Figure 1.
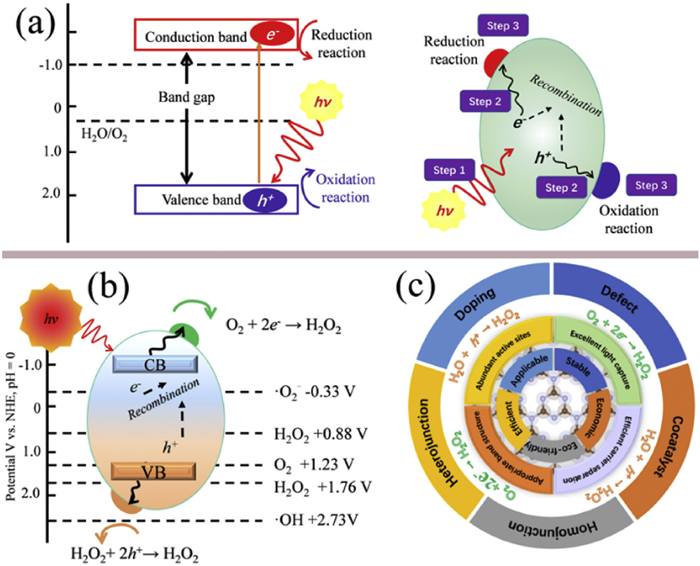
(a) Schematic diagram of the basic principle of photocatalysis. (b) Photocatalytic preparation of H2O2 reaction. (c) The modification strategies schematic of g-C3N4.
Solar-driven hydrogen peroxide production on designed g-C3N4: Strategies, mechanisms, and perspectives
Zhimin Yuan , Xingling Zhao , Xianglin Zhu , Kaili Wang , Ya-Qian Lan , Zaiyong Jiang

Hydrogen peroxide is recognized as a green oxidant with broad applicability across diverse fields, including wastewater treatment, biomedicine, organic synthesis, chemical sensing, and energy conversion technologies such as fuel cells [1-6]. Its appeal lies in its environmentally benign decomposition pathway, yielding only water and oxygen [7-11]. With the advancement of green chemistry and carbon neutrality goals, H2O2 has not only gained prominence as a clean oxidant but is also regarded as a promising energy carrier and electrocatalytic medium within renewable energy systems [12-17]. However, the current industrial production of H2O2 predominantly relies on the anthraquinone redox process, which involves organic solvents, anthraquinone carriers, high-pressure hydrogen, and multiple mass-transfer steps [18-20]. This method suffers from complexities in operation, high energy consumption, and significant environmental impact. In response to the demand for green synthesis and decentralized applications, several alternative strategies have been proposed, including the direct synthesis from hydrogen and oxygen, electrocatalytic oxygen reduction, and plasma-assisted processes [21-23]. Nonetheless, these approaches often encounter limitations such as safety concerns, low Faradaic efficiency, or complicated reactor design [24-26]. Hence, there is a pressing need to develop novel H2O2 synthesis systems that operate without hazardous gases, under mild conditions, with low energy input, and through clean reaction pathways [27,28].
In recent years, photocatalytic H2O2 production has emerged as an attractive alternative, driven by solar energy and utilizing benign reactants such as O2 and H2O under ambient conditions [29-32]. In a typical photocatalytic process (Fig. 1a), the photocatalyst absorbs photons with energy equal to or exceeding its band gap, electrons in the valence band are excited to the conduction band, forming electron-hole pairs [33-37]. These unstable pairs then need to separate and migrate to the catalyst’s surface, meaning efficient separation is critical to avoid non-productive recombination that dissipates energy as heat or light. Once on the surface, the electrons react with electron acceptors, while holes oxidize donors; these radical species further drive target redox reactions, with minimizing electron-hole recombination being key to enhancing catalytic efficiency [38-42]. For the H2O2 generation, the photoexcited electrons migrate to the conduction band to reduce oxygen molecules into H2O2, while the holes can be consumed by sacrificial agents or water oxidation, thereby maintaining charge balance and enabling redox coupling. A critical aspect of this process lies in achieving selective two-electron oxygen reduction (2e− ORR) to form H2O2, while avoiding competing pathways such as the four-electron reduction to water or further reduction and decomposition of reactive intermediates.
The photocatalytic synthesis of H2O2 can proceed via two principal routes: the oxygen reduction reaction (ORR) and the water oxidation reaction (WOR) [43-46]. Upon exposure to light, the photocatalyst generates electron-hole pairs, where the photogenerated electrons drive the reduction of O2 to H2O2, and the photogenerated holes facilitate the oxidation of H2O to H2O2. For both ORR and WOR to proceed efficiently, the photogenerated charge carriers must possess appropriate redox potentials that meet the thermodynamic criteria of the respective reactions.
The reduction of O2 in photocatalytic systems typically proceeds via the following pathways [47-50]:
(a) Oxygen reduction reaction (ORR) pathways
Two-electron reduction (2e− ORR):
|
|
(1) |
One-electron reduction (1e− ORR):
|
|
(2) |
|
|
(3) |
The competing reaction in this process is:
|
|
(4) |
(b) Water oxidation reaction (WOR) pathways [51,52]
Two-electron water oxidation (2e− WOR):
|
|
(5) |
One-electron water oxidation (1e− WOR):
|
|
(6) |
|
|
(7) |
The main competing reaction in this process is:
|
|
(8) |
The ORR involved in H2O2 synthesis proceeds via a two-electron transfer mechanism, which can be conceptualized either as a direct two-electron process (Eq. 1) or as a sequential two-step single-electron process ((2), (3)). In the latter pathway, the superoxide radical anion (·O2−) serves as a key intermediate species. Notably, the redox potential of the O2/H2O2 couple (0.68 V versus the normal hydrogen electrode, NHE) is more positive than that of the O2/·O2− couple (-0.33 V vs. NHE), suggesting, from a thermodynamic standpoint, a greater likelihood for the direct two-electron pathway to predominate. During photocatalytic O2 reduction, a competing reaction exists wherein O2 undergoes further reduction to water (H2O) via photogenerated electrons (Eq. 4). Given that the redox potential of the O2/H2O couple (1.23 V vs. NHE) exceeds that of the O2/H2O couple, the four-electron ORR pathway is thermodynamically more favorable.
Similarly, the WOR on photocatalysts can proceed through multiple electron transfer routes, including four-electron, two-electron, and one-electron pathways (Eqs. 5-7). The two-electron WOR pathway can be further subdivided into an indirect two-step single-electron transfer mechanism (Eqs. 6 and 7) and a direct one-step two-electron process (Eq. 5). Thermodynamically, the direct two-electron pathway is preferred; however, kinetically, the indirect mechanism tends to dominate. For effective H2O2 generation via the two-electron WOR route, the valence band edge of the photocatalyst must be positioned more positively than + 1.76 V versus NHE to satisfy thermodynamic criteria. In contrast, the four-electron WOR pathway is thermodynamically more advantageous, thereby favoring oxygen evolution over H2O2 production.
Photocatalysts typically demonstrate low adsorption energies for oxygen, which facilitates the desorption of reaction products [53]. Overall, the two-step single-electron transfer mechanism is kinetically more favorable. Concurrently, O2 readily interacts with photogenerated holes to form singlet oxygen species (1O2, ·O2⁻/1O2 0.34 V vs. NHE), resulting in a reduction of H2O2 yield. Analogous to the two-electron ORR, the actual efficiency of the two-electron WOR is governed by the interplay between thermodynamic driving forces and kinetic energy barriers. This reaction can proceed via two potential pathways: a direct two-electron transfer or a stepwise single-electron transfer, with the stability of intermediate species critically determining the preferred reaction route.
The formation and stability of key intermediates such as superoxide radicals (·O2−), hydroperoxyl species (*OOH), and singlet oxygen (1O2), alongside oxygen adsorption and activation behaviors, charge separation efficiency, and interfacial reaction kinetics, collectively exert a decisive influence on the rate and selectivity of H2O2 production. It is imperative to modulate the electron density at the catalyst’s active sites to ensure that the bond energies align with the energy barriers required for efficient two-electron transfer, thereby minimizing side reactions associated with excessively strong or weak bonding. Furthermore, in photocatalytic systems, promoting the directional transfer of photogenerated charge carriers-electrons to O2 in ORR or holes to H2O in WOR-while suppressing charge recombination is essential to maintain sufficient charge availability for two-electron processes. An ideal photocatalyst should exhibit an appropriate band structure, a high density of surface-active sites, favorable oxygen affinity, and controllable electron transfer pathways to optimize performance.
In the domain of photocatalytic research, the efficient photocatalyst is one of the important cores. But the developments of semiconductor photocatalysts exhibit high efficiency, robust stability, and the capability to utilize a broad spectrum of solar radiation remains a significant and ongoing challenge. A variety of photocatalysts have been extensively studied, demonstrating promising performance in specific reactions; these include metal oxides [54-62], sulfides [63-68], oxynitrides [69], metal-organic frameworks (MOF) [70-74], covalent organic frameworks (COF) [75-77], and polymer [78,79], etc. Nevertheless, they still share several fundamental limitations, such as restricted visible-light absorption, rapid recombination of photogenerated charge carriers, low quantum yield, and inadequate long-term chemical stability [60,61,80]. These issues significantly hinder their practical applications on a larger scale.
A prototypical example is titanium dioxide (TiO2), one of the most extensively investigated photocatalysts. With a bandgap of approximately 3.2 eV, TiO2 predominantly absorbs ultraviolet light, which accounts for less than 5% of the solar spectrum, and exhibits minimal photocatalytic activity under visible light [81-87]. Although strategies such as elemental doping, composite formation, and surface sensitization have been employed to enhance its light-harvesting capabilities, these modifications cannot fully surmount the fundamental absorption limitations imposed by its wide bandgap. Photostability constitutes another critical issue. Narrow-bandgap materials such as cadmium sulfide (CdS) and tantalum oxynitride (TaON), which possess favorable band alignments for visible-light absorption, are prone to photo-corrosion and chemical degradation [88]. For example, under illumination, photogenerated holes in CdS can oxidize sulfide ions to sulfate, causing irreversible structural damage [89]. Similarly, certain nitride-based photocatalysts experience surface oxidation or lattice instability upon prolonged light exposure, leading to rapid declines in catalytic activity over successive cycles. Additionally, phenomena such as light-induced leaching of active species and surface reconstruction further undermine the structural integrity and long-term durability of these materials.
Multielectron processes, such as overall water splitting, impose more rigorous demands on photocatalysts [90-93]. The photocatalytic material must not only provide active sites for both proton reduction and water oxidation but also effectively reduce the substantial overpotentials associated with hydrogen and oxygen evolution reactions [94-96]. These requirements establish stringent criteria concerning the band structure, charge transport characteristics, and surface reaction kinetics of photocatalysts. At present, only a limited number of systems concurrently fulfill the criteria of broad spectral absorption, efficient charge separation, and exceptional stability [97,98]. Consequently, progress in this domain necessitates the design and synthesis of novel photocatalytic materials that can harness a wider spectrum of solar radiation, promote effective charge separation and transfer, and sustain durable performance under chemically demanding conditions. Pursuing this approach is critical for realizing efficient, stable, and scalable solar energy conversion technologies.
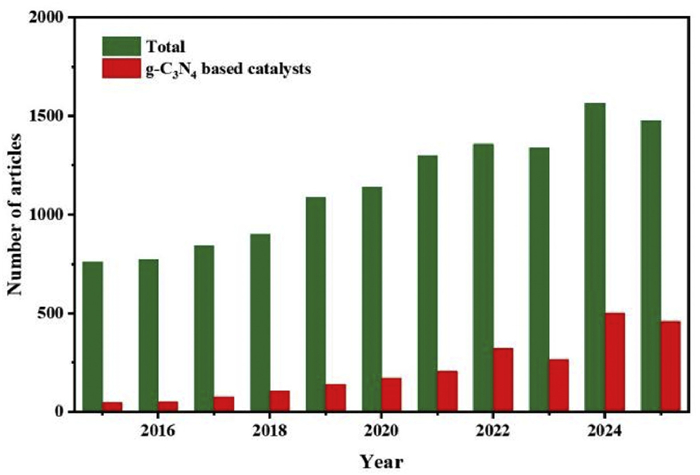
Recently, g-C3N4 has garnered considerable interest as a potential photocatalyst for H2O2 synthesis and become a hot research topic in recent years (Fig. 2), due to its unique two-dimensional layered structure, metal-free composition, ability to absorb visible light, and adjustable electronic properties [99-102]. The performance comparison of some modified g-C3N4-based photocatalysts for H2O2 production and relevant references are shown in Table 1. The conduction band potential of g-C3N4 is approximately -1.3 V vs. NHE [103], which thermodynamically facilitates the oxygen reduction reaction (Fig. 1b). Additionally, the intrinsic g-C3N4 material absorbs mainly in the UV–visible region, yet its visible-light response remains insufficient, restricting its photocatalytic efficiency [104,105]. Therefore, improving photocatalysts capable of effectively utilizing visible light is crucial for enhancing solar-energy conversion. Although nearly 48% of solar irradiation lies below 700 nm, the limit constrains the theoretical conversion efficiency, meaning that extending the absorption range alone does not guarantee higher photocatalytic performance. Furthermore, the practical utilization of g-C3N4 is impeded by several inherent drawbacks, including a high exciton binding energy that hampers effective charge carrier separation, limited oxygen adsorption capacity, a low density of states within the conduction band region, a scarcity of catalytically active sites, and the propensity of H2O2 to decompose under reaction conditions.
 DownLoad:
CSV
DownLoad:
CSV
| Authors (year) | Catalyst | Modification strategy | Light source | H2O2 production activity | Enhancement factor (vs. pristine CN) | AQY/wavelength | Electron donor | Ref. |
| Yang et al. (2023) | A0.05CN | Carbon doping & surface -OH | Visible (λ ≥ 420 nm) | 3.6 mmol g−1 h−1 | 3.5 × | Not available | Pure water | [130] |
| Zhang et al. (2024) | s-BxCN | Carbon doping, N vacancies, -C≡N | Visible light | 54.2 µmol L−1 h−1 | Not available | Not available | Pure water | [131] |
| Xu et al. (2025) | KLCN | Lignin-derived carbon ring embedding & N defects | Visible light | 266.82 µmol/L | 6.93 × | Not available | Isopropanol | [132] |
| Wei et al. (2018) | OCN-500 | Oxygen doping (C-O-C/—OH) | Visible light | 730 µmol / 5h | 3.5 × | 28.5%@365 nm, 10.2%@420 nm | Isopropanol | [133] |
| Yu et al. (2023) | PCN-D | Dual-site P doping (bay & corner) | Visible light | 413.85 µmol L−1 h−1 | > 6 × | Not available | Isopropanol | [134] |
| Ma et al. (2024) | BCN-450 | B doping (Frustrated Lewis Pairs) | Visible light | 51008 µmol L−1 g−1 h−1 | 12.4 × | Not available | Ethanol | [135] |
| Zhang et al. (2023) | CN-1.0 | S doping & N vacancies | Xenon lamp | 293.2 µmol/L / 4 h | 2.4 × | Not available | Ethanol | [136] |
| Liu et al. (2021) | B-CNT | B/P/S co-doping, nanotube | Simulated sunlight | 1830.98 µmol/L / 4 h | 2 × | Not available | Isopropanol | [137] |
| Habarugira et al. (2024) | 10SD-CN | Na doping & N defects | Visible light | 297.2 µmol L−1 h−1 | 9.8 × | 1.29%@420 nm | Isopropanol | [138] |
| Wu et al. (2020) | ACNN | K⁺/Na⁺ doping & N vacancies | Visible (λ ≥ 420 nm) | 10.2 mmol g−1 h−1 | 89.5 × | 30.7%@429 nm | Isopropanol | [139] |
| Chen et al. (2021) | 5Cv@g-C3N4 | Cyano defects & Na doping, porous | Visible (λ ≥ 420 nm) | 7.01 mmol L−1 h−1 | 220 × | 9.58%@420 nm | Ethanol | [140] |
| Zhang et al. (2020) | AKMT | S/K co-doping | Visible light | 4.1 mmol/L / 3 h | Not available | 100%@450 nm | Ethanol | [145] |
| Wang et al. (2024) | Na1.2-P0.2-CN | P/Na co-doping | Visible light | 3001.64 µmol g−1 L−1 / 100 min | 62 × | Not available | Isopropanol | [146] |
| Wu et al. (2022) | GCN | Carbon defects, flower-like | Xenon lamp (λ > 420 nm) | 21.59 mmol L−1 g−1 h−1 | 5 × | Not available | Isopropanol | [149] |
| Zhang et al. (2022) | Nv-C≡N-CN | N vacancies & -C≡N groups | Visible (λ ≥ 420 nm) | 3093 µmol g−1 h−1 | 8.7 × | 36.2%@400 nm | Isopropanol | [152] |
| Li et al. (2023) | SCN2 | S doping, -NH2/-CH3 edge groups | Visible (λ ≥ 420 nm) | 416.7 µmol g−1 h−1 | 10 × | 31.6%@365 nm | Pure water | [150] |
| Zeng et al. (2025) | BPMC-Vs | B/P co-doping & N defects | Visible (λ ≥ 420 nm) | 650.7 µmol g−1 h−1 | 9.9 × | 39.1%@420 | Isopropanol | [151] |
| Chen et al. (2023) | CNT | Crystallinity enhancement (Molten salt) | AM 1.5 | 2.48 mmol g−1 h−1 | 5.8 × | 22%@ 00 nm | Isopropanol | [153] |
| Zhong et al. (2024) | CN-K(1:6) | K doping, High crystallinity | LED | 7.8 mmol L−1 h−1 | 220 × | 5.17%@420 nm | Isopropanol | [154] |
| Li et al. (2024) | BTOCN2 | Bi4Ti3O12/g-C3N4 S-scheme | Simulated sunlight | 1650 µmol g−1 h−1 | 2.8 × | 0.36%@420 nm | Methanol | [173] |
| Zhang et al. (2023) | 3CuInS2/PCN | CuInS2/PCN S-scheme | Visible light | 1247.6 µmol g−1 h−1 | 11.6 × | 16.0%@420 nm | Isopropanol | [174] |
| Chen et al. (2023) | homo-CN | Homojunction (OH-rich/OH-defective) | Xenon lamp (λ ≥ 420 nm) | 508.4 µmol L−1 L−1 | 4.5 × | 2.1%@500 nm | Ethanol | [179] |
| Zhang et al. (2024) | CN-NH4-NaK | Homojunction (Order-Disorder) | Visible (λ ≥ 420 nm) | 16675 µmol g−1 h−1 | 158 × | 34.3%@395 nm, 28.4%@420 nm | Isopropanol | [181] |
| Foo et al. (2025) | CCN-550 | Homojunction (PHI/PTI), K+/Li+, -C≡N | Visible (λ ≥ 420 nm) | 5838.91 µmol L−1 h−1 | 31.7 × | 11.57%@420 nm | Benzyl alcohol | [184] |
| Hou et al. (2024) | CoOx-NvCN | N vacancies & CoOx clusters | Visible (λ ≥ 420 nm) | 244.8 µmol L−1 h−1 | 12.1 × | 5.73%@420 nm | Pure water | [190] |
| Jing et al. (2024) | CoOx-BCN-FeOOH | B doping, FeOOH & CoOx clusters | Visible (λ ≥ 420 nm) | 0.34 mmol g−1 h−1 | 30 × | 8.36%@420 nm | Pure water | [191] |
| Zhang et al. (2023) | Ni-SAPs/PuCN | Ni single-atom (Ni-N3) | Visible (λ ≥ 420 nm) | 342.2 µmol L−1 h−1 | 20.7 × | 10.9%@420 nm | Pure water | [143] |
| Su et al. (2021) | Sb-SAPC15 | Sb single-atom (Sb-N) | Visible (λ > 420 nm) | 6.2 mg L−1 h−1 | 248 × | 17.6%@420 nm | Pure water | [144] |
From view of the structure, the layered structure held together by weak van der Waals forces provides poor in-plane conductivity and promotes rapid electron–hole recombination, further exacerbated by abundant intrinsic defect states associated with lone-pair nitrogen atoms that act as charge-trapping centers. The low specific surface area of bulk g-C3N4 (< 10 m2/g) leads to a scarcity of catalytically active sites and weak adsorption of O2 and the OOH* intermediate, which slows the kinetics of the 2e− oxygen reduction pathway required for selective H2O2 formation. In addition, its conduction band is sufficiently negative to trigger competing 4e− ORR or promote further reduction and decomposition of the generated H2O2, while the valence band position (~1.4 V vs. RHE) is not positive enough to thermodynamically drive the 2e⁻ water oxidation pathway, preventing a synergistic oxidation contribution to H2O2 accumulation [106-109]. The combination of restricted visible-light absorption, short carrier lifetimes, limited active-site density, weak O2 activation capability, and competing redox pathways collectively results in the inherently low H2O2 yield of g-C3N4, making rational structural modification essential for achieving high photocatalytic efficiency.
While the fundamental mechanisms provide a theoretical foundation, translating these principles into efficient photocatalytic systems requires deliberate material design. To enhance the photocatalytic performance of g-C3N4, researchers have developed multi-dimensional optimization strategies focusing on its electronic structure and interface chemistry (Fig. 1c). Approaches such as defect engineering, heteroatom doping, single-atom loading, π-π interaction modification, junction construction, colloidal dispersion, and multi-interface synergy have been proposed [110-113]. These strategies not only improve the light absorption capacity and charge carrier separation efficiency of g-C3N4, but also regulate the O2 adsorption mode, the formation and stability of *OOH intermediates, proton transfer pathways, and H2O2 desorption behavior from the intrinsic structural level, thereby significantly boosting catalytic activity and product selectivity [114-117]. Particularly in recent years, the H2O2 production rate of high-performance g-C3N4 catalysts has increased from several hundred µmol g−1 h−1 in the early stage to 10–50 mmol g−1 h−1, with quantum efficiency exceeding 30%, indicating that photocatalytic H2O2 synthesis is becoming increasingly mature for practical application. Nonetheless, current systems continue to face significant challenges, including limited universality in structural regulation, difficulties in in situ observation of reaction intermediates, inadequate long-term stability, and obstacles to large-scale fabrication, all of which impede the commercial advancement of photocatalytic H2O2 technologies.
In response, this paper centers on g-C3N4 and provides a systematic review and in-depth analysis of the evolution of reaction mechanisms, material design strategies, and performance optimization pathways for photocatalytic H2O2 production in recent years. The scope of this work encompasses: (1) Fundamental reaction pathways and surface dynamic processes involved in photocatalytic H2O2 generation; (2) the effects of intrinsic and modified structural regulation of g-C3N4 on catalytic performance; (3) key challenges and limitations in current research; and (4) prospects for future material design, device integration, and application development. This study aims to offer a theoretical foundation and research reference to facilitate the development of a new generation of efficient, environmentally friendly, and cost-effective photocatalytic H2O2 technologies.
Doping represents a straightforward yet effective approach for optimizing the performance of g-C3N4 in photocatalytic application. Introducing metal or non-metal dopants can significantly modulate the electronic structure of the material, thereby improving its catalytic activity [118-125]. Such modifications induce profound changes in the electronic configuration through interactions between dopant atoms and the carbon/nitrogen framework [126-129]. Specifically, doping narrows the band gap by introducing in-gap states or facilitating orbital hybridization, while also tuning the energies of the valence band maximum (VBM) and conduction band minimum (CBM) to optimize redox potentials. Furthermore, the density of states near the Fermi level is altered, enhancing charge carrier density and conductivity. Notably, the reconstruction of the surface electronic structure promotes the adsorption of reactants and reduces the activation energy barriers for key reaction intermediates. Based on the nature of the incorporated elements, doping strategies for g-C3N4 can be categorized into non-metal doping, metal doping, and co-doping. Each approach contributes synergistically to enhance light absorption, facilitate charge separation, and improve interfacial catalytic efficiency. These mechanisms have been corroborated through comprehensive experimental analyses, including spectroscopic characterization, transient photoelectrochemical techniques, and detailed reaction pathway studies.
Carbon doping is commonly achieved by increasing the C/N ratio within the g-C3N4 framework or introducing carbon-rich organic precursors to partially substitute nitrogen sites. This approach enhances the π-electron density and increases the density of states in the conduction band, thereby facilitating photogenerated electron transfer toward the oxygen reduction pathway. Additionally, the incorporation of carbon can lead to the formation of C-terminal defects and non-conjugated groups, which further modulate the conduction band edge-promoting O2 adsorption and stabilizing reactive intermediates. These modifications collectively contribute to an extended charge carrier lifetime.
For example, Yang et al. engineered dual defects-namely carbon doping and surface hydroxyl (-OH) modification-by adjusting the acetylacetone to urea precursor ratio [130]. The optimized sample A0.05CN demonstrated a H2O2 production rate of 3.6 mmol g−1 h−1 under visible light irradiation (λ ≥ 420 nm), which is 3.5 times greater than that of pristine g-C3N4 (Figs. 3a-d). Additionally, the selectivity increased from 60% to 85%. Integrated experimental and theoretical investigations indicated that carbon doping enhanced oxygen adsorption and activation at N2C sites, whereas surface hydroxyl groups promoted charge carrier diffusion and inhibited recombination. The observed narrowing of the band gap to 2.56 eV, the enlarged surface area, and a significant electron transfer number (n ≈ 2.0) collectively corroborate the improved two-step single-electron oxygen reduction mechanism.
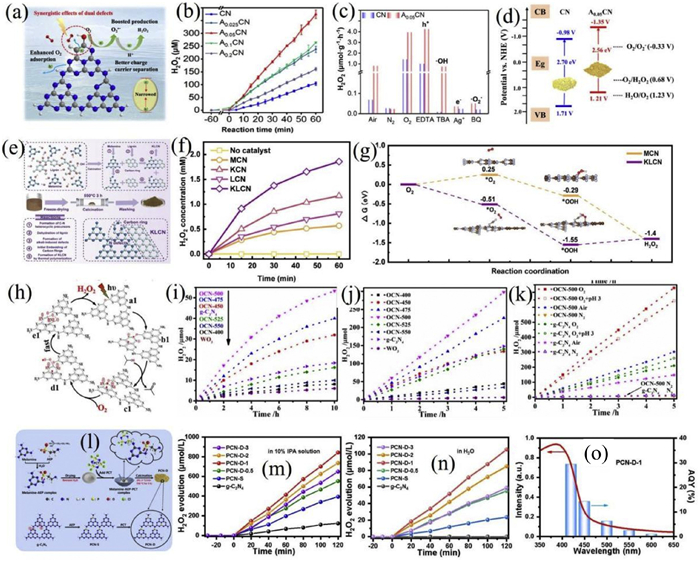
In another approach, Zhang et al. synthesized carbon-doped, defect-rich g-C3N4 (s-B≡CN) via supramolecular pre-assembly and thermal polymerization [131]. Incorporating barbituric acid yielded a nest-like porous structure enriched with N vacancies and –C≡N groups, which broadened visible light absorption and improved charge separation. The material achieved an H2O2 production rate of 108.4 µmol/L within 2 h in pure water, attributed to the synergistic effect of strengthened 2e− ORR (via ·O2−) and hole-driven water oxidation. Furthermore, the in-situ generated H2O2 enabled efficient degradation of 4-chlorophenol via a photo-self-Fenton process, highlighting potential practical applications. Xu et al. employed a co-modification strategy using lignin and KOH to embed carbon rings and induce nitrogen defects (cyano groups and N vacancies) in KLCN (Figs. 3e-g) [132]. This synergistic incorporation resulted in an H2O2 yield 6.93 times greater than that of the unmodified catalyst, with mechanistic studies confirming the critical role of ·O2− intermediates in the two-step single-electron reduction pathway.
Current studies have demonstrated that carbon doping enhances the photocatalytic activity of g-C3N4 through band structure engineering, improved charge carrier kinetics, and enhanced surface adsorption of reactants. A key advantage is the concurrent activation of both oxygen reduction and water oxidation pathways, providing a versatile strategy for designing efficient photocatalytic systems for H2O2 production.
Wei et al. developed an oxygen-enriched carbon nitride polymer (OCN) with enhanced photocatalytic performance for H2O2 generation (Figs. 3h-k) [133]. DFT simulations indicated that incorporating C-O-C groups considerably lowered the energy barrier for forming 1,4-endoperoxide intermediates by 10.5 kcal/mol compared to -NH2 groups, thus favoring selective oxygen reduction. The material, labeled OCN-500, was synthesized through calcination at 500 ℃ in air using dicyandiamide and ammonium paratungstate as precursors. This process introduced C-O-C and -OH functional groups, increased the specific surface area, and narrowed the band gap. Under O2-saturated conditions in pure water, OCN-500 exhibited an H2O2 production rate of 730 µmol over 5 h, representing a 3.5-fold enhancement over unmodified g-C3N4. The catalyst also demonstrated high apparent quantum yields (28.5% at 365 nm and 10.2% at 420 nm) and exceptional stability over 20 h. RDE and EPR analyses corroborated the dominance of the two-electron oxygen reduction pathway (n = 2.54) and the presence of radical intermediates (ROO·/RO·), respectively, with the proposed 1,4-endoperoxide mechanism further supported by DFT.
A dual-site phosphorus doping strategy was introduced by Yu et al. to markedly improve the photocatalytic H2O2 production activity of g-C3N4 (Figs. 3l-o) [134]. Through thermal polymerization, P atoms were selectively incorporated at both bay and corner sites of the heptazine framework, effectively breaking electron localization symmetry and reducing exciton binding energy. This resulted in an exciton dissociation efficiency surge from 0.59% to 11.9%, alongside a 13-fold increase in charge carrier concentration and a 7-fold mobility improvement. The optimized catalyst (PCN-D) achieved an H2O2 generation rate of 413.85 µmol L−1 h−1 under visible light which is 6 times that of pristine g-C3N4 and performed robustly in both pure water and seawater. Mechanistic studies revealed that corner-site P doping enhances O2 adsorption and activation, while bay-site doping facilitates charge delocalization and separation, collectively promoting an efficient 2e− ORR.
Ma et al. employed boron doping to construct frustrated Lewis pairs (FLPs) within the carbon nitride matrix, significantly boosting H2O2 production (Figs. 4a-d) [135]. The FLPs consist of boron Lewis acid sites and nitrogen Lewis base sites adjacent to cyano groups. The introduced B atoms created spin-polarized empty orbitals, improving visible light absorption and charge separation. The FLP structure preferentially activated ethanol via a "push–pull" mechanism rather than direct oxygen reduction. DFT studies indicated a reduction of the ethanol dissociation barrier to 0 eV, leading to H2O2 formation through synergistic action of ·O2− and 1O2 intermediates. In a 10% ethanol solution, the catalyst (BCN-450) attained a remarkable H2O2 production rate of 51008 µmol L−1 g−1 h−1, 12.4 times that of the baseline, with consistent performance over five cycles. In-situ IR spectroscopy confirmed ethanol activation to aldehyde species, underscoring the role of FLPs in altering reaction selectivity and pathway efficiency.
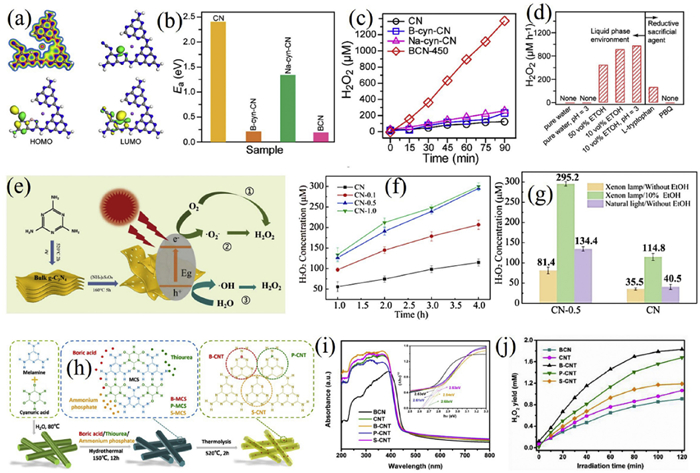
Zhang et al. concurrently introduced sulfur dopants and nitrogen vacancies into g-C3N4 via hydrothermal etching with ammonium persulfate, coupled with morphological control to obtain a porous fluffy architecture (Figs. 4e-g) [136]. This dual-defect system synergistically optimized the electronic structure, and the 27-fold enlargement of specific surface area significantly improved O2 adsorption capacity. The modified catalyst yielded 293.2 µmol/L H2O2 within 4 h, 2.4 times higher than pristine g-C3N4, with high selectivity for the two-electron pathway. Sulfur doping induced a valence band positive shift, while nitrogen vacancies enhanced charge separation. The porous framework further facilitated mass transfer and exposure of active sites. Nevertheless, the system’s reliance on ethanol as a sacrificial agent-causing a 53% activity drop in its absence-highlights a limitation for practical application and underscores the need for developing sacrificial-agent-free configurations.
Liu et al. prepared boron/phosphorus/sulfur-doped g-C3N4 nanotubes (B/P/S-CNT) via a template-free hydrothermal-thermal polymerization synergistic strategy, enabling efficient photocatalytic H2O2 synthesis (Figs. 4h-j) [137]. A melamine-cyanuric acid supramolecular self-assembly was used to form a tubular precursor, which was then thermally polymerized at 520 ℃ to obtain a porous nanotube structure (BET specific surface area was 6.7 times that of bulk g-C3N4). Its hierarchical pores significantly promoted mass transfer and light harvesting. Simultaneously, the co-doping of non-metallic elements B, P, and S optimized the electronic structure, shifting the conduction band negatively by 0.15 eV and improving charge carrier separation efficiency. The optimal B-CNT sample achieved a maximum H2O2 yield of 1830.98 µmol/L, 4 h, twice that of bulk g-C3N4. Experimental results confirmed that the activity difference originated from the regulatory strength of doped elements on the reduction potential and the enhanced light reflection and absorption by the tubular structure. The two-electron oxygen reduction pathway dominated the H2O2 generation process, and isopropanol as a sacrificial agent provided key protons.
Metal doping introduces foreign metal atoms into the g-C3N4 matrix, effectively modulating its electronic structure and surface properties to enhance photocatalytic H2O2 production. Habarugira et al. developed a one-step copolymerization approach using sodium dicyanamide to fabricate sodium-doped carbon nitride with nitrogen vacancies (SD-CN) [138]. The introduced sodium disrupted the triazine unit ordering, leading to a wrinkled morphology. Characterization confirmed the coexistence of cyano groups, nitrogen vacancies, and Na incorporation, with the band gap narrowing to 2.75 eV (Figs. 5a-d). The optimized catalyst exhibited an H2O2production rate of 297.2 µmol L−1 h−1, which is 9.8 times higher than pristine g-C3N4 and obtained an AQE of 1.29% at 420 nm, and 90% stability retention over 8 cycles. Mechanism studies indicated cyano groups aided charge separation, while nitrogen vacancies promoted O2 adsorption, collectively favoring the two-electron ORR pathway as evidenced by ESR and RDE (n = 1.77). Despite these merits, the low specific surface area (3.40 m2/g) and limited anti-interference ability in real water environments necessitate further improvements in mass transfer and practicality.
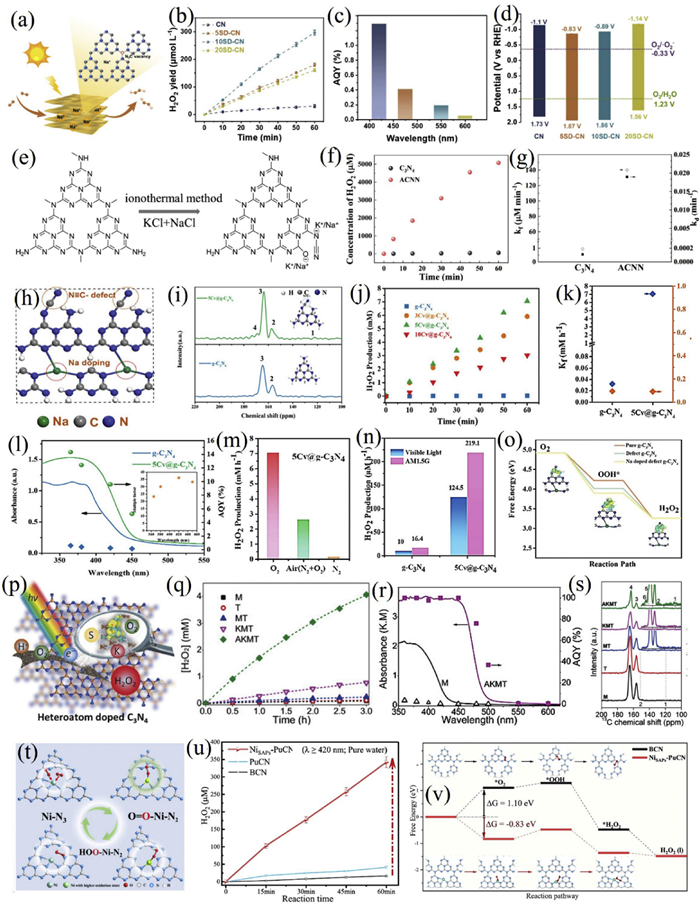
Wu et al. employed a eutectic salt ionothermal etching strategy (NaCl/KCl) to co-incorporate alkali metals (K+/Na+) and nitrogen vacancies into g-C3N4 (Figs. 5e-g) [139]. The dual modification narrowed the band gap from 2.85 eV to 2.63 eV and enhanced charge separation efficiency by 6.2 times. The best-performing ACCN sample achieved a remarkable H2O2 yield of 10.2 mmol g−1 h−1, 89.5 times that of the pristine material, with an apparent rate constant increased by 149 times and an AQE of 30.7% at 429 nm. Nitrogen vacancies functioned as electron traps to facilitate charge separation, while alkali metal doping optimized the band structure via charge compensation from -O⁻ and -C≡N groups. Nevertheless, the system’s dependency on isopropanol as a sacrificial agent and modest cycling stability represent challenges for sustainable application.
Chen et al. designed a cyano-rich, honeycomb-structured carbon nitride (Cv@g-C3N4) via a NaCl-assisted calcination route (Figs. 5h-o) [140]. The material concurrently introduced cyano defects and sodium doping, which reduced the band gap through newly formed defect levels and enhanced O2 adsorption via improved charge polarization. A NaN3-like coordination environment further promoted carrier separation and stabilized superoxide intermediates. Combined with a porous morphology for improved surface area, the catalyst delivered an H2O2 production rate of 7.01 mmol L−1 h−1 under visible light-220 times higher than conventional g-C3N4-with an AQE of 9.58% at 420 nm. It also demonstrated broad pH adaptability (3–12) and good cycling stability, maintaining 5 mmol L−1 h−1 after 5 cycles.
Photocatalytic activity is considered the cumulative result of surface reactions between active sites on the catalyst and reactants. However, identifying the dynamic structural evolution of active sites during photocatalytic surface reactions and clarifying the corresponding catalytic enhancement mechanisms remain major challenges. Single-atom photocatalysts (SAPs), featuring well-defined single-atom active sites and high atomic utilization, serve as ideal catalytic models to explore the structural evolution of active sites and reaction mechanisms in depth [141,142].
Zhang et al. anchored Ni single atoms in a porous carbon nitride matrix (Ni-SAPs/PuCN) with a high loading of 12.5 wt% in Ni-N3 coordination (Figs. 5t-v) [143]. The catalyst delivered an H2O2 production rate of 342.2 µmol L−1 h−1 in pure water, with an AQY of 10.9% and an SCC efficiency of 0.82%. In-situ synchrotron X-ray absorption and Raman spectroscopy revealed the dynamic evolution of Ni sites from Ni-N3 to O1-Ni-N2 and finally to HOO-Ni-N2 during reaction, which reduced the energy barrier for *OOH formation and suppressed O-O bond cleavage. The Ni atoms also optimized the host electronic structure, enhancing charge separation and migration.
In a recent advance, Su et al. developed an antimony single-atom catalyst (Sb-SAPC) with Sb3+ (4d105s2) atoms atomically dispersed in a polymeric carbon nitride framework via Sb-N bonds [144]. The unique electronic configuration prevented midgap states and facilitated charge separation. Under visible light and without sacrificial agents, the catalyst achieved an AQY of 17.6% at 420 nm, an SCC efficiency of 0.61%, and an H2O2 production rate 248 times that of the pristine material. The Sb single atoms adsorbed O2 in an end-on configuration, forming an Sb-OOH intermediate that promotes the 2e− ORR pathway, while simultaneously accelerating the 4e− WOR through hole accumulation on adjacent N atoms. The in-situ produced O2 from WOR was rapidly consumed via ORR, creating a synergistic cycle that enhanced overall kinetics.
Heteroatom co-doping can create synergistic effects between metal and non-metal elements, further optimizing the electronic and catalytic properties of g-C3N4. Zhang et al. constructed a sulfur and potassium co-doped catalyst (AKMT) using thiourea/melamine precursors treated with NaOH/KCl (Figs. 5p-s) [145]. The dopants disrupted the layered structure, generating abundant edge cyano and surface hydroxyl groups. Sulfur was incorporated via C-S and N-S bonds, while potassium formed N-K coordination complexes, collectively red-shifting the absorption edge to 500 nm (band gap 2.52 eV) and positively shifting the valence band to 1.89 eV. Under acidic and O2-saturated conditions, the H2O2 yield reached 4.1 mmol/L within 3 h, with a notable AQE of 100% at 450 nm and 96% electron selectivity. Transient infrared spectroscopy revealed that sulfur doping extended the lifetime of shallowly trapped electrons and directed deeply trapped electrons toward oxygen reduction. The catalyst also demonstrated potential for practical disinfection applications through in situ H2O2 generation in tap water, though activity decay in alkaline conditions remains an issue.
In a related study, Wang et al. reported a one-pot synthesis of phosphorus-sodium co-doped -C3N4 (Na1.2-P0.2-CN) [146]. Phosphorus substitution formed N-P-N bonds, while sodium intercalation induced cyano group formation and valence band shifts. DFT simulations illustrated that charge density redistribution established a built-in electric field, and transition state calculations confirmed a reduced energy barrier for *O2 formation. With isopropanol as a sacrificial agent, the H2O2 yield reached 3001.64 µmol g−1 L−1 within 100 min, surpassing the undoped catalyst by 62 times.
Defects and vacancies, as crucial regulatory units of the electronic structure and surface properties of catalysts, exert a concentration- and type-dependent dual influence on catalytic activity [147]. Typically, precise regulation of defect/vacancy type, concentration, and distribution to balance activity, selectivity, and stability remains a research focus in catalyst engineering. Surface defect engineering is an effective approach to enhance the photocatalytic efficiency of carbon nitride by addressing its inherent limitations, such as restricted visible-light absorption and rapid charge carrier recombination [59,148]. Introducing surface defects can achieve band structure modulation via defect-induced energy levels that extend light absorption into the near-infrared region and facilitate carrier separation. Defect sites serve as active centers can optimize reactant adsorption and electron migration, enhancing reaction kinetic.
Wu et al. employed an oxalic acid etching-assisted self-assembly method to synthesize flower-like carbon nitride with carbon defects (GCN) (Figs. 6a-d) [149]. Using melamine in DMSO followed by etching and calcination at 550 ℃, a porous microstructure with a diameter of 1.2 µm was obtained. The resulting material exhibited a reduced C/N ratio (0.78) and a 1.8-fold stronger EPR signal, confirming enhanced carbon defects that improved O2 adsorption and promoted the two-electron oxygen reduction pathway, as evidenced by an electron transfer number of 2.50 in RDE tests. The H2O2 production rate reached 21.59 mmol L−1 g−1 h−1, five times that of bulk g-C3N4. When applied in a self-Fenton system, it efficiently degraded 2,4-dichlorophenol with a rate constant of 0.070 min−1. Mechanism studies revealed that carbon defects increased carrier separation efficiency, raising the photocurrent density by 300% and the photoluminescence quenching rate by 60%. The in-situ generated H2O2 reacted with Fe2+ to produce ·OH, which was confirmed by ESR, and the system showed over 90% stability in tap water across four cycles, demonstrating practicality for pollutant degradation.
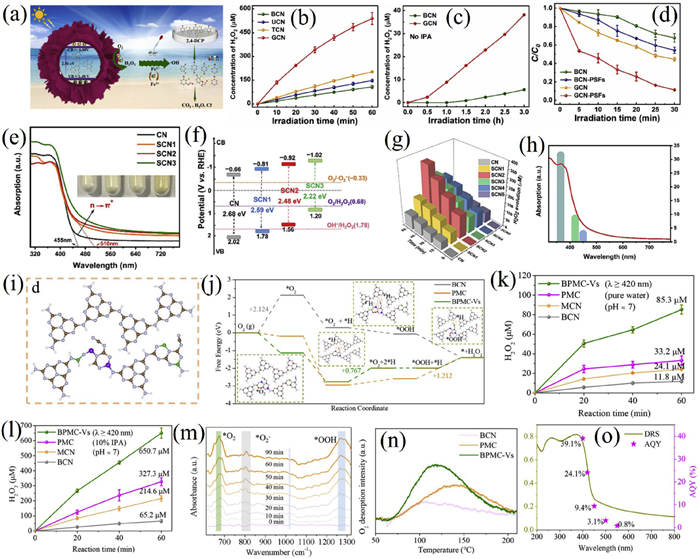
Nitrogen defects also play a critical role in modulating the electronic and catalytic properties of g-C3N4. Li et al. optimized the electronic structure and spin density distribution of carbon nitride (SCNx) via a combined strategy of sulfur doping and edge group modification, enabling efficient photocatalytic H2O2 synthesis without sacrificial agents (Figs. 6e-h) [150]. The material exhibited a high surface area (163 m2/g), a negatively shifted conduction band (-0.92 V), and a narrowed band gap (2.48 eV). Without sacrificial agents, it achieved an H2O2 production rate of 416.7 µmol g−1 h−1 under visible light (λ ≥ 420 nm), 10 times that of pristine g-C3N4, with an apparent quantum yield of 31.6% at 365 nm. In-situ IR and theoretical analyses revealed that edge functionalization enhanced O2 adsorption and reduced the energy barrier of *OOH formation, underscoring the role of spin density localization in promoting reaction kinetics. The H2O2 solution (624.8 µmol/L) generated by this catalyst in pure water could efficiently degrade rhodamine B (5 mg/L, > 95% removal within 5 min) and inactivate Escherichia coli, providing a new insight for the design of spin-regulated photocatalysts.
Zeng et al. constructed a carbon nitride with multiple active sites (BPMC-Vs) via a synergistic strategy of boron-phosphorus dual doping and nitrogen defects, significantly enhancing the efficiency of photocatalytic H2O2 synthesis (Figs. 6i-o) [151]. The cooperation of P/B atoms and defects increased local proton coverage and reduced the *OOH energy barrier from 1.121 eV to 0.767 eV. Under visible light and sacrificial agent-free conditions, the catalyst achieved an H2O2 production rate of 465 µmol g−1 h−1 driven by visible light (λ ≥ 420 nm), with a solar-to-chemical conversion efficiency of 0.33% and a selectivity of 95.2%. Key contributions included enhanced proton adsorption (via P and -C≡N), improved O2 activation (via B and N vacancies), and facilitated charge separation, as confirmed by photoelectrochemical and spectroscopy results.
Zhang et al. developed a dual-defect catalyst (Nv-C≡N-CN) incorporating both nitrogen vacancies and cyano groups (Figs. 7a-g) [152]. This combination resulted in an electron-rich structure with localized charge distribution, significantly enhancing light absorption, charge separation, and H2O2 production, which reached 3093 µmol g−1 h−1 with an apparent quantum yield of 36.2% at 400 nm. The nitrogen vacancies facilitated O2 adsorption and activation, while the cyano groups promoted proton adsorption, collectively improving H2O2 formation kinetics.
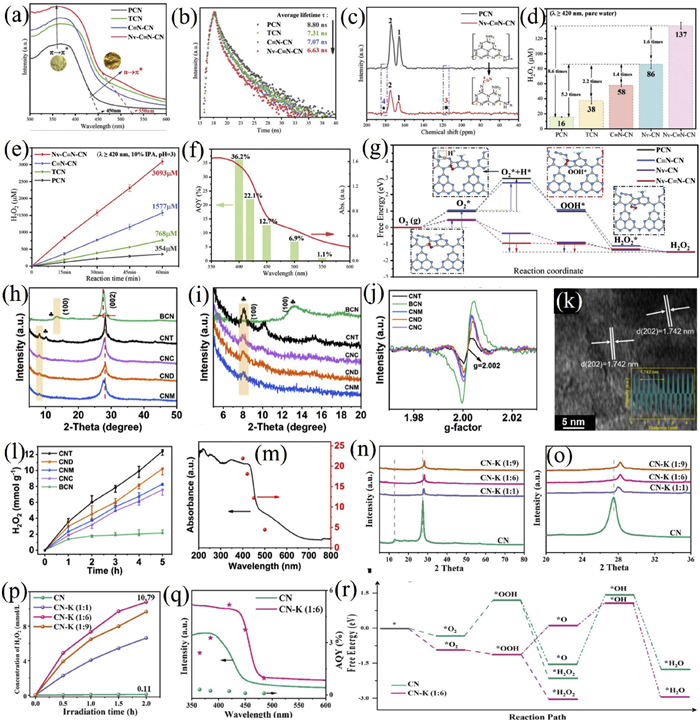
Enhancing bulk crystallinity represents another essential route to improve photocatalytic performance by reducing internal defects and suppressing charge recombination. Highly crystalline materials possess ordered atomic arrangements and fewer trap states, thereby promoting the migration of photogenerated carriers to surface reaction sites.
Chen et al. proposed an anti-defect engineering strategy to synthesize high-crystallinity carbon nitride (CNT) via a molten salt method (Figs. 7h-m) [153]. The material showed well-defined lattice fringes (1.742 nm) and a defect concentration 1.8 times lower than conventional g-C3N4. These structural merits led to a carrier separation rate three times higher, a 76% reduction in radiative recombination, and an extended carrier lifetime of 3.27 ns. The H2O2 production rate attained 2.48 mmol g−1 h−1 with an AQE of 22% at 400 nm 5.8 times higher than that of traditional carbon nitride. Rotating Disk Electrode (RDE) tests confirmed enhanced selectivity for the two-electron oxygen reduction pathway (electron transfer number n = 1.73), and the conduction band potential shifted negatively to -1.23 V (vs. NHE), promoting O2 reduction kinetics. The in-situ generated H2O2 further enabled efficient tetracycline degradation via a Fenton-like process, achieving 84% removal within 3 h.
Zhong et al. synthesized a potassium-doped highly crystalline g-C3N4 photocatalyst (CN-K) using a KI-assisted recrystallization strategy. The as-prepared CN-K exhibited higher interplanar crystallinity, a narrower band gap, and smaller particle size (Figs. 7n-r) [154]. Potassium atoms acted as electron acceptors, shifting the conduction band negatively by 0.35 eV and enhancing the O2 adsorption energy to -0.93 eV. The CN-K (1:6) sample achieved a photocatalytic H2O2 production rate of 7.8 mmol L−1 h−1, 220 times higher than that of traditional carbon nitride.
In summary, defect engineering-whether through introducing beneficial surface defects or suppressing bulk defects-offers powerful strategies for enhancing charge behavior and surface reactions, providing a robust foundation for designing highly efficient photocatalysts.
Heterojunctions boost catalytic activity through a built-in electric field that separates charges efficiently, while band bending guides charge transfer and blocks recombination [155-160]. New interfacial active sites lower reaction barriers, and tailored electronic structures optimize reactant adsorption, collectively enhancing performance [161-166]. In photocatalytic filed the catalyst composed of two or more semiconductors with tailored band alignments will enhance photocatalytic activity primarily through the formed built-in electric field at the interface, which can significantly suppress non-productive charge recombination [105,167-172]. The combining of a narrow-bandgap semiconductor enables more efficient utilization of solar energy compared to single semiconductors. Finally, the separated electrons and holes retain sufficient reducing and oxidizing capabilities respectively, meeting the energy requirements for target reactions that a single catalyst might fail to achieve. These combined effects collectively boost the overall photocatalytic performance.
Li et al. successfully constructed a Bi4Ti3O12/g-C3N4 (BTOCN) inorganic/organic S-scheme heterojunction photocatalyst via an electrospinning-assisted ultrasonic assembly strategy; this composite system formed a tightly contacted S-scheme heterojunction through interface engineering, which significantly optimized carrier kinetic behavior, prolonged carrier lifetime, and notably enhanced the generation of ·O2⁻ active intermediates and the two-electron oxygen reduction pathway (Figs. 8a-e) [173]. Under simulated sunlight irradiation, the optimized BTOCN2 sample achieved an H2O2 production rate of 260 µmol g−1 h−1 in pure water, 2.8 times higher than that of single g-C3N4, with an apparent quantum yield of 0.36% at 420 nm, and retained 93% of its activity after 10 cycles.
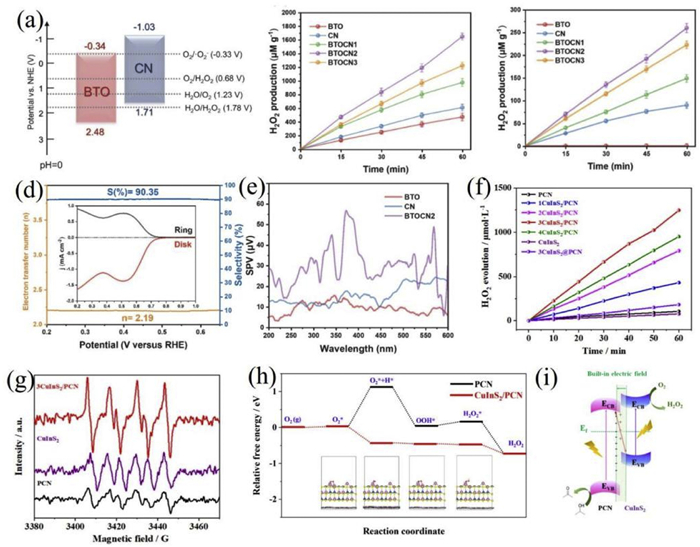
In another work, Zhang et al. in-situ grew copper indium sulfide (CuInS2) nanosheets on the surface of polymeric carbon nitride (PCN) via a low-temperature hydrothermal method, successfully constructing a CuInS2/PCN photocatalyst with an S-scheme heterojunction structure (Figs. 8f-i) [174]. The well-dispersed CuInS2 and tight interfacial contact induced electron transfer from CuInS2 to PCN due to their Fermi level difference, forming a built-in electric field. Under visible light, recombination between PCN electrons and CuInS2 holes preserved electrons with high reducing ability, contributing to an H2O2 production rate of 1247.6 µmol g−1 h−1 in 5% isopropanol (AQY = 16.0% at 420 nm), which is 11.6 times and 16.0 times that of individual PCN and CuInS2, respectively.
Homojunctions, composed of the same material with varying phases or morphologies, offer superior lattice matching and lower charge migration barriers than heterojunctions, showing great potential in facilitating charge separation for photocatalytic applications [175-178]. For instance, in a study by Chen et al., a carbon nitride homojunction (homo-CN) was constructed via a hydroxyl-induced strategy (Figs. 9a-d) [179]. By in-situ growing a hydroxyl-rich carbon nitride (HR-CN) nanolayer on hydroxyl-defective carbon nitride (HD-CN) nanosheets, the synergistic optimization of bulk charge separation and surface reverse reactions was achieved. This design adjusted the band structure by regulating hydroxyl concentration, creating a gradient driving force to facilitate electron-hole separation and extending the light absorption edge. Surface hydroxyl groups provided additional electron-accepting bands, enabling efficient extraction of electrons from the bulk to the surface; meanwhile, they enhanced the adsorption energy of *OOH intermediates and increased the overpotential of the 4-electron oxygen reduction reaction, making the 2e− ORR kinetically more favorable and thus inhibiting H2O2 decomposition. Performance tests showed that the H2O2 production rate of this homojunction was 4.5 times that of pristine carbon nitride, with an apparent quantum yield of 2.1% at 500 nm in pure water.
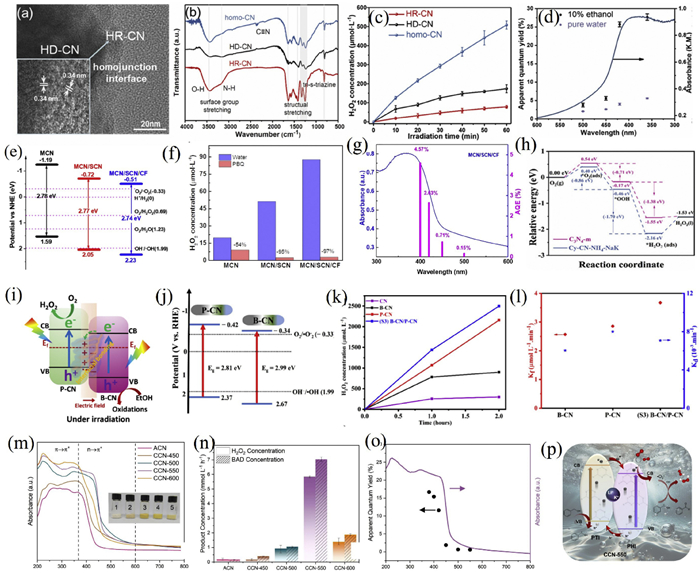
Zhou and his collaborators constructed a novel dual-channel carbon nitride homojunction composite catalyst by incorporating cellulose-derived carbon nanofibers (CF) into the homojunction formed by carbon nitride nanotubes (MCN) and carbon nitride nanosheets (SCN) (Figs. 9e-g) [180]. The MCN/SCN/CF composite exhibited a dual-channel H2O2 synthesis mechanism: ORR on the conduction band and WOR on the valence band. The unique structure, coupled with improved light absorption and charge separation, resulted in an H2O2 generation rate of 136.9 µmol L−1 h−1 without sacrificial agents, with an AQY of 4.57% and solar-to-chemical efficiency of 0.57%, which was 5 times that of MCN alone.
Zhang’s team first fragmented bulk g-C3N4 into smaller pieces (CN-NH4) using a soft template strategy, then subjected them to molten salt treatment for directional splicing, resulting in a homojunction (CN-NH4-NaK) with multiple ordered-disordered interfaces (Fig. 9h) [181]. The material featured coexisting crystalline and amorphous regions, abundant cyano groups, and a built-in electric field that enhanced charge separation. It achieved an exceptional H2O2 production rate of 16675 µmol g−1 h−1 and over 91% selectivity across a wide pH range, which is 158 times that of pristine g-C3N4. The crystalline domains facilitated H2O2 production via a two-step single-electron ORR, while the amorphous regions utilized in-situ oxygen from water oxidation through a direct two-electron pathway.
In Khan et al.’s study, an S-scheme homojunction (B-CN/P-CN) based on boron-doped carbon nitride (B-CN) and phosphorus-doped carbon nitride (P-CN) was constructed for efficient photocatalytic H2O2 synthesis (Figs. 9i-l) [182]. The work function difference [183] drove interfacial charge transfer, forming a built-in electric field, while B-P bonds acted as charge migration bridges. The optimal composite showed an H2O2 production rate of 2199.5 µmol L−1 h−1 under 365 nm irradiation, outperforming pristine CN by 2-8 times.
Foo et al. synthesized a heptazine/triazine-based crystalline homojunction (CCN-X) via one-step ionothermal polymerization (Figs. 9m-p) [184]. KCl and LiCl promoted the formation of cyano/hydroxy groups and triazine units, respectively, while K+ and Li+ intercalation enhanced interlayer charge transport. The catalyst simultaneously produced H2O2 and benzaldehyde with high yields (5838.91 and 7041.32 µmol L−1 h−1, respectively), 100% selectivity, and an AQY of 11.57% at 420 nm.
In photocatalytic H2O2 synthesis, the efficient formation of H2O2 relies critically on the activation of O2 or H2O molecules through well-designed surface sites that lower reaction energy barriers and steer selectivity toward the desired two-electron pathway [185,186]. Beyond intrinsic strategies such as doping and defect engineering, which introduce electron-rich or electron-deficient centers, recent efforts have focused on constructing extrinsic active sites via anchored clusters or single atoms [187-189]. These approaches preserve the structural integrity of carbon nitride while improving bulk charge migration and interfacial reactivity through synergistic effects.
For instance, Hou et al. constructed a photocatalyst (CoOx-NvCN) co-modified with surface nitrogen vacancies and CoOx nanoclusters on polymeric carbon nitride (CN) via a two-step method to enhance the performance of 2e− ORR for H2O2 production (Figs. 10a-d) [190]. The nitrogen vacancies optimized the electronic structure to facilitate charge separation, while the CoOx nanoclusters served as high-activity sites that enhanced O2 adsorption and reduced the energy barrier for *OOH formation. This dual modification resulted in a catalyst capable of producing H2O2 at a rate of 244.8 µmol L−1 h−1 in pure water without sacrificial agents, with an apparent quantum yield (AQY) of 5.73% at 420 nm and a solar-to-chemical conversion (SCC) efficiency of 0.47%, ranking among the top-performing carbon nitride-based systems.
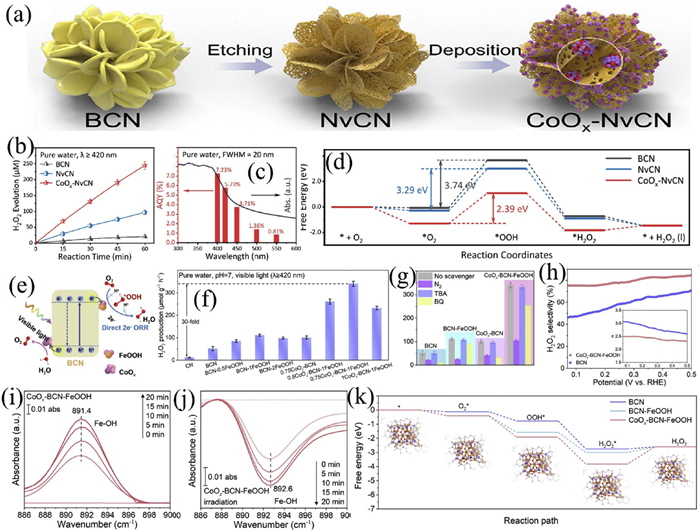
In another study, Jing’s team designed a boron-doped carbon nitride (BCN) photocatalyst (CoOx-BCN-FeOOH) modified with dual active sites: Coordinatively unsaturated FeOOH and CoOx clusters (Figs. 10e-k) [191]. The boron doping optimized the band structure, while the CoOx clusters promoted water oxidation and extended charge lifetime, and the unsaturated FeOOH clusters facilitated electron acceptance and O2 activation. The catalyst achieved an H2O2 production rate of 0.34 mmol g−1 h−1 (30 times that of pristine g-C3N4, an SCC efficiency of 0.75% and an AQY of 8.36% at 420 nm). Notably, the FeOOH sites enabled Pauling-type O2 adsorption, stabilized peroxide intermediates, and increased selectivity for the 2e− ORR pathway to 85%.
Due to its distinctive electronic structure, responsiveness to visible light, and metal-free composition, g-C3N4 holds significant promise in the photocatalytic synthesis of H2O2. Comprehensive investigations into oxygen reduction pathways-including the two-electron 2e− ORR, ·O2− mediated pathway, and 1O2-mediated pathway-have elucidated that the formation and stability of critical intermediates such as ·O2−, *OOH, and 1O2 are pivotal determinants of H2O2 selectivity. The photocatalytic performance of g-C3N4 is collectively influenced by its band structure, surface proton coverage, and the nature of its defects.
Advancements in the mechanistic understanding have facilitated the implementation of various modification strategies, including defect engineering, elemental doping, construction of heterojunctions and homojunctions, and surface-active site modification. These approaches effectively tailor the band structure, enhance light absorption, promote charge carrier separation, optimize oxygen adsorption and activation, stabilize reactive intermediates, suppress H2O2 decomposition, improve charge separation efficiency and mass transfer, and reinforce interfacial reaction kinetics and pathway selectivity.
The synergistic integration of these modifications has substantially improved the light-harvesting capability, charge separation efficiency, and surface reaction kinetics of g-C3N4, resulting in a marked enhancement of its H2O2 production rate under visible light irradiation. Certain systems have demonstrated remarkable stability and applicability across diverse conditions, including pure water, broad pH ranges, and seawater. These advancements have elevated H2O2 production rates from trace amounts reported in early studies to the millimole per gram per hour scale, with AQY surpassing 30%, thereby underscoring the potential for transitioning from laboratory-scale research to practical applications.
The band gap of g-C3N4, approximately 2.7 eV, constrains its capacity to fully harness visible light, particularly limiting its ability to absorb long-wavelength photons. This limitation impedes further enhancement of solar energy conversion efficiency. Although strategies such as doping, defect engineering, and heterojunction construction have yielded improvements, significant challenges persist due to pronounced electron-hole recombination within the bulk material and at interfaces. Notably, the efficiency of hole utilization in systems devoid of sacrificial agents requires urgent enhancement. Additionally, the competition among multiple reaction pathways-such as 4e− ORR and H2O2 decomposition-remains difficult to suppress entirely. The in-situ observation of the dynamic behavior of critical intermediates is also challenging, complicating the rational design of catalysts with high selectivity. Furthermore, most current modification approaches depend on complex precursor designs, high-temperature polymerization processes, and template-assisted synthesis, which collectively hinder the scalable production of g-C3N4 materials with uniform structure and stable performance. Moreover, the majority of investigations have been conducted under controlled laboratory conditions, with limited systematic evaluation of the effects of real-world factors such as impurities in natural water sources, pH variability, and fluctuating light conditions on catalytic performance.
Future progress in g-C3N4-based photocatalytic hydrogen peroxide synthesis will necessitate interdisciplinary collaboration and systematic design strategies, aiming for synergistic advancements across three domains: Material development, mechanistic understanding, and device engineering. At the material level, integrating theoretical modeling with in situ dynamic characterization techniques is essential to rationally engineer surface structures that exhibit narrowed band gaps, enhanced charge separation, and multi-site synergistic effects. Such design will facilitate precise control over oxygen adsorption, proton transfer, and H2O2 desorption processes. Regarding light energy utilization, the development of advanced composite systems is imperative to extend light absorption capabilities into the visible and near-infrared spectra. From the perspectives of synthesis and application, it is critical to establish green, low-temperature, and scalable fabrication methods.
In addition, increasing efforts in computational analysis and materials informatics provide useful guidance for the rational design and scalable preparation of g-C3N4-based photocatalysts for hydrogen peroxide production. By systematically examining the relationships among precursor chemistry, polymerization conditions, structural modification strategies, and resulting material properties, it becomes possible to identify key compositional and structural features that are beneficial for efficient H2O2 generation. This perspective is particularly relevant to g-C3N4 systems, where small variations in synthesis parameters, calcination temperature, atmosphere, or post-treatment methods, can lead to pronounced differences in crystallinity, defect distribution, surface chemistry, and electronic structure. A clearer understanding of these structure-property relationships may help guide targeted material optimization and reduce excessive empirical trial-and-error. With further integration of computational analysis and experimental synthesis, g-C3N4 photocatalysts with more controllable structures, improved reproducibility, and better scalability are expected to be developed, which will be beneficial for advancing photocatalytic hydrogen peroxide production toward practical applications. Concurrently, efforts should focus on comprehensive validation and long-term stability assessments of photocatalysis-Fenton coupling systems, flow reactors, and performance under actual water body conditions. Ultimately, these endeavors aim to transition this technology from laboratory-scale research to large-scale, sustainable production and environmental remediation applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zhimin Yuan: Writing – original draft. Xingling Zhao: Supervision. Xianglin Zhu: Formal analysis. Kaili Wang: Data curation. Ya-Qian Lan: Conceptualization. Zaiyong Jiang: Writing – review & editing.
This work was financially supported by the National Natural Science Foundation of China (Nos. 22572148, 22502145, 22402151), the Natural Science Foundation of Shandong Province, China (No. ZR2023MB049).
X. Zhang, Y. Wan, Y. Wen, et al., Nat. Catal. 8 (2025) 465–475. doi: 10.1038/s41929-025-01335-4
Z. Tian, C. Han, Y. Zhao, et al., Nat. Commun. 12 (2021) 2039. doi: 10.1038/s41467-021-22394-8
Z. Yuan, H. Miao, Z. Jiang, et al., Mol. Catal. 577 (2025) 114962.
C. Luo, H. Xi, Y.Q. Feng, et al., Energy Convers. Manage. 267 (2022) 115914. doi: 10.1016/j.enconman.2022.115914
C. Zhou, Y. Song, Z. Wang, et al., J. Environ. Chem. Eng. 11 (2023) 110138. doi: 10.1016/j.jece.2023.110138
H. Jiang, L. Wang, X. Yu, et al., Chem. Eng. J. 466 (2023) 143129. doi: 10.1016/j.cej.2023.143129
S. Huo, D. Necas, F. Zhu, et al., Chem. Eng. J. 406 (2021) 127160. doi: 10.1016/j.cej.2020.127160
L. Liu, Z. Zhang, J. Wang, et al., Energy 168 (2019) 946–952. doi: 10.1016/j.energy.2018.11.132
S. Tong, D. Chen, P. Mao, et al., J. Hazard. Mater. 424 (2022) 127622. doi: 10.1016/j.jhazmat.2021.127622
X. Li, Y. Wan, F. Deng, et al., Chin. Chem. Lett. 36 (2025) 111418. doi: 10.1016/j.cclet.2025.111418
Z. Cao, T. Zhou, X. Ma, et al., ACS Sustain. Chem. Eng. 8 (2020) 11007–11015.
J. Liu, M. Zhou, K. Jin, et al., Battery Energy 3 (2024) 20230049. doi: 10.1002/bte2.20230049
Y. Shiraishi, Y. Ueda, A. Soramoto, et al., Nat. Commun. 11 (2020) 3386. doi: 10.1038/s41467-020-17216-2
T. Gu, J. Ge, A. Li, et al., Int. J. Hydrogen Energy 49 (2024) 764–774. doi: 10.1016/j.ijhydene.2023.07.187
Z. Liu, X. Liu, L. Tang, et al., Chin. Chem. Lett. 37 (2026) 112024. doi: 10.1016/j.cclet.2025.112024
C. Wang, X. Xie, F. Qiu, et al., Chin. Chem. Lett. 37 (2026) 111604. doi: 10.1016/j.cclet.2025.111604
Z. Liu, L. Wang, P. Liu, et al., Food Chem. 357 (2021) 129753. doi: 10.1016/j.foodchem.2021.129753
X. Sun, J. Yang, X. Zeng, et al., Angew. Chem. Int. Ed. 63 (2024) e202414417. doi: 10.1002/anie.202414417
S.M. Chen, L.K. Chen, N. Tian, et al., ACS Catal. 14 (2024) 16664–16672. doi: 10.1021/acscatal.4c04779
K. Wang, M. Wang, Q. Lei, et al., Mol. Catal. 580 (2025) 115121.
Y. Chen, C. Zhen, Y. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202407163. doi: 10.1002/anie.202407163
L. Lin, Z. Sun, H. Chen, et al., Acta Phys. Chim. Sin. 40 (2024) 2305019. doi: 10.3866/PKU.WHXB202305019
N. Bhuvanendran, S. Ravichandran, Q. Xu, et al., Int. J. Hydrogen Energy 47 (2022) 7113–7138. doi: 10.1016/j.ijhydene.2021.12.072
M. Dan, R. Zhong, S. Hu, et al., Chem. Catal. 2 (2022) 1919–1960.
S. Gao, H. Yang, D. Rao, et al., Chem. Eng. J. 433 (2022) 134460. doi: 10.1016/j.cej.2021.134460
W. Li, N. Bhuvanendran, W. Zhang, et al., Int. J. Hydrogen Energy 47 (2022) 24807–24816. doi: 10.1016/j.ijhydene.2022.05.227
Z. Liu, Y. Bian, G. Dawson, et al., Chin. Chem. Lett. 36 (2025) 111272. doi: 10.1016/j.cclet.2025.111272
K. Liu, Z. Wang, Y. Xie, et al., Chin. Chem. Lett. (2025), doi: 10.1016/j.cclet.2025.111701.
Y. An, T. Cai, W. Jiang, et al., Green Chem. 27 (2025) 10478–10509. doi: 10.1039/d5gc02617b
T. Liu, Z. Pan, J.J.M. Vequizo, et al., Nat. Commun. 13 (2022) 1034. doi: 10.1038/s41467-022-28686-x
Y. Ding, S. Maitra, S. Halder, et al., Matter 5 (2022) 2119–2167. doi: 10.1016/j.matt.2022.05.011
W. Wang, L. Wang, L. Sun, et al., Chem. Eng. J. 477 (2023) 146945. doi: 10.1016/j.cej.2023.146945
H. Zong, G. Zeng, H. Miao, et al., Appl. Surf. Sci. 649 (2024) 158964. doi: 10.1016/j.apsusc.2023.158964
Z. Yuan, B. Zhang, X. Zhu, et al., Adv. Funct. Mater. 35 (2025) 2503531. doi: 10.1002/adfm.202503531
G. Zeng, H. Miao, J. Wu, et al., Chem. Eng. J. 499 (2024) 156367. doi: 10.1016/j.cej.2024.156367
H. Miao, G. Zeng, J. Wu, et al., Int. J. Hydrogen Energy 102 (2025) 963–971. doi: 10.1016/j.ijhydene.2025.01.145
X. Qiu, X. Wang, X. Liu, et al., Chemistry 30 (2024) e202400348. doi: 10.1002/chem.202400348
X. Zhu, H. Zong, C.J.V. Pérez, et al., Angew. Chem. Int. Ed. 62 (2023) e202218694. doi: 10.1002/anie.202218694
S. Shi, H. Zong, Z. Yuan, et al., Inorg. Chem. Front. 12 (2025) 2524–2536. doi: 10.1039/d4qi02990a
X. Zhu, E. Zhou, X. Tai, et al., Angew. Chem. Int. Ed. 63 (2025) e202425439.
H. Miao, J. Wu, X. Luo, et al., Inorg. Chem. 64 (2025) 10290–10301. doi: 10.1021/acs.inorgchem.5c01250
Y. Xu, M.M. Hassan, S. Ali, et al., J. Agric. Food. Chem. 69 (2021) 1667–1674. doi: 10.1021/acs.jafc.0c06513
H. Zhang, J. Liu, Y. Zhang, et al., J. Mater. Sci. Technol. 166 (2023) 241–249. doi: 10.1016/j.jmst.2023.05.030
F. Liu, P. Zhou, Y. Hou, et al., Nat. Commun. 14 (2023) 4344. doi: 10.1038/s41467-023-40007-4
X. Zhang, X. Zhao, P. Zhu, et al., Nat. Commun. 13 (2022) 2880. doi: 10.1038/s41467-022-30337-0
L.J. Li, L.P. Xu, Z.F. Hu, et al., Adv. Funct. Mater. 31 (2021) 2106120. doi: 10.1002/adfm.202106120
A.T. Murray, S. Voskian, M. Schreier, et al., Joule 3 (2019) 2942–2954. doi: 10.1016/j.joule.2019.09.01923.10.001
Y.Y. Qian, Y.L. Han, X.Y. Zhang, et al., Nat. Commun. 14 (2023) 3083. doi: 10.1038/s41467-023-38884-w
Y. Shiraishi, Y. Kofuji, H. Sakamoto, et al., ACS Catal. 5 (2015) 3058–3066. doi: 10.1021/acscatal.5b00408
Y. Shiraishi, S. Kanazawa, Y. Sugano, et al., ACS Catal. 4 (2014) 774–780. doi: 10.1021/cs401208c
J. Ma, X. Peng, Z. Zhou, et al., Angew. Chem. Int. Ed. 61 (2022) e202210856. doi: 10.1002/anie.202210856
X. Hu, Z. Sun, G. Mei, et al., Adv. Energy Mater. 12 (2022) 2201466. doi: 10.1002/aenm.202201466
Q.B. Liao, Q.N. Sun, H.C. Xu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310556. doi: 10.1002/anie.202310556
J. Liu, L. Huang, Y. Li, et al., J. Environ. Chem. Eng. 10 (2022) 106668. doi: 10.1016/j.jece.2021.106668
T. Tang, Z. Yin, J. Chen, et al., Chem. Eng. J. 417 (2021) 128058. doi: 10.1016/j.cej.2020.128058
X.X. Zhang, Y.G. Xiao, S.S. Cao, et al., J. Cleaner Prod. 352 (2022) 131560. doi: 10.1016/j.jclepro.2022.131560
F. Qiao, W. Liu, J. Yang, et al., Int. J. Hydrogen Energy 53 (2024) 840–847. doi: 10.1016/j.ijhydene.2023.11.292
Y. Wei, X. Li, Y. Zhang, et al., Renew. Energy 179 (2021) 756–765. doi: 10.1016/j.renene.2021.07.091
Z. Yuan, X. Zhu, Q. Gao, et al., Molecules 28 (2023) 4057. doi: 10.3390/molecules28104057
X. Pan, F. Kong, M. Xing, Res. Chem. Intermed. 48 (2022) 2837–2855. doi: 10.1007/s11164-022-04748-z
X. Gao, L. Cao, Y. Chang, et al., ACS Sustain. Chem. Eng. 11 (2023) 5597–5607. doi: 10.1021/acssuschemeng.2c07626
Z. Du, Z. Zhou, N. Sun, et al., Chin. Chem. Lett. 37 (2026) 111341. doi: 10.1016/j.cclet.2025.111341
H. Zhang, Q. Su, ACS Omega 10 (2025) 8793–8815. doi: 10.1021/acsomega.4c09487
X. Yan, B. Wang, J. Zhao, et al., Chem. Eng. J. 452 (2023) 139271. doi: 10.1016/j.cej.2022.139271
C. Li, H. Che, Y. Yan, et al., Chem. Eng. J. 398 (2020) 125523. doi: 10.1016/j.cej.2020.125523
S. Yin, L. Sun, Y. Zhou, et al., Chem. Eng. J. 406 (2021) 126776. doi: 10.1016/j.cej.2020.126776
R. Yu, B. Luo, M. Chen, et al., Int. J. Hydrogen Energy 48 (2023) 24285–24294. doi: 10.1016/j.ijhydene.2023.03.167
C. Zhang, M. Tan, X. Lu, et al., Phys. Chem. Chem. Phys. 25 (2023) 24960–24967. doi: 10.1039/d3cp02615a
S. Chen, Y. Qi, T. Hisatomi, et al., Angew. Chem. Int. Ed. 54 (2015) 8498–8501. doi: 10.1002/anie.201502686
C. Li, G. Ding, X. Liu, et al., Chem. Eng. J. 435 (2022) 134740. doi: 10.1016/j.cej.2022.134740
J. Rong, G. Zhu, W.R. Osterloh, et al., Chem. Eng. J. 412 (2021) 127556. doi: 10.1016/j.cej.2020.127556
Z. Xia, R. Yu, H. Yang, et al., Int. J. Hydrogen Energy 47 (2022) 13340–13350. doi: 10.1016/j.ijhydene.2022.02.087
C. Li, X. Liu, Y. Yan, et al., Chem. Eng. J. 410 (2021) 128316. doi: 10.1016/j.cej.2020.128316
L. Yu, W. Fan, N. He, et al., Int. J. Hydrogen Energy 46 (2021) 17741–17750. doi: 10.1016/j.ijhydene.2021.02.194
Y.X. He, J.S. Zhao, Y.T. Sham, et al., ACS Sustain. Chem. Eng. 11 (2023) 17552–17563. doi: 10.1021/acssuschemeng.3c06421
H. Guo, S. Wang, X. Chen, et al., Nat. Synth. 4 (2025) 1610–1620. doi: 10.1038/s44160-025-00880-x
R. Liu, Y. Chen, H. Yu, et al., Nat. Catal. 7 (2024) 195–206. doi: 10.1038/s41929-023-01102-3
X. Liu, X. Yang, X. Ding, et al., Chin. Chem. Lett. 34 (2023) 108148. doi: 10.1016/j.cclet.2023.108148
X. Pan, J. Ji, N. Zhang, et al., Chin. Chem. Lett. 31 (2020) 1462–1473. doi: 10.1016/j.cclet.2019.10.002
Z. Yuan, X. Zhu, X. Gao, et al., Environ. Sci. Ecotechnol. 20 (2024) 100368. doi: 10.1016/j.ese.2023.100368
H. Wang, H. Jiang, P. Huo, et al., J. Environ. Chem. Eng. 10 (2022) 106908. doi: 10.1016/j.jece.2021.106908
T. Wang, X. Liu, Q. Men, et al., Renew. Energy 147 (2020) 856–863. doi: 10.1016/j.renene.2019.09.025
L. Wang, G. Tang, S. Liu, et al., Chem. Eng. J. 428 (2022) 131338. doi: 10.1016/j.cej.2021.131338
D. Tsukamoto, A. Shiro, Y. Shiraishi, et al., ACS Catal. 2 (2012) 599–603. doi: 10.1021/cs2006873
P. Karuppasamy, N.R.N. Nisha, A. Pugazhendhi, et al., J. Environ. Chem. Eng. 9 (2021) 105254. doi: 10.1016/j.jece.2021.105254
C. Jin, S. Rao, J. Xie, et al., Chem. Eng. J. 447 (2022) 137369. doi: 10.1016/j.cej.2022.137369
W. Wang, G. Zhang, Q. Wang, et al., Chin. Chem. Lett. 35 (2024) 109193. doi: 10.1016/j.cclet.2023.109193
Y. Qin, H. Li, J. Lu, et al., Chem. Eng. J. 384 (2020) 123275. doi: 10.1016/j.cej.2019.123275
J. Tao, M. Wang, X. Zhang, et al., Fuel 338 (2023) 127259. doi: 10.1016/j.fuel.2022.127259
S. Wang, J. Zhang, B. Li, et al., J. Environ. Chem. Eng. 10 (2022) 107438. doi: 10.1016/j.jece.2022.107438
J.Y. Yue, Z.X. Pan, P. Yang, et al., ACS Mater. Lett. 6 (2024) 3932–3940. doi: 10.1021/acsmaterialslett.4c01273
Y.Y. Tang, W.M. Wang, J.Q. Ran, et al., Energy Environ. Sci. 17 (2024) 6482–6498. doi: 10.1039/D4EE02505A
C.B. Wu, Z.Y. Teng, C. Yang, et al., Adv. Mater. 34 (2022) 2110266. doi: 10.1002/adma.202110266
Z. Lang, X. Wang, S. Jabeen, et al., Adv. Mater. 37 (2025) e2418942. doi: 10.1002/adma.202418942
W. Wang, L. Wang, L. Sun, et al., Chem. Eng. J. 477 (2023) 146945. doi: 10.1016/j.cej.2023.146945
S. Wu, H.T. Yu, S. Chen, et al., ACS Catal. 10 (2020) 14380–14389. doi: 10.1021/acscatal.0c03359
L. Wang, Y. Hu, J. Xu, et al., Int. J. Hydrogen Energy 48 (2023) 16987–16999. doi: 10.1016/j.ijhydene.2023.01.172
C.C. Qin, X.D. Wu, L. Tang, et al., Nat. Commun. 14 (2023) 5238. doi: 10.1038/s41467-023-40991-7
R. Justinabraham, A. Durairaj, S. Ramanathan, et al., J. Water Process Eng. 44 (2021) 102422. doi: 10.1016/j.jwpe.2021.102422
N. Su, S. Cheng, P. Zhang, et al., Int. J. Hydrogen Energy 47 (2022) 41010–41020. doi: 10.1016/j.ijhydene.2022.09.188
J. Ye, D. Yang, J. Dai, et al., Chem. Eng. J. 431 (2022) 133972. doi: 10.1016/j.cej.2021.133972
W. Chu, S. Li, H. Zhou, et al., J. Environ. Chem. Eng. 10 (2022) 108623. doi: 10.1016/j.jece.2022.108623
Y. Wang, J. Xiong, L. Xin, et al., Chin. Chem. Lett. 36 (2025) 110003. doi: 10.1016/j.cclet.2024.110003
E. Cui, Y. Lu, Z. Li, et al., Chin. Chem. Lett. 36 (2025) 110288. doi: 10.1016/j.cclet.2024.110288
Y. Dai, W. Peng, Y. Ji, et al., J. Food Sci. 89 (2024) 8022–8035. doi: 10.1111/1750-3841.17398
L. Jing, D. Wang, M. He, et al., J. Hazard. Mater. 401 (2021) 123309. doi: 10.1016/j.jhazmat.2020.123309
Q. Wang, G. Zhang, W. Xing, et al., Angew. Chem. Int. Ed. 62 (2023) e202307930. doi: 10.1002/anie.202307930
M. Liu, G. Zhang, X. Liang, et al., Angew. Chem. Int. Ed. 62 (2023) e202304694. doi: 10.1002/anie.202304694
M. Xu, X. Zhao, H. Jiang, et al., J. Environ. Chem. Eng. 9 (2021) 106469. doi: 10.1016/j.jece.2021.106469
C. Feng, X. Ouyang, Y. Deng, et al., J. Hazard. Mater. 441 (2023) 129845. doi: 10.1016/j.jhazmat.2022.129845
W. Chen, Z. -C. He, G. -B. Huang, et al., Chem. Eng. J. 359 (2019) 244–253. doi: 10.1016/j.cej.2018.11.141
Y. Wang, Y. Tian, L. Yan, et al., J. Phys. Chem. C 122 (2018) 7712–7719. doi: 10.1021/acs.jpcc.8b00098
X.J. Lu, C.Z. Yuan, S. Chen, et al., Langmuir 40 (2024) 11067–11077. doi: 10.1021/acs.langmuir.4c00605
Y. Zhang, J.Y. Qiu, B.C. Zhu, et al., Chem. Eng. J. 444 (2022) 136584. doi: 10.1016/j.cej.2022.136584
X.D. Zhang, J.G. Yu, W. Macyk, et al., Adv. Sustain. Syst. 7 (2023) 2200113. doi: 10.1002/adsu.202200113
Y. Xue, Y.T. Wang, Z.H. Pan, et al., Angew. Chem. Inter. Ed. 60 (2021) 10469–10480. doi: 10.1002/anie.202011215
Y. Yang, J.J. Liu, M.L. Gu, et al., Appl. Catal. B: Environ. 333 (2023) 122780. doi: 10.1016/j.apcatb.2023.122780
S. Zhao, T. Chen, H. Li, et al., Chem. Eng. J. 472 (2023) 145115. doi: 10.1016/j.cej.2023.145115
Z. Sun, W. Wang, Q. Chen, et al., J. Mater. Chem. A 8 (2020) 3160–3167. doi: 10.1039/c9ta13012h
J. Lu, Y. Zeng, X. Ma, et al., Polymers 11 (2019) 828. doi: 10.3390/polym11050828
J. Chen, X. Zhu, Z. Jiang, et al., Inorg. Chem. Front. 9 (2022) 103–110. doi: 10.1039/D1QI01137E
H. Che, G. Che, P. Zhou, et al., Chem. Eng. J. 382 (2020) 122870. doi: 10.1016/j.cej.2019.122870
P. Sun, Z. Mo, J. Zhang, et al., Chem. Eng. J. 478 (2023) 147337. doi: 10.1016/j.cej.2023.147337
J. Wei, Y. Zhang, W. Peng, et al., J. Food Compos. Anal. 144 (2025) 107675. doi: 10.1016/j.jfca.2025.107675
E. Han, T. Gao, T. Wang, et al., J. Food Compos. Anal. 143 (2025) 107597. doi: 10.1016/j.jfca.2025.107597
B. Wang, L. Yang, F. Yuan, et al., Electrochim. Acta 439 (2023) 141681. doi: 10.1016/j.electacta.2022.141681
J. Qi, Q. Li, M. Huang, et al., Colloid. Surf. A 683 (2024) 132998. doi: 10.1016/j.colsurfa.2023.132998
J. Wu, C. Li, H. Dong, et al., Renew. Energy 157 (2020) 660–669. doi: 10.1016/j.renene.2020.04.086
S. Shi, H. Zong, Z. Yuan, et al., Inorg. Chem. Front. 12 (2025) 2524–2536. doi: 10.1039/d4qi02990a
J. Yang, Z. Ji, S. Zhang, ACS Appl. Energy Mater. 6 (2023) 3401–3412. doi: 10.1021/acsaem.2c04124
Z. Zhang, P. Luo, L. Gan, et al., Appl. Surf. Sci. 649 (2024) 159118. doi: 10.1016/j.apsusc.2023.159118
Y. Xu, G. Wang, W. Li, et al., Chem. Eng. J. 503 (2025) 158655. doi: 10.1016/j.cej.2024.158655
Z. Wei, M. Liu, Z. Zhang, et al., Energy Environ. Sci. 11 (2018) 2581–2589. doi: 10.1039/c8ee01316k
G. Yu, K. Gong, C. Xing, et al., Chem. Eng. J. 461 (2023) 142140. doi: 10.1016/j.cej.2023.142140
L. Ma, Y. Gao, B. Wei, et al., ACS Catal. 14 (2024) 2775–2786. doi: 10.1021/acscatal.3c05360
H. Zhang, Y. Zhu, Y. Sun, et al., J. Environ. Chem. Eng. 11 (2023) 111122. doi: 10.1016/j.jece.2023.111122
Y. Liu, Y. Zheng, W. Zhang, et al., J. Mater. Sci. Technol. 95 (2021) 127–135. doi: 10.1016/j.jmst.2021.03.025
F.N. Habarugira, D. Yao, W. Miao, et al., Chin. Chem. Lett. 35 (2024) 109886. doi: 10.1016/j.cclet.2024.109886
S. Wu, H. Yu, S. Chen, et al., ACS Catal. 10 (2020) 14380–14389. doi: 10.1021/acscatal.0c03359
L. Chen, C. Chen, Z. Yang, et al., Adv. Funct. Mater. 31 (2021) 2105731. doi: 10.1002/adfm.202105731
C. Chu, Q. Zhu, Z. Pan, et al., Proc. Natl. Acad. Sci. U. S. A. 117 (2020) 6376–6382. doi: 10.1073/pnas.1913403117
Y.B. Li, F.X. Xiao, Chem. Sci. 16 (2025) 2661–2672. doi: 10.1039/d4sc06256f
X. Zhang, H. Su, P. Cui, et al., Nat. Commun. 14 (2023) 7115. doi: 10.1038/s41467-023-42887-y
Z. Teng, Q. Zhang, H. Yang, et al., Nat. Catal. 4 (2021) 374–384. doi: 10.1038/s41929-021-00605-1
P. Zhang, Y. Tong, Y. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 16209–16217. doi: 10.1002/anie.202006747
Y. Wang, M. Wang, X. Zhang, et al., J. Catal. 440 (2024) 115807. doi: 10.1016/j.jcat.2024.115807
X. Zhu, J. Yang, X. Zhu, et al., Chem. Eng. J. 422 (2021) 129888. doi: 10.1016/j.cej.2021.129888
T. Zhou, Z. Cao, X. Tai, et al., Polymers 14 (2022) 1510. doi: 10.3390/polym14081510
Y. Wu, J. Chen, H. Che, et al., Appl. Catal. B: Environ. 307 (2022) 121185. doi: 10.1016/j.apcatb.2022.121185
Y. Li, W. Wang, Y. Wang, et al., Chem. Eng. Sci. 282 (2023) 119333. doi: 10.1016/j.ces.2023.119333
W. Zeng, Y. Dong, X. Ye, et al., ACS Catal. 15 (2025) 6036–6045. doi: 10.1021/acscatal.5c01205
X. Zhang, P. Ma, C. Wang, et al., Energy Environ. Sci. 15 (2022) 830–842. doi: 10.1039/d1ee02369a
J. Chen, W. Gao, Y. Lu, et al., ACS Appl. Nano Mater. 6 (2023) 3927–3935. doi: 10.1021/acsanm.3c00092
W. Zhong, D. Zheng, Y. Ou, et al., Acta Phys. Chim. Sin. 40 (2024) 2406005. doi: 10.3866/pku.whxb202406005
H. Miao, G. Zeng, H. Zong, et al., J. Environ. Chem. Eng. 12 (2024) 113496. doi: 10.1016/j.jece.2024.113496
W. Yang, M. Gao, Y. Zhang, et al., J. Food Compos. Anal. 136 (2024) 106738. doi: 10.1016/j.jfca.2024.106738
S. Meng, D. Liu, Y. Li, et al., J. Agric. Food Chem. 70 (2022) 13583–13591. doi: 10.1021/acs.jafc.2c05910
H. Wu, S. Yu, Y. Wang, et al., Int. J. Hydrogen Energy 45 (2020) 30142–30152. doi: 10.1016/j.ijhydene.2020.08.112
S. Vignesh, P. Eniya, M. Srinivasan, et al., J. Environ. Chem. Eng. 9 (2021) 105996. doi: 10.1016/j.jece.2021.105996
Y. Cui, J. Zheng, Z. Wang, et al., J. Environ. Chem. Eng. 9 (2021) 106666. doi: 10.1016/j.jece.2021.106666
Y. Yang, H. Wang, W. Qin, et al., J. Colloid Interface Sci. 561 (2020) 298–306. doi: 10.1016/j.jcis.2019.10.102
Q. Wu, Q. Gao, L. Sun, et al., Chin. J. Catal. 42 (2021) 482–489. doi: 10.1016/S1872-2067(20)63663-4
X. Xu, S. Chen, P. Chen, et al., J. Colloid Interface Sci. 674 (2024) 624–633. doi: 10.1364/jocn.517147
Q. Wu, J. Li, T. Wu, et al., ChemElectroChem 6 (2019) 1996–1999. doi: 10.1002/celc.201900094
Y. Zuo, M. Huang, W. Sheng, et al., Int. J. Hydrogen Energy 47 (2022) 917–927. doi: 10.1016/j.ijhydene.2021.10.064
X. Zhu, H. Miao, J. Chen, et al., Inorg. Chem. Front. 9 (2022) 2252–2263. doi: 10.1039/d2qi00311b
X. Wang, Y. Duan, X. Zhao, et al., Mol. Catal. 573 (2025) 114854.
X. Yu, W. Zhang, L. Ma, et al., Green Chem. 27 (2025) 731–742. doi: 10.1039/d4gc05278a
Q. Su, C. Zuo, M. Liu, et al., Molecules 28 (2023) 5576. doi: 10.3390/molecules28145576
S. Ma, L.G. Pan, T. You, et al., J. Agric. Food Chem. 69 (2021) 4874–4882. doi: 10.1021/acs.jafc.1c00141
X. Du, W. Du, J. Sun, et al., Food Chem. 385 (2022) 132731. doi: 10.1016/j.foodchem.2022.132731
S. Meng, D. Liu, Y. Li, et al., J. Agric. Food. Chem. 70 (2022) 13583–13591. doi: 10.1021/acs.jafc.2c05910
K. Li, C. Liu, J. Li, et al., Acta Phys. Chim. Sin. 40 (2024) 2403009. doi: 10.3866/pku.whxb202403009
Y. Zhang, X. Chen, Y. Ye, et al., J. Catal. 419 (2023) 9–18. doi: 10.1016/j.jcat.2023.01.021
Z. Yu, X. Yue, J. Fan, et al., ACS Catal. 12 (2022) 6345–6358. doi: 10.1021/acscatal.2c01563
J. Qiu, Y. Wu, S. Jiang, et al., ACS Energy Lett. 8 (2023) 4173–4178. doi: 10.1021/acsenergylett.3c01779
Y. Lei, W. Si, Y. Wang, et al., ACS Appl. Mater. Interfaces 15 (2023) 6726–6734. doi: 10.1021/acsami.2c18694
X. Li, Z. Jiang, S. Shi, et al., J. Photoch. Photobio. A 471 (2026) 116709. doi: 10.1016/j.jphotochem.2025.116709
Q. Chen, C. Lu, B. Ping, et al., Appl. Catal. B: Environ. 324 (2023) 122216. doi: 10.1016/j.apcatb.2022.122216
J. Zhou, T. Shan, F. Zhang, et al., Adv. Fiber Mater. 6 (2024) 387–400. doi: 10.1007/s42765-023-00354-9
F. He, Y. Lu, Y. Wu, et al., Adv. Mater. 36 (2024) e2307490. doi: 10.1002/adma.202307490
S. Khan, M.A. Qaiser, W.A. Qureshi, et al., ACS Appl. Mater. Interfaces 17 (2025) 6249–6259. doi: 10.1021/acsami.4c17246
H.Z. Zhang, J.J. Liu, Y. Zhang, et al., J. Mater. Sci. Technol. 166 (2023) 241–249. doi: 10.1016/j.jmst.2023.05.030
J.J. Foo, S.F. Ng, S.H. Kok, et al., Chem. Eng. J. 505 (2025) 158992. doi: 10.1016/j.cej.2024.158992
Z.M. Zhou, M.H. Sun, Y.B. Zhu, et al., Appl. Catal. B: Environ. 334 (2023) 122862. doi: 10.1016/j.apcatb.2023.122862
T. Yang, Y. Chen, Y.C. Wang, et al., ACS Appl. Mater. Interfaces 15 (2023) 8066–8075. doi: 10.1021/acsami.2c20506
Q. Ji, X. Yu, L. Chen, et al., Ind. Crop. Prod. 172 (2021) 114064. doi: 10.1016/j.indcrop.2021.114064
J. Liang, H. Li, L. Chen, et al., Ind. Crop. Prod. 193 (2023) 116214. doi: 10.1016/j.indcrop.2022.116214
L. Yu, Z. Mo, X. Zhu, et al., Green Energy Environ. 6 (2021) 538–545. doi: 10.1016/j.gee.2020.05.011
J. Hou, K. Wang, X. Zhang, et al., ACS Catal. 14 (2024) 10893–10903. doi: 10.1021/acscatal.4c00334
P. Liu, T. Liang, Y. Li, et al., Nat. Commun. 15 (2024) 9224. doi: 10.1038/s41467-024-53482-0

Figure 1 (a) Schematic diagram of the basic principle of photocatalysis. (b) Photocatalytic preparation of H2O2 reaction. (c) The modification strategies schematic of g-C3N4.

Figure 2 The number of articles on photocatalytic H2O2 generation searched from web of science from 2015 to 2025.

Figure 3 (a) Schematic diagram of reaction mechanism. (b) The H2O2 production under visible light irradiation. (c) Effects of reactive species scavengers. (d) Schematic illustration of ORR and WOR in CN and A0.05CN. Reproduced with permission [130]. Copyright 2023, American Chemical Society. (e) The preparation technical route. (f) Photocatalytic production of H2O2 under simulated sunlight. (g) Free energy diagrams of ORR steps for MCN and KLCN. Reproduced with permission [132]. Copyright 2025, Elsevier B.V. (h) The mechanism of OCN model (C-O-C) photocatalytic O2 reduction. (i) The photocatalytic generation of H2O2 in O2 saturated pure water under visible light. (j) The photocatalytic generation of H2O2 with 10% isopropyl alcohol under visible light irradiation with air atmosphere. (k) The photocatalytic generation of H2O2 with 10% isopropyl alcohol under different atmosphere and pH under visible light irradiation. Reproduced with permission [133]. Copyright 2018, Royal Society of Chemistry. (l) Schematic diagram of the PCN-D sample synthesis. (m) Time course of photocatalytic H2O2 generation of PCN-D samples in 10% IPA solution, and (n) in pure water. (o) Wavelength-dependent AQY of PCN-D-1 sample. Reproduced with permission [134]. Copyright 2023, Elsevier B.V.

Figure 4 (a) Electron density, spin density, HOMO, and LUMO of BCN. (b) Energy barriers (Ea) of the rate-determining steps on various samples. (c) Time-dependent photocatalytic H2O2 production of different CN-based samples. (d) H2O2 production rate of BCN-450 with different reductive scavengers. Reproduced with permission [135]. Copyright 2024, American Chemical Society. (e) The possible mechanism for H2O2 production over CN-0.5 catalyst, (f) H2O2 generation rates of CN, CN-0.1, CN-0.5, and CN-1.0 catalysts. (g) Concentration of the produced H2O2 under different conditions. Reproduced with permission [136]. Copyright 2023, Elsevier B.V. (h) Schematic illustration for catalyst fabrication. (i) The optical properties of samples. (j) Photocatalytic H2O2 generation performance of different samples. Reproduced with permission [137]. Copyright 2021, Elsevier B.V.

Figure 5 (a) Schematic illustration of SD-CN catalyst structure and H2O2 generation. (b) H2O2 production rates of different SD-CN and CN samples. (c) Apparent quantum yield (AQY) of 10SD-CN, and (d) the electronic band structure. Reproduced with permission [138]. Copyright 2024, Elsevier B.V. (e) Schematic illustration of the preparation procedure. (f) H2O2 production of ACNN and C3N4 under visible light irradiation. (g) Formation (kf) and decomposition (kd) kinetics constant of H2O2 over ACNN and C3N4. Reproduced with permission [139]. Copyright 2020, American Chemical Society. (h) Schematic of proposed structure of Cv@g-C3N4. (i) The solid-state nuclear magnetic resonances (NMR) spectra 13C of g-C3N4 and 5Cv@g-C3N4. (j) Catalytic performance of Cv@g-C3N4. (k) Rate constant over g-C3N4 and 5Cv@g-C3N4. (l) The apparent quantum yield (AQY). (m) The H2O2 production rate with different conditions. (n) H2O2 generation rate in the O2-saturated pure water. (o) The free energy results. Reproduced with permission [140]. Copyright 2021, Wiley. (p) Proposed reaction mechanism of AKMT catalyst. (q) H2O2 production performance at pH 3. (r) The AQY of M and AKMT samples. (s) Solid-state nuclear magnetic resonance (NMR) spectra of 13C. Reproduced with permission [145]. Copyright 2020, Wiley. (t) Schematic diagram of Ni site structure evolution in the photocatalytic H2O2 generation. (u) The time course of H2O2 production activities in pure water. (v) Free energy profiles for photocatalytic H2O2 evolution reactions. Reproduced with permission [143]. Copyright 2023, Springer.

Figure 6 (a) Possible degradation mechanism. (b) Photocatalytic H2O2 generation performance in the presence of IPA. (c) H2O2 concertation of BCN and GCN without IPA under visible light irradiation. (d) Photocatalytic decomposition curves. Reproduced with permission [149]. Copyright 2022, Elsevier B.V. (e) UV-visible DRS spectra of CN and SCNx samples. (f) Estimated band structure. (g) H2O2 production under visible light irradiation. (h) AQY for photocatalytic H2O2 generation by SCN2. Reproduced with permission [150]. Copyright 2023, Elsevier B.V. (i) Structure model of BPMC-Vs. (j) Free energy results of different samples, time course of H2O2 production performance in (k) pure water, and (l) 10% IPA. (m) In situ FT-IR result under light radiation. (n) TPD-O2 profiles of BCN, PMC, and BPMC-Vs. (o) The wavelength-dependent AQY results. Reproduced with permission [151]. Copyright 2022, American Chemical Society.

Figure 7 (a) UV-vis DRS results and (b) time-resolved photoluminescence of the samples. (c) Solid-state 13C MAS NMR spectra. (d) H2O2 generation of different samples in pure water. (e) Time course of H2O2 production. (f) The wavelength-dependent AQY. (g) Free energy calculation of H2O2 evolution reaction. Reproduced with permission [152]. Copyright 2022, Royal Society of Chemistry. (h, i) XRD patterns. (j) ESR g-factor tests. (k) HRTEM image of CNT. (l) Photocatalytic H2O2 production performance. (m) Wavelength dependent AQE of CNT. Reproduced with permission [153]. Copyright 2023, American Chemical Society. (n, o) XRD data, (p) photocatalytic H2O2 production activities, (q) apparent quantum yield for CN and CN-K. (r) Calculated free energy. Reproduced with permission [154]. Copyright 2024, Chinese Physical Society.

Figure 8 (a) Band structures of BTO and CN. (b) Photocatalytic H2O2 production in 10 vol% methanol aqueous solution and (c) pure water. (d) Calculated electron transfer numbers, H2O2 selectivity and RRDE curves of BTOCN2. (e) Surface photovoltage results. Reproduced with permission [173]. Copyright 2024, Chinese Physical Society. (f) Photocatalytic H2O2 evolution with different catalysts. (g) ESR signals of DMPO-·O2⁻ under visible light irradiation. (h) Free energy profiles. (i) Schematic illustration of S-scheme. Reproduced with permission [174]. Copyright 2023, Elsevier B.V.

Figure 9 (a) HRTEM of homo-CN. (b) FT-IR spectroscopy, (c) photocatalytic H2O2 of homo-CN, HD-CN and HR-CN. (d) AQY of homo-CN in pure water and 10% ethanol. Reproduced with permission [179]. Copyright 2023, Elsevier B.V. (e) Energy band structure diagrams. (f) H2O2 evolution rate with water or 0.1 mmol/L PBQ. (g) AQE of MCN/SCN/CF. Reproduced with permission [180]. Copyright 2024, Wiley. (h) Free energy diagrams through an indirect two-step single-electron ORR, Reproduced with permission [181]. Copyright 2024, Wiley. (i) The S-scheme charge transfer mechanism. (j) Band structure of the samples. (k) Photocatalytic H2O2 generation. (l) kf and kd constant of P-CN/B-CN. Reproduced with permission [182]. Copyright 2025, American Chemical Society. (m) UV-vis measurement and photos of the catalysts. (n) Photocatalytic performance of ACN and CCN-X. (o) AQY results. (p) Schematic of the photogenerated charge transmission. Reproduced with permission [184]. Copyright 2025, Elsevier B.V.

Figure 10 (a) Schematic illustration of the sample synthesis. (b) Photocatalytic H2O2 generation rate. (c) Wavelength-dependent AQY of CoOx-NvCN. (d) Free energy diagrams of O2 reduction reaction. Reproduced with permission [190]. Copyright 2024, American Chemical Society. (e) Schematic of the charge transfer and H2O2 generation. (f) Photocatalytic H2O2 generation performance. (g) H2O2 generation rates under different reaction conditions and the influence of different sacrificial agents. (h) H2O2 selectivity characterization. (i) In situ DRIFTS of O2 adsorption over CoOx-BCN-FeOOH. (j) In-situ DRIFTS of CoOx-BCN-FeOOH during reaction. (k) Free energy diagrams. Reproduced with permission [191]. Copyright 2024, Springer.
Table 1. Performance comparison of some modified g-C3N4-based photocatalysts for H2O2 production.
| Authors (year) | Catalyst | Modification strategy | Light source | H2O2 production activity | Enhancement factor (vs. pristine CN) | AQY/wavelength | Electron donor | Ref. |
| Yang et al. (2023) | A0.05CN | Carbon doping & surface -OH | Visible (λ ≥ 420 nm) | 3.6 mmol g−1 h−1 | 3.5 × | Not available | Pure water | [130] |
| Zhang et al. (2024) | s-BxCN | Carbon doping, N vacancies, -C≡N | Visible light | 54.2 µmol L−1 h−1 | Not available | Not available | Pure water | [131] |
| Xu et al. (2025) | KLCN | Lignin-derived carbon ring embedding & N defects | Visible light | 266.82 µmol/L | 6.93 × | Not available | Isopropanol | [132] |
| Wei et al. (2018) | OCN-500 | Oxygen doping (C-O-C/—OH) | Visible light | 730 µmol / 5h | 3.5 × | 28.5%@365 nm, 10.2%@420 nm | Isopropanol | [133] |
| Yu et al. (2023) | PCN-D | Dual-site P doping (bay & corner) | Visible light | 413.85 µmol L−1 h−1 | > 6 × | Not available | Isopropanol | [134] |
| Ma et al. (2024) | BCN-450 | B doping (Frustrated Lewis Pairs) | Visible light | 51008 µmol L−1 g−1 h−1 | 12.4 × | Not available | Ethanol | [135] |
| Zhang et al. (2023) | CN-1.0 | S doping & N vacancies | Xenon lamp | 293.2 µmol/L / 4 h | 2.4 × | Not available | Ethanol | [136] |
| Liu et al. (2021) | B-CNT | B/P/S co-doping, nanotube | Simulated sunlight | 1830.98 µmol/L / 4 h | 2 × | Not available | Isopropanol | [137] |
| Habarugira et al. (2024) | 10SD-CN | Na doping & N defects | Visible light | 297.2 µmol L−1 h−1 | 9.8 × | 1.29%@420 nm | Isopropanol | [138] |
| Wu et al. (2020) | ACNN | K⁺/Na⁺ doping & N vacancies | Visible (λ ≥ 420 nm) | 10.2 mmol g−1 h−1 | 89.5 × | 30.7%@429 nm | Isopropanol | [139] |
| Chen et al. (2021) | 5Cv@g-C3N4 | Cyano defects & Na doping, porous | Visible (λ ≥ 420 nm) | 7.01 mmol L−1 h−1 | 220 × | 9.58%@420 nm | Ethanol | [140] |
| Zhang et al. (2020) | AKMT | S/K co-doping | Visible light | 4.1 mmol/L / 3 h | Not available | 100%@450 nm | Ethanol | [145] |
| Wang et al. (2024) | Na1.2-P0.2-CN | P/Na co-doping | Visible light | 3001.64 µmol g−1 L−1 / 100 min | 62 × | Not available | Isopropanol | [146] |
| Wu et al. (2022) | GCN | Carbon defects, flower-like | Xenon lamp (λ > 420 nm) | 21.59 mmol L−1 g−1 h−1 | 5 × | Not available | Isopropanol | [149] |
| Zhang et al. (2022) | Nv-C≡N-CN | N vacancies & -C≡N groups | Visible (λ ≥ 420 nm) | 3093 µmol g−1 h−1 | 8.7 × | 36.2%@400 nm | Isopropanol | [152] |
| Li et al. (2023) | SCN2 | S doping, -NH2/-CH3 edge groups | Visible (λ ≥ 420 nm) | 416.7 µmol g−1 h−1 | 10 × | 31.6%@365 nm | Pure water | [150] |
| Zeng et al. (2025) | BPMC-Vs | B/P co-doping & N defects | Visible (λ ≥ 420 nm) | 650.7 µmol g−1 h−1 | 9.9 × | 39.1%@420 | Isopropanol | [151] |
| Chen et al. (2023) | CNT | Crystallinity enhancement (Molten salt) | AM 1.5 | 2.48 mmol g−1 h−1 | 5.8 × | 22%@ 00 nm | Isopropanol | [153] |
| Zhong et al. (2024) | CN-K(1:6) | K doping, High crystallinity | LED | 7.8 mmol L−1 h−1 | 220 × | 5.17%@420 nm | Isopropanol | [154] |
| Li et al. (2024) | BTOCN2 | Bi4Ti3O12/g-C3N4 S-scheme | Simulated sunlight | 1650 µmol g−1 h−1 | 2.8 × | 0.36%@420 nm | Methanol | [173] |
| Zhang et al. (2023) | 3CuInS2/PCN | CuInS2/PCN S-scheme | Visible light | 1247.6 µmol g−1 h−1 | 11.6 × | 16.0%@420 nm | Isopropanol | [174] |
| Chen et al. (2023) | homo-CN | Homojunction (OH-rich/OH-defective) | Xenon lamp (λ ≥ 420 nm) | 508.4 µmol L−1 L−1 | 4.5 × | 2.1%@500 nm | Ethanol | [179] |
| Zhang et al. (2024) | CN-NH4-NaK | Homojunction (Order-Disorder) | Visible (λ ≥ 420 nm) | 16675 µmol g−1 h−1 | 158 × | 34.3%@395 nm, 28.4%@420 nm | Isopropanol | [181] |
| Foo et al. (2025) | CCN-550 | Homojunction (PHI/PTI), K+/Li+, -C≡N | Visible (λ ≥ 420 nm) | 5838.91 µmol L−1 h−1 | 31.7 × | 11.57%@420 nm | Benzyl alcohol | [184] |
| Hou et al. (2024) | CoOx-NvCN | N vacancies & CoOx clusters | Visible (λ ≥ 420 nm) | 244.8 µmol L−1 h−1 | 12.1 × | 5.73%@420 nm | Pure water | [190] |
| Jing et al. (2024) | CoOx-BCN-FeOOH | B doping, FeOOH & CoOx clusters | Visible (λ ≥ 420 nm) | 0.34 mmol g−1 h−1 | 30 × | 8.36%@420 nm | Pure water | [191] |
| Zhang et al. (2023) | Ni-SAPs/PuCN | Ni single-atom (Ni-N3) | Visible (λ ≥ 420 nm) | 342.2 µmol L−1 h−1 | 20.7 × | 10.9%@420 nm | Pure water | [143] |
| Su et al. (2021) | Sb-SAPC15 | Sb single-atom (Sb-N) | Visible (λ > 420 nm) | 6.2 mg L−1 h−1 | 248 × | 17.6%@420 nm | Pure water | [144] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: