引用本文:
解庆范, 郭妙莉, 陈延民. 邻氨基酚Schiff碱钴、镍双核配合物:合成、结构和抗癌活性及量化计算[J]. 无机化学学报,
2018, 34(2): 309-316.
doi:
10.11862/CJIC.2018.028

Citation: XIE Qing-Fan, GUO Miao-Li, CHEN Yan-Min. Binuclear Cobalt(Ⅱ) and Nickel(Ⅱ) Complexes with o-Aminophenol Schiff Base: Syntheses, Structures, Antitumor Activity and Quantum Chemistry Calculation[J]. Chinese Journal of Inorganic Chemistry, 2018, 34(2): 309-316. doi: 10.11862/CJIC.2018.028
Citation: XIE Qing-Fan, GUO Miao-Li, CHEN Yan-Min. Binuclear Cobalt(Ⅱ) and Nickel(Ⅱ) Complexes with o-Aminophenol Schiff Base: Syntheses, Structures, Antitumor Activity and Quantum Chemistry Calculation[J]. Chinese Journal of Inorganic Chemistry, 2018, 34(2): 309-316. doi: 10.11862/CJIC.2018.028
邻氨基酚Schiff碱钴、镍双核配合物:合成、结构和抗癌活性及量化计算
摘要:
分别采用水热法和溶剂挥发法合成了一个五配位的钴双核配合物[Co2(H2O)2(L1)2(4,4'-bipy)](1)和一个镍双核配合物[Ni2(L2)2(4,4'-bipy)](2)(H2L1=邻氨基酚缩5-溴水杨醛,H2L2=邻氨基酚缩水杨醛),并经红外光谱和紫外光谱以及热重分析进行了表征。X射线单晶衍射分析表明1的晶体结构属三斜晶系P1空间群;2属于三方晶系R3空间群。采用MTT法初步研究了配体和配合物的体外抗癌活性,结果表明,1和2对人肝肿瘤细胞HEPG2和人结肠癌细胞SW620均具有增殖抑制作用。同时利用量子化学计算对配合物2进行了理论计算。
English
Binuclear Cobalt(Ⅱ) and Nickel(Ⅱ) Complexes with o-Aminophenol Schiff Base: Syntheses, Structures, Antitumor Activity and Quantum Chemistry Calculation
Abstract:
A five-coordinated binuclear cobalt(Ⅱ) complex with the formula[Co2(H2O)2(L1)2(4, 4'-bipy)] (1) (H2L1=2-hydroxybenzene (2-hydroxyl-5-nitro-phenyl-methyl) amine) was hydrothermally synthesized and a binuclear nickel(Ⅱ) complex of general formula[Ni2(L2)2(4, 4'-bipy)] (2) (H2L2=2-hydroxybenzene (2-hydroxyl-phenyl-methyl) amine) synthesized by using evaporation method, and the comlexes were characterized by IR, UV-Vis spectra and TGA. The X-ray diffraction analyses reveal that 1 crystallizes in the triclinic space group P1 while 2 in trigonal space group R3. The antitumor activity in vitro of ligands and complexes were measured by MTT method, and the results show that the complexes 1 and 2 possess inhibiting effects to HEPG2 and SW620 cancer cells lines. The quantum chemical calculation for 2 was performed by means of Gaussian 09 program at UB3LYP/6-31G(d) basis set. CCDC: 1440518, 1; 1432468, 2.
-
过去数十年关于Schiff碱的过渡金属配合物的研究一直非常活跃,一个重要的原因是它们在抑菌、抗病毒、消炎和抗肿瘤等方面的药理作用使之具有潜在的应用前景[1-4]。另一方面,过渡金属广泛存在于许多生物酶中,作为酶的活性中心发挥着重要的作用[5-6]。利用简单的有机配体合成新型的配合物,是分子结构设计的重要策略之一,合成的关键在于金属离子和配体的选择以及反应条件[7-8]。Schiff碱具有双齿、三齿和多齿等多种类型,有助于形成单核、双核和多核等各种类型的配合物,在配合物的分子设计和合成中可以发挥更多的想象力。近年来,非贵金属配合物作为抗肿瘤药物的研究取得快速发展[9],研究较多的是铜的配合物[10-11]。事实上,有些锌、镉、钴、镍等的金属配合物也表现出良好的抗肿瘤活性[12-19]。我们分别以Schiff碱邻氨基酚缩5-硝基水杨醛(H2L1)和邻氨基酚缩水杨醛(H2L2)为第一配体,4,4'-联吡啶为第二配体合成了一种五配位的钴双核配合物[Co2(H2O)2(L1)2(4,4'-bipy)] (1)和一种大平面的双核镍配合物[Ni2(L2)2(4,4'-bipy)] (2),本文主要报导它们的晶体结构和抗癌活性,并分析了它们的光谱特征和热稳定性,对镍配合物应用Gaussian 09程序,采用密度泛函方法(DFT),在UB3LYP/6-31G(d)水平进行了量化计算。
1 实验部分
1.1 仪器与试剂
德国Elmentar Vario EL元素分析仪;美国Nicolet is10型FT-IR红外光谱仪;上海美普达UV-1800PC型紫外-可见分光光度计;德国塞驰STA 409 PC型综合热分析仪;美国Varian CARY/ Eclipse型荧光分光光度计;日本理学Rigaku Saturn724 CCD单晶衍射仪。所用试剂均为市售分析纯试剂。
1.2 配合物的合成
1.2.1 配合物1的合成
将大约10 mmol的邻氨基酚和等量的5-硝基水杨醛置于20 mL无水乙醇中,75 ℃下水浴加热回流3 h,冷却、过滤,并用无水乙醇洗涤,得到黄色粉末状配体H2L1,真空干燥备用。取0.2 mmol乙酸钴、0.2 mmol H2L1、0.1 mmol 4,4'-联吡啶、6 mL水、4 mL乙醇和1 mL DMF,置于内衬聚氟乙烯不锈钢自动升压反应釜中,于120 ℃恒温3 d,得到1的暗红色块状晶体。对C36H28Co2N6O10的元素分析计算值(%):C 52.57,H 3.43,N 10.22;测试值(%):C 52.48,H 3.51,N 10.16。IR(cm-1):H2L1:1 616s,1 593,1 545s,1 520,1 316vs,1 240,1 201,831,793, 743s;1:1 609 vs,1 581,1 527,1 472s,1 440,1 302,1 217,830,815,737s。
1.2.2 配合物2的合成
将10 mmol邻氨基酚与1.2 mL水杨醛溶于20 mL无水乙醇,并加1 mL乙酸,在75 ℃下水浴加热回流3 h,冷却、过滤,并用无水乙醇洗涤,得到黄色粉末状配体H2L2,真空干燥备用。在5 mL DMF和15 mL乙醇的混合溶剂中,加入0.4 mmol乙酸镍、0.2 mmol 4,4'-联吡啶、0.4 mmol配体H2L2和0.4 mmol乙酸钠,于60 ℃下加热回流2 h,析出橙色粉末。过滤后用DMF重结晶,滤液静置1周后析出2的橙色棒状单晶。对C36H26N4Ni2O4的元素分析计算值(%):C 62.12,H 3.77,N 8.05;测试值(%):C 62.15,H 3.70,N 8.11。IR(cm-1):H2L2:1 628vs,1 601,1 548, 1 507,1 454,1 321,1 269,1 219,879,824,754vs;2:1 605vs,1 582,1 527,1 481vs,1 457,1 319,1 217,846,813,737。
1.3 晶体结构测试
分别选取0.40 mm×0.32 mm×0.05 mm (1)和0.38 mm×0.30 mm×0.22 mm (2)的单晶置于Rigaku Saturn 724 CCD单晶衍射仪上,用经石墨单色器单色化的Mo Kα射线(λ=0.071 073 nm),以ω扫描方式收集单晶衍射数据用于单晶结构分析。衍射数据和晶胞参数用SAINT程序[20]还原,全部强度数据均经Lp因子校正,并进行了经验吸收校正。晶体结构由直接法解出,对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正,有机物上的氢原子由理论加氢法得到,上水分子上的氢原子通过差值图中找出。结构精修分别采用SHELXL-97[21]和Olex2程序包[22]。主要晶体数据列于表 1。
 表 1
配合物的晶体数据
Table 1.
Crystallographic data for the complexes
表 1
配合物的晶体数据
Table 1.
Crystallographic data for the complexes
Complex 1 2 Empirical formula C36H28Co2N6O10 C36H26N4Ni2O4 Formula weight 822.50 696.03 Temp erature/K 293(2) 293(2) Crystal system Triclinic Trigonal Space group P1 R3 a/nm 0.547 17(11) 3.743 7(11) b/nm 1.155 5(2) 3.743 7(11) c/nm 1.420 9(3) 0.604 90(12) α/(°) 104.63(3) β/(°) 98.75(3) γ/(°) 102.51(3) Volume/nm3 0.828 0(3) 7.342(3) Z 1 9 Dc/(Mg·m-3) 1.650 1.417 Absorption coefficient/mm-1 1.074 1.199 F(000) 420 3 222 Range of θ/(°) 3.04~25.01 2.88~25.01 Limiting indices (h, k, l) -6~6, -13~13, -16~16 -44~44, -44~43, -7~7 Reflection collected 13 760 32 457 Unique reflection 2 917(Rint=0.051 4) 2 875(Rint=0.110 5) Data, restraint, parameter 2 918, 0, 245 2 875, 3, 208 Goodness-of-fit on F2 1.040 1.115 Final R indices [I>2σ(I)] R1=0.042 8, wR2=0.112 3 R1=0.096 6, wR2=0.205 7 R indices (all data) R1=0.051 7, wR2=0.117 8 R1=0.159 3, wR2=0.234 8 Largest diff. peak and hole/(e·nm-3) 553 and -440 1 121 and -621 CCDC:1440518,1;1432468,2。
1.4 体外抗癌活性的检测
采用MTT法,参照文献[23]方法,考查了配合物对人肝癌细胞HEPG2和人结肠癌细胞SW620的体外增殖抑制作用,以同一药物不同浓度对抑制率作图,根据线回归方程求出半数抑制浓度IC50。
2 结果与讨论
2.1 晶体结构
2.1.1 配合物1的晶体结构
配合物1的分子结构见图 1,主要键长和键角列于表 2。1属三斜晶系P1空间群,它由2个中心离子Co2+、2个Schiff碱配体L12-、1个4,4'-联吡啶和2个配位水组成,是一种中心对称的双核配合物。其中,中心离子的配位环境呈现四方锥几何构型[CoN2O3]。Co2+的这种配位构型在许多配合物中都曾出现过,这主要源自该离子的d7电子构型。Schiff碱L12-以三齿螯合配位,它提供的2个氧原子和1个亚胺基的氮原子与联吡啶的1个氮原子构成锥底,Co2+偏离锥底平面约0.021 nm,配位水位于锥顶,Co-O键长为0.195 8(2)~0.207 8(2) nm,Co-N键长0.205 1(2)~0.209 8(3) nm,键角为81.30(9)°~171.43(10)°。配体H2L1配位时酚羟基脱除质子,C1-O1和C13-O2键长分别为0.135 3(4)和0.129 5(4) nm,比正常的C-O单键的略短,说明酚羟基氧原子与苯环存在共轭作用。借助Diamond 3.2计算出L12-的2个苯环(C1~C6和C8~C13)的二面角为11°,4,4'-联吡啶的整个分子呈现共平面,其吡啶环(C14~C18, N3)与L12-的硝基苯酚片段(N1, C8~C13, O2)的二面角为73.5°,吡啶环与苯酚片段(O1,C1~C6)的二面角是65°。
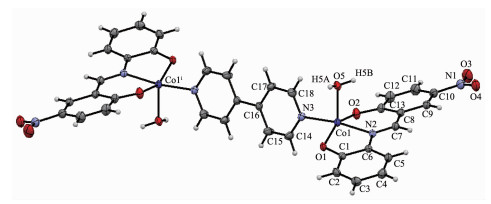 图1
配合物1的分子结构
Figure1.
Molecular structure of 1
表 2
配合物1的主要键长和键角
Table 2.
Selected bond lengths (nm) and bond angles (°) for the complex 1
图1
配合物1的分子结构
Figure1.
Molecular structure of 1
表 2
配合物1的主要键长和键角
Table 2.
Selected bond lengths (nm) and bond angles (°) for the complex 1
Co1-O2 0.195 8(2) Co1-O1 0.201 1(2) Co1-N2 0.205 1(2) Co1-O5 0.207 8(2) Co1-N3 0.209 8(3) O1-C1 0.135 3(4) N2-C6 0.140 9(4) N2-C7 0.128 7(4) O2-C13 0.129 5(4) O2-Co1-O1 155.03(10) O5-Co1-N3 95.25(9) O1-Co1-N3 96.27(9) O2-Co1-O5 100.38(10) O2-Co1-N2 89.35(9) O1-Co1-N2 81.30(9) O2-Co1-N3 89.56(10) O1-Co1-O5 103.23(9) N2-Co1-O5 93.31(9) N2-Co1-N3 171.43(10)
表 3
配合物1中的氢键参数
Table 3.
Parameters of hydrogen bonds in the complex 1
D-H…A d(D-H)/nm d(H…A)/nm d(D…A)/nm ∠DHA/(°) 05-H5A…O1ⅱ 0.086 0.239 0.281 02 111 05-H5B…O1ⅲ 0.082 0.189 0.269 41 157 C7-H7…O4 0.093 0.243 0.333 90 164 Symmetry codes: ⅱ1-x, 1-y, 1-z; ⅲ1+x, y, z 配位水的氢原子H5A和H5B分别与另外2个相邻的配合物基元的酚羟基氧原子O1形成强烈的氢键,O5-H5A…O1ⅱ和O5-H5B…O1ⅲ氢键键长分别为0.238 nm和0.189 nm,键角111°和156°。由于氢键的作用,配合物联接形成梯形层状超分子(图 2),而层与层之间C7-H7与硝基的氧原子O4之间存在弱的氢键(0.312 9 nm)。这些氢键共同稳定了配合物的晶体结构。
 图2
配合物1中的氢键
Figure2.
Hydrogen bands in the complex 1
图2
配合物1中的氢键
Figure2.
Hydrogen bands in the complex 1
2.1.2 配合物2的晶体结构
配合物2也是一种双核结构,属于三方晶系,R3空间群,它由2个中心离子Ni2+、1个4,4'-联吡啶和2个Schiff碱L22-组成(图 3),主要键长和键角列于表 4。在2中H2L2脱除羟基质子,同样以-2价配体形式采用三齿螯合方式与Ni(Ⅱ)配位,Ni(Ⅱ)的配位数为4,处于平面四边形的配位环境,键角为82.50(3)°~177.90(3)°。整个分子的所有原子几乎完全共平面,呈现巨大的平面结构,2个苯酚片段(O1,C1~C6和O2,C13~C8)的二面角为1.8°,Schiff碱L22-的分子平面与吡啶环(N2,C14~C18)的二面角为3.9°。由此可见,2的分子内存在很强共轭作用,从而导致各种化学键均比一般的Schiff碱镍配合物的化学键要短。Ni-O键长为0.183 6(6)~0.179 3(6) nm,Ni-N键长为0.192 9(8)~0.194 9(5) nm。键长也明显比1中化学键Co-O或Co-N短,亚胺基C=N键长0.109 3(12) nm明显比2中相应的键(0.128 7(4) nm)短得多。相邻的配合物分子相互平行,吡啶环之间存在较强的π-π相互作用,质心间距0.335 8 nm(图 4)。由于分子间的π-π相互作用,配合物堆积形成一种一维超分子链。
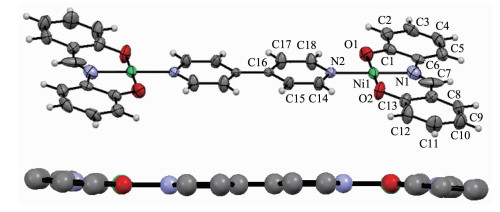 图3
配合物2的分子结构
Figure3.
Molecular structure of the complex 2
表 4
配合物2的主要键长、键角和扭转角
Table 4.
Selected bond lengths (nm), bond angles and torsion angles (°) for the complex 2
图3
配合物2的分子结构
Figure3.
Molecular structure of the complex 2
表 4
配合物2的主要键长、键角和扭转角
Table 4.
Selected bond lengths (nm), bond angles and torsion angles (°) for the complex 2
Ni1-O1 0.183 6(6) Ni1-O2 0.179 3(6) Ni1-N1 0.192 9(8) Ni1-N2 0.194 9(5) C7-N1 0.109 3(12) C6-N1 0.149 2(11) O1-C1 0.132 2(8) O2-C13 0.131 1(9) O2-Ni1-O1 177.9(3) O2-Ni1-N1 98.9(3) O1-Ni1-N1 82.5(3) O2-Ni1-N2 89.0(2) O1-Ni1-N2 89.6(3) N1-Ni1-N2 171.7(3) N1-Ni1-O2-C13 0.5(5) N2-Ni1-O1-C1 178.0(5) O1-Ni1-N2-C18 -2.4(6) N1-Ni1-O2-C13 2.3(7) N2-Ni1-O2-C13 -175.5(7) O2-Ni1-N2-C14 0.4(6) C15ⅰ-C16ⅰ-C16-C15 180 N1-C6-C1-O1 3.5(10) O2-Ni1-N1-C7 -1.4(9) Symmetry codes: ⅰ-x+1/3, -y+2/3, -z+5/3 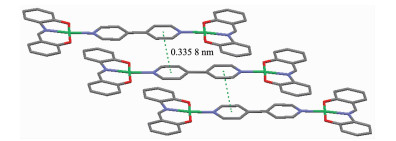 图4
配合物2晶体中的芳环π-π堆积作用
Figure4.
π-π stacking interactions in the crystal of complex 2
图4
配合物2晶体中的芳环π-π堆积作用
Figure4.
π-π stacking interactions in the crystal of complex 2
2.2 电子吸收光谱
以DMF为溶剂,将配合物配制成浓度大约为5 μmol·L-1的溶液,并以DMF为参比液,用UV-1800PC型紫外-可见分光光度计在200~600 nm范围扫描,结果见图 5。H2L1在294、364和434 nm处的吸收分别来自配体的π→π*电子跃迁、n→π*电子跃迁和分子内的电荷转移跃迁(ILCT);与Co(Ⅱ)形成配合物1后π→π*电子跃迁蓝移(Δλ=24 nm)至270 nm,n→π*电子跃迁则消失,而ILCT蓝移(Δλ=20 nm)至414 nm处,发生蓝移的原因可能是配体的共平面程度减小,共轭作用减弱。H2L2的电子吸收特征与H2L1存在较大差别,π→π*电子跃迁出现在264 nm处,而348 nm处极强的ILCT吸收带掩盖了n→π*电子跃迁。形成配合物2后,配体的共平面程度增大,π→π*电子跃迁红移至270 nm;与H2L2比较可见,ILCT吸收带消失,而在432 nm处出现了1个新的吸收带,根据量化计算结果,可以将它指认为配体H2L2到配体4,4'-bipy的电荷转移跃迁(LLCT)。
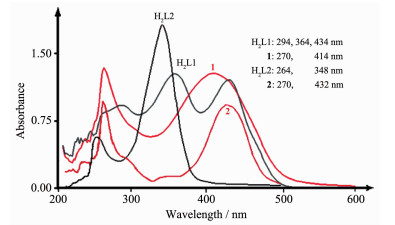 图5
配合物的紫外-可见光谱
Figure5.
UV-Vis spectra of the complexes
图5
配合物的紫外-可见光谱
Figure5.
UV-Vis spectra of the complexes
2.3 配合物的热稳定性
在N2气氛中,以10 ℃·min-1升温速率观察了配合物的热稳定性(图 6)。1和2的热分解行为均分2阶段完成,1在127~160 ℃失去2个配位水,失重4.84%(计算值4.38%);305 ℃开始配合物骨架崩塌,有机物快速分解并挥发,在305~339 ℃快速失重52.41%,之后基本恒重,至900 ℃残重39.37%。2在276~333 ℃失重22.02%,相当于失去1个4,4'-联吡啶(计算值22.71%),第二阶段在398~419 ℃失重33.11%,至900 ℃残重40.02%。
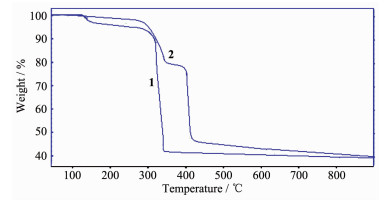 图6
配合物的热重分析图
Figure6.
TGA curves of the complexes
图6
配合物的热重分析图
Figure6.
TGA curves of the complexes
2.4 抗肿瘤活性
在无菌的条件下,取对数生长期人结肠癌细胞SW620和人肝癌细胞HEPG细胞,2.5 g·L-1胰酶消化,调整细胞密度为2.5×104 mL-1,培养时间为24 h,用全自动酶标仪在570 nm处测定它的吸光度值(OD值)并计算细胞的增殖抑制率。根据不同浓度对肿瘤细胞生长抑制率作图(图 7和8),结果表明,配体H2L1和H2L2均不具有抗肿瘤活性;而配合物1和2均表现出一定的抗肿瘤活性,配合物的抗肿瘤活性可能来自金属离子配位的不饱和性和双核结构以及大型的平面结构,这些因素均有利于配合物分子与DNA发生作用。相比较而言,镍配合物的活性略高于钴配合物,1和2对人结肠癌细胞SW620的半数抑制浓度IC50分别为13.75和9.96 μg·mL-1, 对人肝癌细胞HEPG2的IC50分别为18.75和11.51 μg·mL-1。
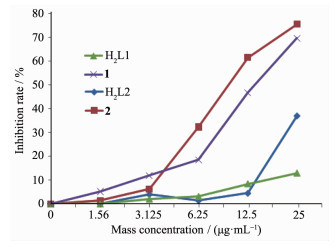 图7
配合物对SW620的抑制作用
Figure7.
Inhibition effects of the complexes on cell SW620
图7
配合物对SW620的抑制作用
Figure7.
Inhibition effects of the complexes on cell SW620
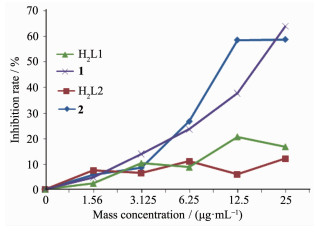 图8
配合物对HEPG2的抑制作用
Figure8.
Inhibition effects of the complexes on cell HEPG2
图8
配合物对HEPG2的抑制作用
Figure8.
Inhibition effects of the complexes on cell HEPG2
2.5 量子化学计算
用Gaussian 09程序包[24],采用密度泛函理论(DFT)[25],在B3LYP水平对C,H,O,N原子用6-31G(d)基组,Ni原子用lanl2dz基组,将分子分为吡啶C5N、Schiff碱C13NO2和金属原子Ni三部分,计算了分子的碎片对前线分子轨道的贡献,同时计算了配合物2的Wiberg键级,结果列于表 5和6。
表 5
配合物2的前线分子轨道能量和分子碎片对该分子轨道贡献
Table 5.
Frontier molecular orbital energy and molecular fragment contribution to the molecular orbitals of complex 2
Orbital Energy/eV Contribution of molecular fragment/% C5N C13NO2 Ni LUMO+1 -1.734 96.4 0.0 0.0 LUMO -2.563 96.2 0.0 0.0 HOMO -5.127 0.0 86.6 8.4 HOMO-1 -5.132 0.0 86.5 8.5
表 6
配合物2的Wiberg键级
Table 6.
Wiberg bond order of the complex 2
Ni1-O1 0.548 C6-N1 1.053 C4-C5 1.457 C9-C10 1.516 Ni1-O2 0.512 C7-N1 1.527 C5-C6 1.359 C10-C11 1.334 Ni1-N1 0.496 C1-C2 1.317 C6-C1 1.26 C11-C12 1.514 Ni1-N2 0.367 C2-C3 1.471 C7-C8 1.218 C12-C13 1.284 C1-O1 1.141 C3-C4 1.384 C8-C9 1.293 C14-N2 1.348 C13-O2 1.204 C14-C15 1.449 C15-C16 1.373 C16-C17 1.374 C17-C18 1.448 C18-N2 1.346 C8-C13 1.216 由表 5可见,在最高占据分子轨道HOMO中,电子云主要分布在Schiff碱配体C13NO2(86.6%),而在最低空轨道LUMO中电子云主要集中在吡啶配体(96.2%),说明电子由HOMO向LUMO跃迁时,主要发生的是电子由Schiff碱配体向吡啶配体的电荷转移跃迁,即LLCT电子跃迁(最大吸收波长计算值为483 nm,实验值为432 nm)。
由于共轭作用,芳环上的电子云均一化,导致C-C/N的平均键级(1.367)小于1.500,而酚羟基C1-O1,C13-O2和C7-C8的键级分别为1.141、1.204和1.218,大于单键键级。Ni1-O1、Ni1-O2、Ni1-N1、Ni1-N2的键级分别为0.548、0.512、0.496和0.367,表明配位能力酚羟基 > 亚胺基 > 吡啶基,热分解时Ni1-N2将优先断裂,这从理论上证明了2的热分解第一阶段的失重属于脱除4,4'-联吡啶行为。
-
-
[1]
Rollas S, Küçükgüzcl S G. Molecules, 2007, 12:1910-1939 doi: 10.3390/12081910
-
[2]
孙晓红, 白燕, 刘源发, 等.化学学报, 2008, 68(8):788-792 http://www.cqvip.com/QK/91047X/201008/34040656.htmlSUN Xiao-Hong, BAI Yan, LIU Yua-Fa, et al. Acta Chim. Sinica, 2008, 68(8):788-792 http://www.cqvip.com/QK/91047X/201008/34040656.html
-
[3]
Suvarapu L N, Seo Y K, Baek S O, et al. Eur. J. Chem., 2012, 9(3):1288-1304
-
[4]
Hu G Q, Hou L L, Xie S Q, et al. Chin. J. Chem., 2008, 26(6):1145-1149 doi: 10.1002/cjoc.v26:6
-
[5]
梁芳珍, 杜鸣, 任建成.无机化学学报, 1999, 15(3):393-396 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=19990320&flag=1LIANG Fang-Zhen, DU Ming, REN Jian-Cheng. Chinese J. Inorg. Chem., 1999, 15(3):393-396 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=19990320&flag=1
-
[6]
Garbutcheon-singh K B, Grant M P, Harper B P, et al. Curr. Top. Med. Chem., 2011, 11:521-542 doi: 10.2174/156802611794785226
-
[7]
Huang Y Q, Chen H Y, Li Z G, et al. Inorg. Chim. Acta, 2017, 466:71-77 doi: 10.1016/j.ica.2017.05.039
-
[8]
Huang Y Q, Wan Y, Chen H Y, et al. New J. Chem., 2016, 40(9):7587-7595 doi: 10.1039/C6NJ01231K
-
[9]
Jungwirth U, Kowol C R, Keppler B K, et al. Antioxid. Redox Signaling, 2011, 15(4):1085-1127 doi: 10.1089/ars.2010.3663
-
[10]
Li X, Fang C, Zong Z, et al. Inorg. Chim. Acta, 2015, 432:198-207 doi: 10.1016/j.ica.2015.04.012
-
[11]
Zhang Y H, Zhang L, Liu L, et al. Inorg. Chim. Acta, 2010, 363(2):289-293 doi: 10.1016/j.ica.2009.08.017
-
[12]
Dai C H, Mao F L. J. Struct. Chem., 2013, 54(3):624-629 doi: 10.1134/S0022476613030244
-
[13]
Chen C L, Zhu X F, Li M X, et al. Russ. J. Coord. Chem., 2011, 37(6):435-438 doi: 10.1134/S1070328411050034
-
[14]
Akbayeva D N, Gonsalvi L, Oberhauser W, et al. Chem. Commun., 2003:264-265 http://europepmc.org/abstract/MED/12585422
-
[15]
Chellaian J D, Johnson J. Spectrochim. Acta A, 2014, 127:396-404 doi: 10.1016/j.saa.2014.02.075
-
[16]
Wang R M, He N P, Song P F, et al. Pure Appl. Chem., 2009, 81(12):2397-2405 http://www.freepatentsonline.com/article/Pure-Applied-Chemistry/216339273.html
-
[17]
Adhikary J, Kundu P, Dasgupta S, et al. Polyhedron, 2015, 101:93-102 doi: 10.1016/j.poly.2015.07.055
-
[18]
闵睿, 范晓瑞, 周攀, 等.无机化学学报, 2014, 30(8):1771-1777 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140805&flag=1MIN Rui, FAN Xiao-Rui, ZHOU Pan, et al. Chinese J. Inorg. Chem., 2014, 30(8):1771-1777 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140805&flag=1
-
[19]
Kareem A, Zafar H, Sherwani A, et al. J. Mol. Struct., 2014, 1075:17-25 doi: 10.1016/j.molstruc.2014.06.073
-
[20]
Bruker AXS Inc., SAINT Ver.8.34A, Madison, WI, USA, 2013.
-
[21]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2008, A64:112-122 http://www.scienceopen.com/document/vid/9001a01f-495e-41c1-a9ff-560bde2378a2
-
[22]
Dolomanov O V, Bourhis L J, Gildea R J, et al. J. Appl. Cryst., 2009, 42:339-341 doi: 10.1107/S0021889808042726
-
[23]
高平章, 郑敏敏, 陈雅心, 等.无机化学学报, 2016, 32(9):1572-1578 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160911&flag=1GAO Ping-Zhang, ZHENG Min-Min, CHEN Ya-Xin, et al. Chinese J. Inorg. Chem., 2016, 32(9):1572-1578 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160911&flag=1
-
[24]
Frisch M J, Trucks G W, Schlegel H B. Gaussian 09, Revision A.. 02, Gaussian Inc.[J]. Wallingford CT, 2009, : .
-
[25]
Dreizler R M, Gross E U K. Density Functional Theory. Heidelberg:Springer-Verlag, 1990.
-
[1]
-

图 1 配合物1的分子结构
Figure 1 Molecular structure of 1
30% probability displacement ellipsoids; Symmetry codes:ⅰ-x, -y, -z

图 2 配合物1中的氢键
Figure 2 Hydrogen bands in the complex 1
Symmetry codes: ⅱ1-x, 1-y, 1-z; ⅲ1+x, y, z

图 3 配合物2的分子结构
Figure 3 Molecular structure of the complex 2
30% probability displacement ellipsoids
表 1 配合物的晶体数据
Table 1. Crystallographic data for the complexes
Complex 1 2 Empirical formula C36H28Co2N6O10 C36H26N4Ni2O4 Formula weight 822.50 696.03 Temp erature/K 293(2) 293(2) Crystal system Triclinic Trigonal Space group P1 R3 a/nm 0.547 17(11) 3.743 7(11) b/nm 1.155 5(2) 3.743 7(11) c/nm 1.420 9(3) 0.604 90(12) α/(°) 104.63(3) β/(°) 98.75(3) γ/(°) 102.51(3) Volume/nm3 0.828 0(3) 7.342(3) Z 1 9 Dc/(Mg·m-3) 1.650 1.417 Absorption coefficient/mm-1 1.074 1.199 F(000) 420 3 222 Range of θ/(°) 3.04~25.01 2.88~25.01 Limiting indices (h, k, l) -6~6, -13~13, -16~16 -44~44, -44~43, -7~7 Reflection collected 13 760 32 457 Unique reflection 2 917(Rint=0.051 4) 2 875(Rint=0.110 5) Data, restraint, parameter 2 918, 0, 245 2 875, 3, 208 Goodness-of-fit on F2 1.040 1.115 Final R indices [I>2σ(I)] R1=0.042 8, wR2=0.112 3 R1=0.096 6, wR2=0.205 7 R indices (all data) R1=0.051 7, wR2=0.117 8 R1=0.159 3, wR2=0.234 8 Largest diff. peak and hole/(e·nm-3) 553 and -440 1 121 and -621  下载: 导出CSV
下载: 导出CSV
表 2 配合物1的主要键长和键角
Table 2. Selected bond lengths (nm) and bond angles (°) for the complex 1
Co1-O2 0.195 8(2) Co1-O1 0.201 1(2) Co1-N2 0.205 1(2) Co1-O5 0.207 8(2) Co1-N3 0.209 8(3) O1-C1 0.135 3(4) N2-C6 0.140 9(4) N2-C7 0.128 7(4) O2-C13 0.129 5(4) O2-Co1-O1 155.03(10) O5-Co1-N3 95.25(9) O1-Co1-N3 96.27(9) O2-Co1-O5 100.38(10) O2-Co1-N2 89.35(9) O1-Co1-N2 81.30(9) O2-Co1-N3 89.56(10) O1-Co1-O5 103.23(9) N2-Co1-O5 93.31(9) N2-Co1-N3 171.43(10)
下载: 导出CSV
表 3 配合物1中的氢键参数
Table 3. Parameters of hydrogen bonds in the complex 1
D-H…A d(D-H)/nm d(H…A)/nm d(D…A)/nm ∠DHA/(°) 05-H5A…O1ⅱ 0.086 0.239 0.281 02 111 05-H5B…O1ⅲ 0.082 0.189 0.269 41 157 C7-H7…O4 0.093 0.243 0.333 90 164 Symmetry codes: ⅱ1-x, 1-y, 1-z; ⅲ1+x, y, z
下载: 导出CSV
表 4 配合物2的主要键长、键角和扭转角
Table 4. Selected bond lengths (nm), bond angles and torsion angles (°) for the complex 2
Ni1-O1 0.183 6(6) Ni1-O2 0.179 3(6) Ni1-N1 0.192 9(8) Ni1-N2 0.194 9(5) C7-N1 0.109 3(12) C6-N1 0.149 2(11) O1-C1 0.132 2(8) O2-C13 0.131 1(9) O2-Ni1-O1 177.9(3) O2-Ni1-N1 98.9(3) O1-Ni1-N1 82.5(3) O2-Ni1-N2 89.0(2) O1-Ni1-N2 89.6(3) N1-Ni1-N2 171.7(3) N1-Ni1-O2-C13 0.5(5) N2-Ni1-O1-C1 178.0(5) O1-Ni1-N2-C18 -2.4(6) N1-Ni1-O2-C13 2.3(7) N2-Ni1-O2-C13 -175.5(7) O2-Ni1-N2-C14 0.4(6) C15ⅰ-C16ⅰ-C16-C15 180 N1-C6-C1-O1 3.5(10) O2-Ni1-N1-C7 -1.4(9) Symmetry codes: ⅰ-x+1/3, -y+2/3, -z+5/3
下载: 导出CSV
表 5 配合物2的前线分子轨道能量和分子碎片对该分子轨道贡献
Table 5. Frontier molecular orbital energy and molecular fragment contribution to the molecular orbitals of complex 2
Orbital Energy/eV Contribution of molecular fragment/% C5N C13NO2 Ni LUMO+1 -1.734 96.4 0.0 0.0 LUMO -2.563 96.2 0.0 0.0 HOMO -5.127 0.0 86.6 8.4 HOMO-1 -5.132 0.0 86.5 8.5
下载: 导出CSV
表 6 配合物2的Wiberg键级
Table 6. Wiberg bond order of the complex 2
Ni1-O1 0.548 C6-N1 1.053 C4-C5 1.457 C9-C10 1.516 Ni1-O2 0.512 C7-N1 1.527 C5-C6 1.359 C10-C11 1.334 Ni1-N1 0.496 C1-C2 1.317 C6-C1 1.26 C11-C12 1.514 Ni1-N2 0.367 C2-C3 1.471 C7-C8 1.218 C12-C13 1.284 C1-O1 1.141 C3-C4 1.384 C8-C9 1.293 C14-N2 1.348 C13-O2 1.204 C14-C15 1.449 C15-C16 1.373 C16-C17 1.374 C17-C18 1.448 C18-N2 1.346 C8-C13 1.216
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1298
- HTML全文浏览量: 174
 下载:
下载:
