Scheme 1.
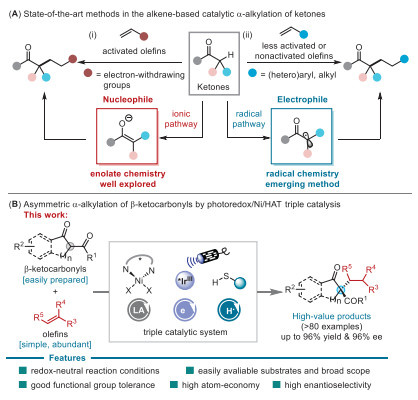
Background and project design.

The construction of all-carbon quaternary stereocenters represents a pivotal and enduring challenge in the realm of synthetic organic chemistry, given their ubiquitous presence in biologically active natural products and pharmaceuticals and the profound steric constraints they impose during bond formation. While significant strides have been made in enantioselective catalysis, the direct α-alkylation of carbonyl compounds using simple, non-activated olefins as coupling partners has remained a particularly stubborn frontier. This difficulty primarily stems from a fundamental polarity mismatch in conventional ionic reaction pathways, where the nucleophilic enolate and the typically electrophile-deficient, non-activated alkene are inherently mismatched. In this context, radical-based strategies that leverage electrophilic α-carbonyl radicals present an elegant and conceptually distinct solution, as these open-shell intermediates can readily add to the electron-rich π-bond of olefins (Scheme 1A) [1]. Early pioneering work laid the foundational principles for this approach. For instance, in 2021, Funes-Ardoiz, Ye, and their team reported a catalytic α-alkylation of active methylene compounds with non-activated alkenes, driven by blue-LED light and orchestrated through a dual hydrogen atom transfer (HAT) catalytic manifold [2]. This was followed in 2023 by Yamashita, Kobayashi, and colleagues, who developed a system employing 4CzIPN as a photoredox catalyst and LiSPh as a multifunctional Lewis acid/Brønsted base/HAT catalyst for similar transformations [3]. Despite these remarkable advances, a critical limitation persisted: the development of a general and highly enantioselective catalytic platform, especially one capable of accommodating sterically demanding substrates to forge these congested stereocenters, was still conspicuously scarce [4].
It is against this backdrop that the recent work by Chen and colleagues constitutes a transformative leap forward. They have ingeniously reported a groundbreaking triple-catalytic system that seamlessly integrates the complementary powers of photoredox catalysis, chiral nickel catalysis, and HAT catalysis. This synergistic platform successfully enables the direct, intermolecular, and highly enantioselective α-alkylation of β-ketocarbonyl compounds with an exceptionally broad range of simple olefins. The scope impressively encompasses both less-activated variants (such as styrenes) and the more challenging non-activated alkenes (including alkyl olefins and vinyl ethers) [5], as detailed in Scheme 1B.
The true brilliance of this methodology lies in its elegantly synchronized, redox-neutral catalytic cycle. This orchestrated process operates under remarkably mild conditions, room temperature and irradiation by blue light, and without the need for any external base or stoichiometric oxidant, highlighting its atom economy and operational simplicity. Within this cycle, each catalyst performs a meticulously designed role: the iridium photocatalyst (Ir(ppy)₂dtbbpyPF₆) harvests light energy to generate critical radical species; the chiral Ni(acac)2/bis(oxazoline) complex acts as a stereodetermining Lewis acid, not only activating the β-ketocarbonyl substrate but also exerting exquisite enantiocontrol over the bond-forming event; and the sterically hindered 2,4,6-triisopropylbenzenethio (TRIP-SH) mediates the key hydrogen atom transfer steps, shuttling hydrogen atoms to propagate the radical chain and terminate the cycle. The confluence of these three catalytic modes collectively enables a transformation that was previously unattainable, achieving outstanding levels of enantioselectivity and setting a new benchmark for the field. This work not only provides a powerful synthetic tool but also serves as an inspiring blueprint for the future development of multi-catalytic enantioselective processes involving radical intermediates.
The reaction demonstrates a remarkably broad substrate scope, underscoring its generality (Scheme 2). A wide array of olefins from electronically diverse styrenes (including challenging ortho-substituted and hetero-aromatic variants) to disubstituted alkenes that generate contiguous stereocenters are all competent coupling partners (1-4). Most notably, the protocol successfully engages challenging non-activated alkyl olefins, with terminal alkenes and those bearing functional groups (halides, alcohols, ketones, esters) being smoothly incorporated (5-8), thus providing versatile synthetic handles for downstream applications. However, this reaction exhibits limited compatibility with linear ketone substrates.
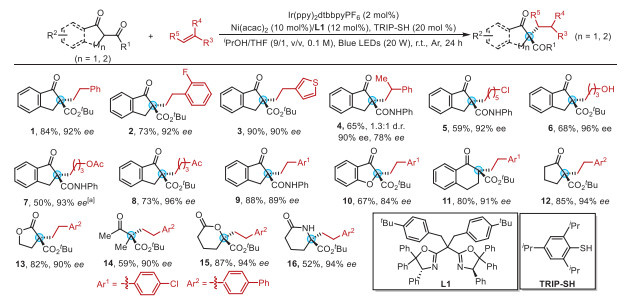
Furthermore, the scope of β-ketocarbonyl compounds is equally impressive. A wide array of cyclic substrates, including β-indanone esters, tetralones, and various aliphatic cyclic β-ketoesters/lactones, underwent smooth alkylation to afford products with high enantioselectivity (9-14). Furthermore, product 12 was readily converted into the corresponding six-membered lactone 15 and lactam 16 via Baeyer–Villiger oxidation and Beckmann rearrangement, both occurring without racemization, attesting to the robustness and synthetic utility of the newly constructed chiral scaffolds.
In conclusion, Chen and colleagues have successfully established a robust and versatile platform for direct asymmetric radical α-alkylation, marking a significant methodological leap in synthetic chemistry. By ingeniously integrating photoredox, nickel, and HAT catalysis into a synergistic triple-catalytic system, they have addressed a persistent challenge: the use of readily available, unactivated olefins as effective coupling partners. This innovative approach bypasses traditional limitations associated with stereocontrol in radical reactions, enabling efficient and highly enantioselective formation of α-quaternary carbon centers. The methodology offers a practical and general route to valuable enantioenriched ketones-structural motifs prevalent in natural products and pharmaceuticals. Beyond its immediate synthetic utility, this work sets an inspiring precedent for the future design of multi-catalytic systems that merge diverse activation modes to achieve challenging enantioselective transformations under mild conditions.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Cui Xin: Writing – original draft. Wei-Min He: Writing – review & editing.
Y. Yamashita, S. Kobayashi, Chem. Asian J. 19 (2024) e202400319.
G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz, J. Ye, J. Am. Chem. Soc. 143 (2021) 11251-11261. doi: 10.1021/jacs.1c05852
Y. Yamashita, Y. Ogasawara, T. Banik, S. Kobayashi, J. Am. Chem. Soc. 145 (2023) 23160-23166. doi: 10.1021/jacs.3c07436
L.J. Li, J.C. Zhang, W.P. Li, et al., J. Am. Chem. Soc. 146 (2024) 9404-9412. doi: 10.1021/jacs.4c01842
X.S. Zhou, Z.Q. Li, W.Y. Qu, et al., Angew. Chem. Int. Ed. 64 (2025) e202424915. doi: 10.1002/anie.202424915

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: