3 Structure determination
Single-crystal X-ray diffraction data for 1 and 2 were recorded on a Bruker D8 QUEST CMOS diffractometer with graphite-monochromated Mo Kα radiation (λ=0.071 073 nm) at 293 K. The structures were solved with the direct method of SHELXS-97 and refined with full-matrix least-squares techniques using the SHELXL-97 program[16-17]. The non-hydrogen atoms of the complexes were refined with anisotropic temperature parameters. The hydrogen atoms attached to carbons were generated geometrically. In addition, there is disorder in complexes 1 and 2. Crystallo-graphic parameters and the data collection statistics for structures 1 and 2 are given in Table 1. Selected bond lengths and bond angles are listed in Table 2.
Table 1

 下载:
导出CSV
下载:
导出CSV
Formula C56H44Cd2Cl4N8O8 C56H44Cl4N8O8Zn2 Formula weight 1 323.59 1 229.53 Crystal system Monoclinic Monoclinic Space group P21/n P21/n a / nm 0.981 35(16) 0.963 3(5) b / nm 2.871 0(5) 2.844 5(5) c / nm 1.009 11(17) 0.999 9(5) β /(°) 94.056(2) 95.045(5) Volume / nm3 2.836 0(8) 2.729(2) Z 2 2 Dc/ (g·cm-3) 1.550 1.496 θ range / (°) 1.42~26.05 1.43~25.35 F(000) 1 328 1 256 Reflection collected, unique [I > 2σ(I)] 15 459, 5 582, 4 726 14 068, 4 981, 3 722 Goodness-of-fit on F2 1.059 1.030 Rint 0.019 8 0.035 7 R1, wR2 [I > 2σ(I)] 0.027 5, 0.063 7 0.042 1, 0.106 2 Table 2
Table 2. Selected bond lengths (nm) and bond angles (°) for 1 and 2下载:
导出CSV
1 Cd(1)-O(1) 0.234 1(15) Cd(1)-O(2) 0.23 8 5(2) Cd(1)-O(3) 0.220 44(17) Cd(1)-N(1) 0.221 2(2)) Cd(1)-N(4A) 0.226 0(2) O(3)-Cd(1)-N(1) 130.42(8) O(3)-Cd(1)-N(4A) 93.91(7)) N(1)-Cd(1)-N(4A) 111.51(8) O(3)-Cd(1)-O(1) 111.9(2) N(1)-Cd(1)-O(1) 111.8(2) N(4A)-Cd(1)-O(1) 85.8(3) O(3)-Cd(1)-O(2) 94.79(8) N(1)-Cd(1)-O(2) 93.62(9) N(4A)-Cd(1)-O(2) 137.48(8) O(1)-Cd(1)-O(2) 52.5(3) 2 Zn(1)-O(2) 0.200 3(3) Zn(1)-O(3) 0.196 2(2) Zn(1)-N(1) 0.200 2(3) Zn(1)-N(4A) 0.204 4(3) O(3)-Zn(1)-N(1) 122.23(11) O(3)-Zn(1)-O(2) 117.86(13) N(1)-Zn(1)-O(2) 107.50(13) O(1')-Zn(1)-N(4A) 125.3(6) N(1)-Zn(1)-N(4A) 111.59(11) O(2)-Zn(1)-N(4A) 95.02(12) Symmetry codes: A: -x, -y, 1-z for 1; A: 2-x, -y, 1-z for 2. CCDC: 1829184, 1; 1828811, 2.
2. Results and discussion
2.1 Description of the structure
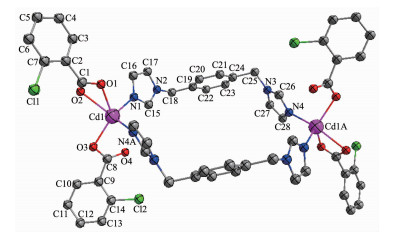
The single-crystal X-ray diffraction study reveals that complexes 1 and 2 are isomorphic. They all crystallize in monoclinic space group P21/n and feature zero-dimensional structure. Here, only the structure of complex 1 is described. The structure of complex 1 is shown in Fig. 1, the coordination of Cd ion can be described as a distorted tetragonal-pyramidal geometry. Each Cd(Ⅱ) ion in 1 is coordinated by three carboxylate oxygen atoms from two different cba ligands, and two nitrogen atom donors from two bridging bix ligands. The Cd-O bond distances are in the range of 0.232 84(19)~0.264 4(2) nm, and those of Cd-N are from 0.225 80(18) to 0.226 57(19) nm. The N(O)-Cd-O(N) angles fall in the 51.51(7)°~137.88(8)° range.
Figure 1
 Figure 1. Molecular structure of complex 1
Figure 1. Molecular structure of complex 1H atoms were omitted for clarity; Ellipsoids at 30% probability; Symmetry codes: A:-x, -y, 1-z
In complex 1, the bix ligands take cis-conformation bridging mode with a dihedral angle between the two imidazole rings of 59.09°, and the cba ligand coordinates to Cd ions through three carboxylic oxygen atoms in a monodentate and bidentate fashion, which gives rise to a double-nuclear subunit (Fig. 1). It should be mentioned that in the unit of 1 one type of 26-membered ring is formed (N(1)-Cd(1)-N(4) 111.52°, Cd…Cd 1.246 3 nm).
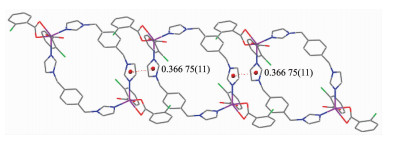
Further investigation of the crystal packing of complex 1 suggests that there are persistent π-π interactions in complex 1 between imidazole rings of bix ligands. The centroid-to-centroid distance between adjacent ring is 0.377 37(17) nm for N3C26N4C28C27 and N3′C26′N4′C28′C27′ (Symmetry codes: -1-x, -y, -z) imidazole rings. The perpendicular distance is 0.366 75(11) nm for N3C26N4C28C27 and N3′C26′N4′C28′C27′ (Symmetry codes: -1-x, -y, -z) imidazole rings. Therefore, through π-π interactions, complex 1 is further extended into a three-dimensional supramolecular framework (Fig. 2).
Figure 2
 Figure 2. View of the 3D supramolecular structure of complex 1 formed by π-π interactions
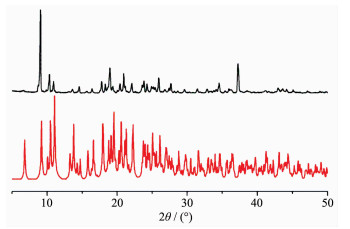
Figure 2. View of the 3D supramolecular structure of complex 1 formed by π-π interactionsTo investigate whether the analyzed crystal structure is truly representative of the bulk materials, X-ray powder diffraction (PXRD) technology has been performed for complex 1 at room temperature (Fig. 3). The main peak positions observed are in good agreement with the simulated ones. Although minor differences can be found in the positions, widths, and intensities of some peaks, the bulk synthesized materials and analyzed crystal can still be considered as homogeneous. The differences may be due to the preferred orientation of the powder samples[18-19].
Figure 3
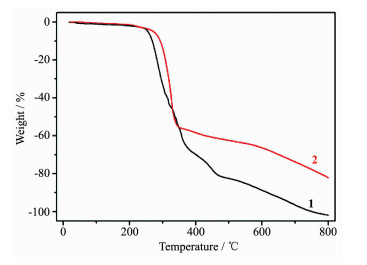
2.2 Thermal analysis
To characterize the thermal stability of complexes 1 and 2, TG curves have been obtained from crystalline samples in the flowing nitrogen atmosphere at a heating rate of 10 ℃·min-1. As depicted in Fig. 4, the TG curve of 1 shows that the complex is stable up to 240 ℃, and then decompose upon further heating. The TG curve of 2 shows that the complex is stable up to 230 ℃, and then decompose upon further heating.
Figure 4
2.3 Photoluminescent property
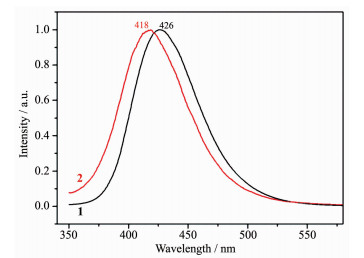
Luminescence property is very significant in photochemistry and photophysics[20-21]. The solid-state photoluminescence spectra of 1 and 2 were measured at room temperature (Fig. 5). Excited by 325 nm, complex 1 exhibits blue emission with the maximum peak at 418 nm. Complex 2 gives blue photoluminescence with an emission maximum at 426 nm upon excitation at 301 nm. In order to study the nature of these emission bands, we first analyzed the photoluminescence properties of free bix, Hcba ligands, and confirmed that they do not emit any luminescence in the range of 400~800 nm. Therefore, on the basis of the previous literature[22], the emission band could be vested to the emission of ligand-to-metal charge transfer (LMCT).
Figure 5
3. Theoretical calculation
All calculations in this work were carried out with the Gaussian09 program[23]. The parameters of the molecular structure for calculation were all from the experimental data of the complex. Natural bond orbital (NBO) analysis was performed by density functional theory (DFT)[24] with the PBE0[25-28] hybrid functional and the LANL2DZ basis set[29].
The selected natural atomic charges, natural electron configuration, wiberg bond and NBO bond orders (a.u.) for the complex 1 are shown in Table 3. It is indicated that the electronic configurations of Cd(1) ion, N and O atoms are 5s0.324d9.985p0.30, 2s1.382p4.22~4.23 and 2s1.68~1.712p5.02~5.06, respectively. Based on the above results, one can conclude that the Cd(1) ion coordination with N and O atoms is mainly on 4d, 5s and 5p orbitals. N atoms form coordination bonds with Cd(1) ion using 2s and 2p orbitals. All O atoms supply electrons of 2s and 2p to Cd(1) ion and form the coordination bonds. Therefore, the Cd(1) ion obtained some electrons from two N atoms of bix ligands and three O atoms of cba ligands[30]. Thus, according to valence-bond theory, the atomic net charge distribution and the NBO bond orders of the complex 1 (Table 3) shows the obvious covalent interaction between the coordinated atoms and Cd(1) ion. The differences of the NBO bond orders for Cd-O and Cd-N bonds make their bond lengths different[31], which is in good agreement with the X-ray crystal structure of complex 1.
Table 3
Table 3. Natural atomic charges, natural valence electron configurations, wiberg bond indexes and NBO bond orders for 1下载:
导出CSV
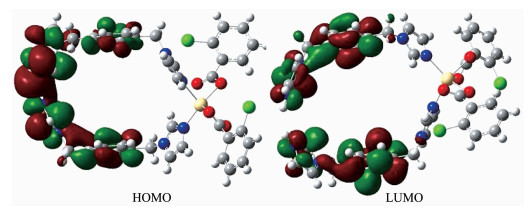
Atom Net charge Electron configuration Bond Wiberg bond index NBO bond order / a.u. Cd(1) 1.388 61 [core]5s0.324d9.985p0.30 O⑴ -0.746 52 [core]2s1.682p5.05 Cd(1)-O(1) 0.160 8 0.343 1 O(2) -0.732 38 [core]2s1.692p5.02 Cd(1)-O(2) 0.160 0 0.180 0 O(3) -0.784 08 [core]2s1.712p5.06 Cd⑴-O(3) 0.210 7 0.232 0 N⑴ -0.627 61 [core]2s1.382p4.23 Cd(1)-N(1) 0.184 3 0.175 0 N(4A) -0.621 45 [core]2s1.382p4.22 Cd(1)-N(4A) 0.172 2 0.247 5 As can be seen from the Fig. 6, lowest unoccupied molecular orbital (LUMO) is mainly composed of π orbits of imidazole rings in bix ligand, whereas highest occupied molecular orbital (HOMO) mainly consists of π orbits of benzene rings in bix ligand. So, ILCT may be inferred from some contours of molecular orbital of complex 1.
Figure 6
-
-
[1]
Noro S, Kitaura R, Kondo M, et al. J. Am. Chem. Soc., 2002, 124:2568-2583 doi: 10.1021/ja0113192
-
[2]
Dybtsev D N, Chun H, Kim K. Angew. Chem., Int. Ed., 2004, 43:50335036 http://europepmc.org/abstract/med/15384114
-
[3]
张中强, 黄如丹, 许颜清, 等.高等学校化学学报, 2008, 29:1528-1531 doi: 10.3321/j.issn:0251-0790.2008.08.007ZHANG Zhong-Qiang, HUANG Ru-Dan, XU Yan-Qing, et al. Chem. J. Chinese Universities, 2008, 29:1528-1531 doi: 10.3321/j.issn:0251-0790.2008.08.007
-
[4]
Ma L, Wu C D, Wanderley M M, et al. Angew. Chem. Int. Ed., 2010, 49:8244-8248 doi: 10.1002/anie.v49:44
-
[5]
Atkins R, Brewer G, Kokot E, et al. Inorg. Chem., 1985, 24:127-134 doi: 10.1021/ic00196a003
-
[6]
Liu G Z, Xin L Y, Wang L Y. CrystEngComm, 2011, 13:3013-3020 doi: 10.1039/c0ce00873g
-
[7]
Yan L, Li C B, Cui D, et al. Chin. J. Struct. Chem., 2015, 34:221-226
-
[8]
Li X L, Liu G Z, Xin L Y. Chin. J. Struct. Chem., 2013, 6:871-876 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=JGHX201306013&dbname=CJFD&dbcode=CJFQ
-
[9]
Zhang Q L, Liu J H, Ren X Z, et al. J. Inorg. Biochem., 2003, 95:194-198 doi: 10.1016/S0162-0134(03)00122-3
-
[10]
Yang J, Ma J F, Liu Y Y, et al. Cryst. Growth Des., 2009, 9:1894-1911 doi: 10.1021/cg801085d
-
[11]
Ye B H, Tong M L, Chen X M. Coord. Chem. Rev., 2005, 249:545-565 doi: 10.1016/j.ccr.2004.07.006
-
[12]
Li X M, Niu Y L, Wang Q W, et al. Chin. J. Struct. Chem., 2010, 7:1122-1126
-
[13]
李国峰, 王亚楠, 王庆伟, 等.无机化学学报, 2014, 11:2577-2583 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20141118&journal_id=wjhxxbcnLI Guo-Feng, WANG Yan-Nan, WANG Qing-Wei, et al. Chinese J. Inorg. Chem., 2014, 11:2577-2583 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20141118&journal_id=wjhxxbcn
-
[14]
Li X M, Sun M, Pan Y R, et al. Chin. J. Struct. Chem., 2016, 2:298-306
-
[15]
李国峰, 王亚楠, 王庆伟, 等.无机化学学报, 2015, 1:183-190 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150127&journal_id=wjhxxbcnLI Guo-Feng, WANG Yan-Nan, WANG Qing-Wei, et al. Chinese J. Inorg. Chem., 2015, 1:183-190 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150127&journal_id=wjhxxbcn
-
[16]
Sheldrick G M. SHELXS 97, Program for the Solution of Crystal Structure, University of Göttingen, Germany, 1997.
-
[17]
Sheldrick G M. SHELXL 97, Program for the Refinement of Crystal Structure, University of Göttingen, Germany 1997.
-
[18]
Gilbert A, Baggott J. Essentials of Molecular Photochemistry. Boca Raton:CRC Press, 1991.
-
[19]
Han Z B, He Y K, Ge C H, et al. Dalton Trans., 2007, 46:3020-3024
-
[20]
Mizukami S, Houjou H, Sugaya K, et al. Chem. Mater., 2005, 17:50-56 doi: 10.1021/cm049744s
-
[21]
Tang C W, Vanslyke S A. Appl. Phys. Lett., 1987, 51:913-915 doi: 10.1063/1.98799
-
[22]
Zheng S L, Chen X M. Aust. J. Chem., 2004, 57:703-712 doi: 10.1071/CH04008
-
[23]
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian09, revision B.09, Gaussian, Inc., Pittsburgh, 2009.
-
[24]
Parr R G, Yang W. Density Functional Theory of Atoms and Molecules. Oxford:Oxford University Press, 1989.
-
[25]
Ernzerhof M, Scuseria G E. J. Chem. Phys., 1999, 110:5029-5036 doi: 10.1063/1.478401
-
[26]
Adamo C, Barone V. J. Chem. Phys., 1999, 110:6158-6170 doi: 10.1063/1.478522
-
[27]
Perdew J P, Burke K, Ernzerhof M. Phys. Rev. Lett., 1996, 77:3865-3868 doi: 10.1103/PhysRevLett.77.3865
-
[28]
Perdew J P, Burke K, Ernzerhof M. Phys. Rev. Lett., 1997, 78:1396-1397
-
[29]
Dunning T H, Hay Jr P J. Modern Theoretical Chemistry. New York:Plenum, 1976:1-28
-
[30]
Wang L, Zhao J, Ni L, et al. J. Inorg. Gen. Chem., 2012, 638:224-230 http://www.ncbi.nlm.nih.gov/pubmed/22372487
-
[31]
李章朋.邢永恒, 张元红, 等.物理化学学报, 2009, 25:741-746 doi: 10.3866/PKU.WHXB20090423LI Zhang-Peng. XING Yong-Heng, ZHANG Yuan-Hong, et al. Acta Phys.-Chim. Sin., 2009, 25:741-746 doi: 10.3866/PKU.WHXB20090423
-
[1]
-
Figure 1 Molecular structure of complex 1
H atoms were omitted for clarity; Ellipsoids at 30% probability; Symmetry codes: A:-x, -y, 1-z
Figure 2 View of the 3D supramolecular structure of complex 1 formed by π-π interactions
Table 1. Crystal data and structure refinement for 1 and 2
Formula C56H44Cd2Cl4N8O8 C56H44Cl4N8O8Zn2 Formula weight 1 323.59 1 229.53 Crystal system Monoclinic Monoclinic Space group P21/n P21/n a / nm 0.981 35(16) 0.963 3(5) b / nm 2.871 0(5) 2.844 5(5) c / nm 1.009 11(17) 0.999 9(5) β /(°) 94.056(2) 95.045(5) Volume / nm3 2.836 0(8) 2.729(2) Z 2 2 Dc/ (g·cm-3) 1.550 1.496 θ range / (°) 1.42~26.05 1.43~25.35 F(000) 1 328 1 256 Reflection collected, unique [I > 2σ(I)] 15 459, 5 582, 4 726 14 068, 4 981, 3 722 Goodness-of-fit on F2 1.059 1.030 Rint 0.019 8 0.035 7 R1, wR2 [I > 2σ(I)] 0.027 5, 0.063 7 0.042 1, 0.106 2  下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond lengths (nm) and bond angles (°) for 1 and 2
1 Cd(1)-O(1) 0.234 1(15) Cd(1)-O(2) 0.23 8 5(2) Cd(1)-O(3) 0.220 44(17) Cd(1)-N(1) 0.221 2(2)) Cd(1)-N(4A) 0.226 0(2) O(3)-Cd(1)-N(1) 130.42(8) O(3)-Cd(1)-N(4A) 93.91(7)) N(1)-Cd(1)-N(4A) 111.51(8) O(3)-Cd(1)-O(1) 111.9(2) N(1)-Cd(1)-O(1) 111.8(2) N(4A)-Cd(1)-O(1) 85.8(3) O(3)-Cd(1)-O(2) 94.79(8) N(1)-Cd(1)-O(2) 93.62(9) N(4A)-Cd(1)-O(2) 137.48(8) O(1)-Cd(1)-O(2) 52.5(3) 2 Zn(1)-O(2) 0.200 3(3) Zn(1)-O(3) 0.196 2(2) Zn(1)-N(1) 0.200 2(3) Zn(1)-N(4A) 0.204 4(3) O(3)-Zn(1)-N(1) 122.23(11) O(3)-Zn(1)-O(2) 117.86(13) N(1)-Zn(1)-O(2) 107.50(13) O(1')-Zn(1)-N(4A) 125.3(6) N(1)-Zn(1)-N(4A) 111.59(11) O(2)-Zn(1)-N(4A) 95.02(12) Symmetry codes: A: -x, -y, 1-z for 1; A: 2-x, -y, 1-z for 2.
下载: 导出CSV
Table 3. Natural atomic charges, natural valence electron configurations, wiberg bond indexes and NBO bond orders for 1
Atom Net charge Electron configuration Bond Wiberg bond index NBO bond order / a.u. Cd(1) 1.388 61 [core]5s0.324d9.985p0.30 O⑴ -0.746 52 [core]2s1.682p5.05 Cd(1)-O(1) 0.160 8 0.343 1 O(2) -0.732 38 [core]2s1.692p5.02 Cd(1)-O(2) 0.160 0 0.180 0 O(3) -0.784 08 [core]2s1.712p5.06 Cd⑴-O(3) 0.210 7 0.232 0 N⑴ -0.627 61 [core]2s1.382p4.23 Cd(1)-N(1) 0.184 3 0.175 0 N(4A) -0.621 45 [core]2s1.382p4.22 Cd(1)-N(4A) 0.172 2 0.247 5
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 458
- HTML全文浏览量: 117
 下载:
下载:
