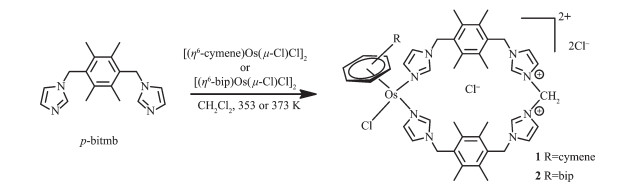
Scheme 1.
Synthesis of mononuclear Os(Ⅱ)-arene metallamacrocyclic complexes 1 and 2 with methylene bridge by a solvothermal reaction

癌症作为导致人类疾病死亡的原因之一,严重威胁着人类的生命健康。治疗癌症的化疗药物主要有顺铂、卡铂和奥沙利铂等铂类药物,然而在后期的使用过程中,这类药物暴露出高毒性、耐药性等问题[1-4]。因此,研究新型的抗癌药物迫在眉睫,对于治疗癌症具有重要意义[5-6]。芳基金属配合物由于其较低的细胞毒性、成键模式的多样性等特点引起了很大的关注,被认为是最有前景的金属抗癌药物之一[7-9]。锇与钌性质相似,金属锇类配合物也可作为潜在的抗癌药物[10-11]。DNA是具有生物遗传功能的分子,影响着细胞的转录与翻译,是许多抗癌药物作用的靶点[12-14]。
我们课题组最近报道了一种合成方法,用来合成含咪唑嗡离子的单核芳基钌配合物RuL2CH2[15]。基于这种合成方法,本文利用1,4-二(1-咪唑基-亚甲基)-2,3,5,6-四甲基苯(p-bitmb)与[(η6-cymene)Os (μ-Cl)Cl]2或[(bip)Os(μ-Cl)Cl]2反应,合成了2种单核芳基锇配合物。通过核磁共振氢谱研究了配合物中亚甲基的来源。用紫外光谱和圆二色谱(CD)研究了配合物与DNA的作用方式及配合物在缓冲溶液中的稳定性,同时还研究了配合物对不同阴离子的响应。
CT-DNA和氘代试剂购自Sigma Aldrich,磷酸缓冲溶液(PBS)等均购自上海生物工程股份有限公司。实验所用溶剂均来自于国药集团,分析纯,未进行进一步纯化。晶体结构用Bruker Smart Apex Ⅱ CCD衍射仪进行测试,1H NMR用Bruck AVANCE 400 MHz/Bruck AVANCE 600 MHz核磁共振波谱仪测定,C、H、N元素分析使用元素分析仪(vario EL Ⅲ,贺力氏公司,德国)测定。圆二色谱(CD光谱)通过英国Applied Photophysics公司生产的Chriascan仪器测定。采用Thermo LCQFLEET电喷雾质谱仪测定配合物的m/z(Source voltage 4.0 kV,流动相为甲醇/乙腈,流速为200 pL·min-1,直接进样),紫外吸收分光光谱仪为Varian Cary 50分光光度计。
将[(p-cymene)OsCl(μ-Cl)]2(0.012 5 mmol,9.8 mg),1,4-二(1-咪唑基-亚甲基)-2, 3, 5,6-四甲基苯(p-bitmb)(0.075 mmol,23.52 mg)加入2.5 mL的二氯甲烷溶液,超声2~5 min,放入装有聚四氟乙烯的内衬的反应釜中,放入80 ℃的烘箱,反应48 h后冷却至室温,得到淡黄色晶体。1H NMR(400 MHz,DMSO-d6):δ 10.13(s,2H),8.48(s,2H),8.24(d,J=1.8 Hz,2H),7.95(s, 2H),7.62~7.29(m,4H),6.37(d,J=14.1 Hz,1H),6.20(d,J=14.1 Hz,1H),5.93(d,J=5.4 Hz,2H),5.76~5.67(m,2H),5.56(d,J=15.1 Hz,2H),5.49~5.39(m,4H),5.34(s,1H),5.31(d,J=2.4 Hz,1H),2.66(s,1H),2.19(s,24H),1.38(s,3H),0.91(d,J=6.9 Hz,6H)。ESI-MS [(p-cymene)Os(L)Cl]3+ m/z:理论值320.91,实测值321.33。元素分析按分子式OsC47H60N8Cl4·CH2Cl2计算,理论值(%):C 49.96, H 5.41, N 9.71;实测值(%):C 50.02;H 6.10;N 9.82。
配合物2的合成与1类似,将[(p-cymene)OsCl(μ-Cl)]2换成[(bip)OsCl(μ-Cl)]2(0.012 5 mmol,10.4 mg),烘箱温度为100 ℃,其余条件不变。1H NMR(600 MHz,DMSO-d6):δ 10.17(s, 2H), 8.35(d,J=1.4 Hz,4H), 7.97(s,2H),7.41~7.22(m,8H),6.49(d,J=14.1 Hz,1H), 6.40(d,J=14.1 Hz,1H),6.29~6.20(m,4H),5.74(t,J=5.0 Hz, 1H), 5.56(d, J=15.0 Hz, 3H), 5.39(dd,J=31.8, 15.0 Hz, 4H), 5.21(d, J=15.0 Hz,2H),2.19(s,24H)。ESI-MS [(bip)Os(L)Cl]3+ m/z:理论值327.57,实测值327.92;{[(bip)Os(L)Cl]Cl}2+ m/z:理论值509.09,实测值509.25;{[(bip)Os(L)Cl]Cl2}+ m/z:理论值1 053.63,实测值1 053.25。元素分析按分子式OsC49H56N8Cl4·2CH2Cl2·H2O计算, 理论值(%):C 47.97, H 4.89, N 8.78;实测值(%):C 47.76,H 5.02,N 9.04。
选取大小为0.1 mm×0.05 mm×0.05 mm配合物1的晶体置于单晶衍射仪上,以经石墨单色器单色化的Mo Kα射线(λ=0.071 073 nm)做入射光源,以ω-2θ扫描方式在296 K下收集衍射点。采用SAINT和SADABS程序进行吸收校正,晶体结构经过直接法解出。所有非H原子都经过各向异性处理,晶体结构通过使用OLEX2 1.2-beta内的SHELXT[16]程序在F2上通过全矩阵最小二乘法进行精修,由于有无序溶剂分子的存在,故采用squeeze对其进行进一步的精修。晶体数据列于表 1。
 下载:
导出CSV
下载:
导出CSV
| Empirical formula | C47H60Cl4N8Os·CH2Cl2 | Limiting indices (h, k, l) | -15~15, 37 ~39, 17 ~17 | |
| Formula weight | 1 154.02 | Reflection collected, unique | 39 549, 11 523 | |
| Crystal system | P21/c | Completeness to θ=25.00°/% | 98.5 | |
| Space group | Monoclinic | θ range/(°) | 1.6~26.7 | |
| a/nm | 1.257 2(3) | μ(Mo Kα)/mm-1 | 2.54 | |
| b/nm | 3.133 1(7) | Rint | 0.051 | |
| c/nm | 1.420 2(3) | Final R indices R1a | 0.103 | |
| β/(°) | 98.790(2) | wR2b [I>2σ(I)] | 0.229 | |
| V/nm3 | 5.528(2) | Refinement method | Full-matrix least Least-squares on F2 | |
| Z | 4 | Data, restraint, parameter | 11 523, 1, 537 | |
| Dc/(g·cm-3) | 1.284 | Goodness-of-fit | 1.19 | |
| F(000) | 2 168 | |||
| a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=|∑w(|Fo|2-|Fc|2)/∑|w(Fo)2|1/2, where w=1/[σ2(Fo2)+(aP)2+bP], P=(Fo2+2Fc2)/3. | ||||
CCDC:1841765,1。
通过紫外光谱测定配合物1和2在缓冲溶液中的稳定性(10 h)[14]。配制浓度为200 μmol·L-1的配合物1和2溶液(VDMSO:VPBS=5:95),用路径为1 cm的石英比色皿,设置波长间隔为1 nm,波长范围为200~600 nm,在室温下每隔1 h记录1次数据。
配制2 mmol·L-1配合物1和2的DMSO溶液,分别用PBS稀释到浓度为200 μmol·L-1(VDMSO:VPBS=5:95)。紫外滴定过程中保持配合物的浓度不变,用双通道进行测试(比色皿的体积为1 mL,厚度为1 cm),即以PBS或者相应浓度的DNA作为空白参照,加入等份的CT-DNA(3.64 mmol·L-1)改变DNA的浓度(每次加1 μL)混合均匀后静置10 min,在200~500 nm的波长范围内记录吸光度值。
配制浓度为100 μmol·L-1的CT-DNA,固定DNA的浓度,配制不同比例的配合物溶液使ccomlex/cDNA的比例r依次为0、0.1、0.2、0.3、0.4,即配合物的浓度依次为0、10、20、30、40 μmol·L-1,在37 ℃下孵化24 h后进行测试。
为了研究单晶结构中亚甲基是否来源于溶剂二氯甲烷,我们将溶剂换成氘代二氯甲烷在相同条件下进行溶剂热反应, 即以[(p-cymene)OsCl(μ-Cl)]2(0.012 5 mmol,9.8 mg),1,4-二(1-咪唑基-亚甲基)-2, 3,5,6-四甲基苯(p-bitmb)(0.075 mmol,23.52 mg)为原料,加入2.5 mL的氘代二氯甲烷溶液,超声2~5 min,放入装有聚四氟乙烯的内衬的反应釜,在80 ℃的烘箱中反应48 h。待冷却到室温以后,收集晶体,洗涤,真空干燥后在DMSO-d6中进行核磁共振研究。
粘度用乌氏粘度计进行测量,保持温度(25±0.1) ℃。测试液配制方法:固定DNA的浓度,逐渐增加配合物浓度的方法配制。测试溶液的相对粘度用下列公式计算[17]:
|
$ \eta = \left( {t - {t_0}} \right)/{t_0} $ |
其中t0为缓冲溶液流经毛细管所需时间,t为DNA溶液或者含不同浓度配合物的DNA溶液流经毛细管所需要的时间。以(η/η0)1/3对比率r(cEB/cDNA或ccomlex/cDNA)作图,η0为未加配合物时DNA的相对粘度。
实验选取了3种细胞株:人卵巢癌细胞(A2780),肺癌细胞(A549)和正常肝脏细胞(L02)。细胞在含10%的血清培养基RPMI1640(Gibco BRL)中37 ℃培养。具体实验方法:将细胞以5 000个每孔的初始密度接种到96孔细胞培养板中,并在培养基中进行培育24 h。加入100 μL阴性和阳性对照以及不同浓度的配合物(VDMSO:V培养基=1:99),在CO2培养箱中培养24 h后,分别向孔中加入20 μL 5 mg·mL-1的MTT溶液,然后孵化4 h,移除96孔板中的培养基,加入150 μL的DMSO,利用酶标仪(Infinite M200,Tecan,Mnnedorf,Switzerland)在590 nm的扫描波长下测试其吸光度值,用SPSSinc软件进行数据处理,计算得到配合物的IC50值。
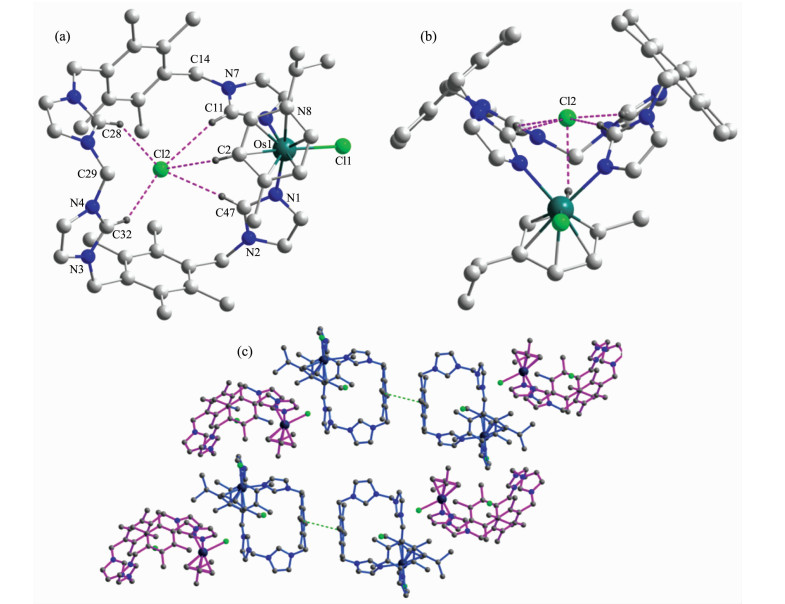
配合物1的晶体结构如图 1所示,主要键长和键角见表 2。配合物1为一个单核锇的结构。中心锇原子与2个p-bitmb配体上的氮原子以及氯原子进行配位,形成了一个以芳基锇为中心的配位端;p-bitmb配体中另一端的咪唑环通过一个亚甲基进行连接,形成了咪唑嗡离子的有机端。最终,配合物1形成一个类似“碗”状的单核金属环状结构。配合物1属于单斜晶系,P21/c空间群。配合物1的空腔内有一个氯离子(Cl2)与配体上的C11、C28、C32、C47以及芳环中C2上的相应氢原子形成氢键(表 3)。配合物1的“碗状”分子在空间上为两种取向,形成了如图 1(c)所示的二维堆积。
 下载:
导出CSV
下载:
导出CSV
| Os1-Cl1 | 0.241 9(3) | Os1-C2 | 0.216 3(8) | Os1-N8 | 0.212 6(10) |
| Os1-C1 | 0.218 0(8) | Os1-N1 | 0.211 9(10) | Os1-C6 | 0.218 2(8) |
| Os1-C4 | 0.215 2(8) | Os1-C5 | 0.216 9(7) | Os1-C3 | 0.214 9(8) |
| N5-C29 | 0.144 5(14) | N4-C29 | 0.147 3(13) | ||
| N8-Os1-Cl1 | 86.1(3) | N1-Os1-Cl1 | 85.6(3) | N1-Os1-N8 | 85.7(4) |
| N5-C29-N4 | 111.3(9) |
下载:
导出CSV
| D-H…A | d(D-H)/nm | d(H…A)/nm | d(D…A)/nm | ∠D-H…A/(°) |
| C11-H11…Cl2 | 0.092 88 | 0.284 2 | 0.375 2 | 166.63 |
| C28-H28…Cl2 | 0.093 06 | 0.254 4 | 0.334 0 | 143.71 |
| C32-H32…Cl2 | 0.092 89 | 0.253 8 | 0.335 2 | 146.45 |
| C47-H47…Cl2 | 0.093 19 | 0.275 4 | 0.365 7 | 163.54 |
| C2-H2…Cl2 | 0.092 93 | 0.257 4 | 0.343 3 | 153.82 |
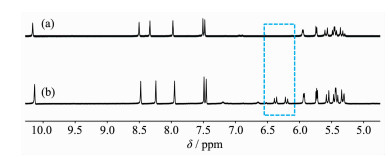
为了验证单核金属环OsLCl(L=(p-bitmb)2CH2)中桥连亚甲基的来源。我们用氘代二氯甲烷代替二氯甲烷,在相同条件下进行溶剂热反应,即在80 ℃的烘箱中反应48 h。待冷却到室温以后,收集晶体,洗涤,干燥样品后进行核磁检测。结果如图 2所示,图 2(a)是以CD2Cl2为溶剂所得配合物的1H NMR,图 2(b)为以CH2Cl2为溶剂所得配合物1的1H NMR。以CH2Cl2为溶剂所得晶体的核磁结果显示在δ 6.449~6.223出现了2个分裂的峰(-CH2-),这可能是由于合成的单核环状配合物1结构的不对称性所导致的[18]。当溶剂为氘代二氯甲烷时,在δ 6.449~6.223处的峰消失。结果表明亚甲基来自于溶剂二氯甲烷。
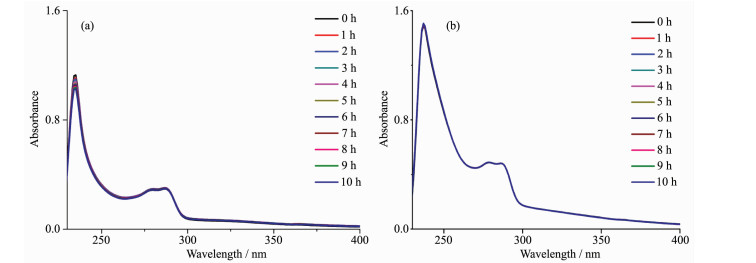
通过紫外光谱研究了配合物在DMSO/PBS溶液(VDMSO/VPBS=5:95)中的稳定性(图 3),配合物1的吸收峰出现在237和279 nm处,分别归属于配合物中金属到配体的电荷跃迁(MLCT)以及配体内部的电荷跃迁(IL)[19]。水解反应10 h以后,配合物1的紫外吸收峰强度几乎未发生变化。配合物2的吸收峰出现在235和287 nm处,分别归属于配合物的MLCT和IL跃迁。在10 h内,配合物的最大吸峰强度几位置未发生明显变化。由上可知,配合物1和2在DMSO/PBS溶液中可以稳定存在。
ccomplex=200 μmol·L-1, VDMSO/VPBS=5:95, 5 mmol·L-1 PBS, pH=7.4
DNA是金属抗癌药的作用靶点之一,在配合物中加入DNA后会引起其紫外吸收强度的变化,使配合物出现增色或者减色现象[20-22]。从图 4可以看出,在200 μmol·L-1的配合物中逐渐加入CT-DNA,配合物1和2的吸光度值都呈现增色效应。
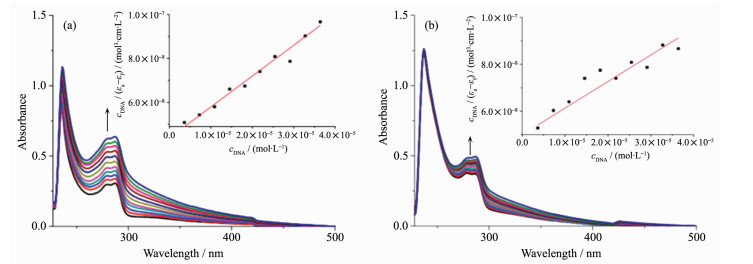
ccomplex=200 μmol·L-1, cDNA=0~36.4 μmol·L-1; Inset: Plot of cDNA/(εa-εf) vs cDNA
为了进一步比较配合物与DNA作用的情况,在紫外滴定实验的基础上通过Benesi-Hildebrand方程式计算配合物DNA的结合常数[23]:
|
$ {c_{{\rm{DNA}}}}/\left( {{\varepsilon _{\rm{a}}} - {\varepsilon _{\rm{f}}}} \right) = {c_{{\rm{DNA}}}}/\left( {{\varepsilon _{\rm{b}}} - {\varepsilon _{\rm{f}}}} \right) + 1/\left[ {{K_{\rm{b}}}\left( {{\varepsilon _{\rm{b}}} - {\varepsilon _{\rm{f}}}} \right)} \right] $ |
其中εa、εb、εf分别为配合物的表观吸收系数,配合物完全结合DNA的吸收系数和纯配合物的吸收系数。根据方程式线性拟合,通过斜率和截距来计算配合物的结合常数Kb。
根据Benesi-Hildebrand方程计算得到配合物1和2与CT-DNA的结合常数,分别为3.22×104 L·mol-1,1.53×104 L·mol-1。配合物1与CT-DNA结合能力大于2的,与已经报道的单核吡啶芳基锇配合物与DNA作用的结合常数大致在同一数量级[24]。
圆二色谱常用来研究配合物与DNA的相互作用,一般B型DNA较为常见,其正吸收峰出现在275 nm附近,与DNA的碱基堆积有关;负峰出现在245 nm附近,与DNA的右手螺旋性质有关[25]。图 5为配合物1和2与CT-DNA相互作用的圆二色谱图。配合物1和2的变化趋势相似,即随着配合物浓度的增大,DNA在275 nm处的正峰信号减弱,说明加入配合物影响了CT-DNA的碱基堆积作用,并且CT-DNA的负峰强度(245 nm附近的峰强度)随着配合物浓度的增大而下降。对配合物1而言,当r=0.2时,CT-DNA信号峰值接近与0,说明1对DNA的碱基堆积和右手螺旋性质产生较大的影响。对于配合物2而言,当r=0.4时,CT-DNA的负峰接近于0,说明配合物2也影响了DNA的右手螺旋性质。上述结果表明,配合物均可以与DNA发生相互作用,减弱DNA的碱基堆积作用并对DNA的右手螺旋性质产生影响。
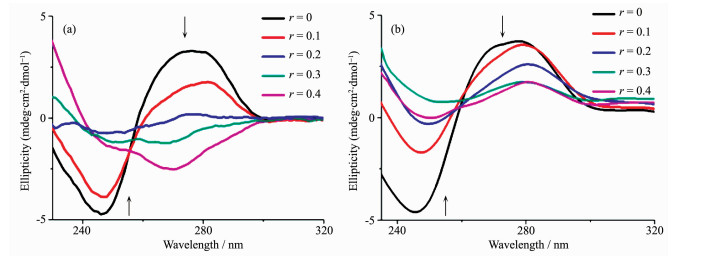
cDNA=100 μmol·L-1, ccomplex=0~40 μmol·L-1, r=ccomplex/cDNA, r=0~0.4
粘度测试是研究配合物与DNA作用方式的方法之一。配合物与CT-DNA以嵌入方式相互作用时,DNA双螺旋链增长,DNA粘度增大;以部分插入的方式作用时,DNA溶液的粘度降低;以静电或沟面方式作用时,DNA溶液的粘度未发生明显变化[26]。溴化乙锭(EB)是典型的DNA嵌入剂,与DNA发生作用时会使其粘度增加。由图 6可知,随配合物1、2浓度的增加,DNA的粘度呈增加的趋势,类似于EB,结果表明配合物以嵌入的方式与DNA结合。
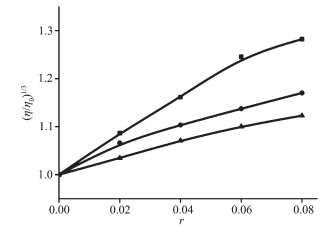
pH=7.4, T=25 ℃, cDNA=100 μmol·L-1, r=cEB/cDNA or ccomplex/cDNA=0, 0.02, 0.04, 0.06, 0.08, respectively
下载:
导出CSV
| μmol·L-1 | |||
| Complex | A2780 | A549 | L02 |
| 1 | 188.89±0.43 | >200 | >200 |
| 2 | 158.23±0.23 | >200 | >200 |
| Cisplatin | 13.84±1.26 | 12.85±0.54 | 2.16±0.25 |
采用MTT法研究了配合物对人卵巢癌细胞(A2780)、肺癌细胞(A549)以及正常的肝脏细胞(L02)的细胞毒性。配合物的IC50结果表明,配合物1和2对癌细胞A2780、A549和正常细胞L02基本没有毒性,其毒性远远低于顺铂。
合成、表征了2种“碗状”单核芳基金属锇配合物。配合物的单晶结构表明芳基锇的Os(Ⅱ)与2个配体的咪唑氮原子以及氯原子配位,2个配体的另一个咪唑基团通过一个亚甲基碳原子连接形成咪唑嗡离子。在氘代试剂中合成配合物并通过核磁共振方法证实桥联亚甲基基团来自于溶剂二氯甲烷。配合物在缓冲溶液中可以稳定存在,并以嵌入的方式与DNA相互作用,影响DNA的螺旋性质及碱基堆积作用。本研究为合成同时具有配位端及咪唑嗡离子端的配合物提供了一个方法。
Cheff D M, Hall M D. J. Med. Chem., 2017, 60:4517-4532 doi: 10.1021/acs.jmedchem.6b01351
Ma L, Ma R, Wang Y, et al. Chem. Commun., 2015, 51:6301-6304 doi: 10.1039/C4CC10409A
Shen D W, Pouliot L M, Hall M D, et al. Pharmacol. Rev., 2012, 64:706-721 doi: 10.1124/pr.111.005637
Wang X, Guo Z J. Chem. Soc. Rev., 2013, 42:202-224 doi: 10.1039/C2CS35259A
Markus G, Michael A J, Bernhard K K. Curr. Med. Chem., 2005, 12:2075-2094 doi: 10.2174/0929867054637626
Wong E, Giandomenico C M. Chem. Rev., 1999, 99:2451-2466 doi: 10.1021/cr980420v
Gasser G, Ott I, Metzler-Nolte N. J. Med. Chem., 2011, 54:3-25 doi: 10.1021/jm100020w
孔亚琼, 李季, 胡晓莹, 等.中国科学:化学, 2017, 47:277-283 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=JBXK201702016&dbname=CJFD&dbcode=CJFQKONG Ya-Qiong, LI Ji, HU Xiao-Ying, et al. Scientia Sinica Chimica, 2017, 47:277-283 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=JBXK201702016&dbname=CJFD&dbcode=CJFQ
Weiss A, Berndsen R H, Dubois M, et al. Chem. Sci., 2014, 5:4742-4748 doi: 10.1039/C4SC01255K
Egger A, Cebrian-Losantos B, Stepanenko I N, et al. Chem. Biodivers., 2008, 5:1588-1593 doi: 10.1002/cbdv.v5:8
Kuhn P S, Buchel G E, Jovanovic K K, et al. Inorg. Chem., 2014, 53:11130-11139 doi: 10.1021/ic501710k
Liu H K, Parkinson J A, Bella J, et al. Chem. Sci., 2010, 1:258-270 doi: 10.1039/c0sc00175a
Liu H K, Sadler P J. Acc. Chem. Res., 2011, 44:349-359 doi: 10.1021/ar100140e
Wang H Y, Qian Y, Wang F X, et al. Eur. J. Inorg. Chem., 2017:1792-1799
Li J, Zhang P, Xu Y, et al. Dalton Trans., 2017, 46:16205-16215 doi: 10.1039/C7DT03374E
Conerney B, Jensen P, Kruger P E, et al. CrystEngComm, 2003, 5:454-458 doi: 10.1039/B311278K
Wang X, Liu S, Weng L H, et al. Chem. Eur. J., 2007, 13:188-195 doi: 10.1002/chem.v13:1
Cohen G, Eisenberg H. Biopolymers, 1969, 8:45-49 doi: 10.1002/(ISSN)1097-0282
Wu Q, Chen T, Zhang Z, et al. Dalton Trans., 2014, 43:9216-9225 doi: 10.1039/C3DT53635A
Govender P, Riedel T, Dyson P J, et al. Dalton Trans., 2016, 45:9529-9539 doi: 10.1039/C6DT00849F
Liu P, Wu B Y, Liu J, et al. Inorg. Chem., 2016, 55:1412-1422 doi: 10.1021/acs.inorgchem.5b01934
Mukhopadhyay S, Gupta R K, Paitandi R P, et al. Organometal., 2015, 34:4491-4506 doi: 10.1021/acs.organomet.5b00475
Krishnamoorthy P, Sathyadevi P, Butorac R R, et al. Dalton Trans., 2012, 41:6842-6854 doi: 10.1039/c2dt30121k
郝元元, 吴琪, 李季, 等.高等学校化学学报, 2018, 4:614-622 doi: 10.7503/cjcu20170634HAO Yuan-Yuan, WU Qi, LI Ji, et al. Chem. J. Chinese Universities, 2018, 4:614-622 doi: 10.7503/cjcu20170634
Jaroslav K, Iva K, Daniel R, et al. Nucleic Acids Res., 2009, 37:1713-1725 doi: 10.1093/nar/gkp026
宋玉民, 杨培菊, 王流芳, 等.化学学报, 2003, 61:1266-1270 http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb200308019SONG Yu-Min, YANG Pei-Ju, WANG Liu-Fang, et al. Acta Chim. Sinica, 2003, 61:1266-1270 http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb200308019

Scheme 1 Synthesis of mononuclear Os(Ⅱ)-arene metallamacrocyclic complexes 1 and 2 with methylene bridge by a solvothermal reaction

图 1 (a) 装载有氯离子的类似“碗状”的单核金属环OsLCl(L=(p-bitmb)2CH2)配合物的单晶结构, (b)具有“碗状”结构的配合物1侧面图, (c)配合物1的二维堆积图
Figure 1 (a) X-ray crystal structure of the "bowl-like" mononuclear OsLCl(L=(p-bitmb)2CH2) metallamacrocycle 1 with a Cl- anion trapped inside the cavity; (b) Side view to show the trapped chloride anion inside the "bowl-like" structure of complex 1; (c) Packing mode of complex 1

图 2 以CD2Cl2 (a)或CH2Cl2 (b)为溶剂通过溶剂热反应得到配合物1的1H NMR谱
Figure 2 1H NMR spectra of 1 in DMSO-d6 solution obtained from the solvothermal reaction in CD2Cl2 (a) or CH2Cl2 (b)

图 3 配合物1和2在DMSO/PBS溶液中的紫外吸收光谱
Figure 3 Absorption spectra of complexes 1 (a) and 2 (b) in DMSO/PBS solution
ccomplex=200 μmol·L-1, VDMSO/VPBS=5:95, 5 mmol·L-1 PBS, pH=7.4

图 4 配合物1 (a)和2 (b)随CT-DNA浓度增加的紫外吸收光谱
Figure 4 UV-Vis absorption spectra of the complexes 1 (a) and 2 (b) in the absence and presence of increasing concentrations of CT-DNA
ccomplex=200 μmol·L-1, cDNA=0~36.4 μmol·L-1; Inset: Plot of cDNA/(εa-εf) vs cDNA

图 5 配合物1 (a)、2 (b)与CT-DNA相互作用的圆二色谱图
Figure 5 CD spectra of CT-DNA in the absence and presence of complexes 1 (a) or 2 (b)
cDNA=100 μmol·L-1, ccomplex=0~40 μmol·L-1, r=ccomplex/cDNA, r=0~0.4

图 6 EB (■)、配合物1 (●)和2 (▲)浓度的增加对DNA粘度的影响
Figure 6 Effect on the relative viscosity of CT-DNA after addition of increasing amounts of ethidium bromide (■), 1 (●) and 2 (▲) in PBS
pH=7.4, T=25 ℃, cDNA=100 μmol·L-1, r=cEB/cDNA or ccomplex/cDNA=0, 0.02, 0.04, 0.06, 0.08, respectively
表 1 配合物1的晶体学数据
Table 1. Crystallographic data for 1
| Empirical formula | C47H60Cl4N8Os·CH2Cl2 | Limiting indices (h, k, l) | -15~15, 37 ~39, 17 ~17 | |
| Formula weight | 1 154.02 | Reflection collected, unique | 39 549, 11 523 | |
| Crystal system | P21/c | Completeness to θ=25.00°/% | 98.5 | |
| Space group | Monoclinic | θ range/(°) | 1.6~26.7 | |
| a/nm | 1.257 2(3) | μ(Mo Kα)/mm-1 | 2.54 | |
| b/nm | 3.133 1(7) | Rint | 0.051 | |
| c/nm | 1.420 2(3) | Final R indices R1a | 0.103 | |
| β/(°) | 98.790(2) | wR2b [I>2σ(I)] | 0.229 | |
| V/nm3 | 5.528(2) | Refinement method | Full-matrix least Least-squares on F2 | |
| Z | 4 | Data, restraint, parameter | 11 523, 1, 537 | |
| Dc/(g·cm-3) | 1.284 | Goodness-of-fit | 1.19 | |
| F(000) | 2 168 | |||
| a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=|∑w(|Fo|2-|Fc|2)/∑|w(Fo)2|1/2, where w=1/[σ2(Fo2)+(aP)2+bP], P=(Fo2+2Fc2)/3. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 配合物1的部分键长和键角
Table 2. Selected bond lengths(nm) and angles (°) for 1
| Os1-Cl1 | 0.241 9(3) | Os1-C2 | 0.216 3(8) | Os1-N8 | 0.212 6(10) |
| Os1-C1 | 0.218 0(8) | Os1-N1 | 0.211 9(10) | Os1-C6 | 0.218 2(8) |
| Os1-C4 | 0.215 2(8) | Os1-C5 | 0.216 9(7) | Os1-C3 | 0.214 9(8) |
| N5-C29 | 0.144 5(14) | N4-C29 | 0.147 3(13) | ||
| N8-Os1-Cl1 | 86.1(3) | N1-Os1-Cl1 | 85.6(3) | N1-Os1-N8 | 85.7(4) |
| N5-C29-N4 | 111.3(9) |
下载: 导出CSV
表 3 配合物1的氢键数据
Table 3. Hydrogen bond geometry of complex 1
| D-H…A | d(D-H)/nm | d(H…A)/nm | d(D…A)/nm | ∠D-H…A/(°) |
| C11-H11…Cl2 | 0.092 88 | 0.284 2 | 0.375 2 | 166.63 |
| C28-H28…Cl2 | 0.093 06 | 0.254 4 | 0.334 0 | 143.71 |
| C32-H32…Cl2 | 0.092 89 | 0.253 8 | 0.335 2 | 146.45 |
| C47-H47…Cl2 | 0.093 19 | 0.275 4 | 0.365 7 | 163.54 |
| C2-H2…Cl2 | 0.092 93 | 0.257 4 | 0.343 3 | 153.82 |
下载: 导出CSV
表 4 配合物1和2的IC50
Table 4. IC50 of complexes 1 and 2
| μmol·L-1 | |||
| Complex | A2780 | A549 | L02 |
| 1 | 188.89±0.43 | >200 | >200 |
| 2 | 158.23±0.23 | >200 | >200 |
| Cisplatin | 13.84±1.26 | 12.85±0.54 | 2.16±0.25 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: