College of Environmental Sciences, Sichuan Agricultural University, Chengdu 611130, China
b.
Sichuan Provincial Engineering Research Center of Agricultural Non-point Source Pollution Control, Sichuan Agricultural University, Chengdu 611130, China
c.
Global Centre for Environmental Remediation (GCER), University of Newcastle, Callaghan, NSW 2308, Australia
d.
Department of Chemical Engineering, University of Patras, Caratheodory 1, University Campus, GR-26504 Patras, Greece
e.
School of Agricultural Sciences, University of Patras, Agrinio 30100, Greece
Received Date:
07 May 2025 Accepted Date:
20 November 2025 Revised Date:
19 September 2025 Available Online:
15 June 2026
Abstract:
Covalent organic frameworks (COFs) with structural designability and functional predictability display great potential as emerging metal-free semiconductor catalysts in the field of photocatalytic degradation for antibiotics. However, the disadvantages of COFs, such as weak interfacial contact with antibiotics, light response limitation, charge-carrier recombination, and low oxygen activation efficiency, limit their practical application in the photocatalytic degradation of antibiotics. On this basis, PyTTA-COF-OH via good crystallinity and hydrophilicity was constructed through the synergistic strategy combining hydroxylation modulation of the surface microenvironment and expansion of the π-conjugated structure. Simultaneously, Surface hydroxyl functionalization of PyTTA-COF-OH not only reduced the band gap width but also enhanced the light absorption in the visible band. Significantly, theoretical calculations indicated that the introduction of hydroxyl groups widened the electrostatic potential difference within the molecular structure, further promoting exciton dissociation and charge separation. PyTTA-COF-OH removed sulfadiazine (SDZ) up to 99% in 10 mg/L SDZ solution. Besides, the results of the interference experiments and the cycling experiments proved that PyTTA-COF-OH had better anti-interference and stability. Based on the mechanistic analysis, the surface hydroxyl–functionalized PyTTA-COF-OH effectively promoted the adsorption of oxygen and the transfer of electrons to the adsorbed oxygen, thus increasing the efficiency of the O2•- generation, aiming at the efficient degradation of antibiotics under the visible light conditions.
Antibiotics as an emerging organic pollutant have obtained much attention from researchers in recent years, due to their long-term presence in the natural aquatic environments, causing potential hazards to aquatic ecosystems and human health [1–3]. Notably, concentrations of antibiotics are extremely low in the aqueous environment, thus resulting in ineffective removal by conventional treatment techniques. For this reason, advanced oxidation processes (AOPs) are increasingly being employed to remove residual antibiotics from the aqueous environment [4–6]. As sunlight is a core source of clean energy in the natural environment, photocatalytic advanced oxidation processes that utilize sunlight as an energy input have been identified as the most sustainable, cost-effective, and potentially advanced technology for the removal of antibiotics from aqueous environments [7–9]. However, traditional metal-semiconductor-based photocatalysts inevitably exhibit leaching of metal ions, which leads to secondary pollution of the aqueous environment [10–12]. In this connection, the development of an efficient and green photocatalyst is particularly important.
Carbon-based photocatalysts have received a great deal of focus in terms of water-safety protection, mainly attributed to the fact that they do not release metal ions to produce the potential hazards of secondary contamination during the catalytic degradation process [13–15]. Among various carbon-based photocatalysts, covalent organic frameworks (COFs) photocatalysts stand out among many carbon-based semiconductors owing to their customizable structure, predictable functionality, and unique framework structures [16–19]. Meanwhile, the ability of COFs to produce active species under light excitation makes them show great potential for application in the field of degrading antibiotics present in aqueous environments [20–22]. However, the weak interfacial contact between COFs and antibiotics results in limited mass transfer of active species and thus reduces degradation efficiency, which was mainly attributed to the very low concentration of antibiotics and the hydrophobicity of COFs in the natural aquatic environment [7,23,24]. In addition, the limited visible light response, severe charge-carrier recombination, and low oxygen activation efficiency of COFs further limit the photocatalytic efficiency and practical applications during the reaction process [25,26]. Based on the above research, the targeted construction of COFs-based photocatalysts with good interfacial contact with antibiotics, strong visible light response, fast charge-carrier separation, and high oxygen activation efficiency is a potential development trend for the degradation of antibiotics in aqueous environments.
Relevant studies have shown that enhancing the hydrophilicity of photocatalysts significantly facilitates the mass transfer process, improves the utilization of photogenerated charge carriers, and enhances the catalyst-antibiotic interactions, which effectively promotes the efficiency of photocatalytic degradation for antibiotics [27–29]. Based on this finding, improving the surface properties of COFs through hydroxylation of the surface microenvironment is considered an effective strategy that enhances interfacial contact with antibiotics, increases charge carrier mobility, and promotes oxygen activation. Remarkably, the presence of hydroxyl functional groups further encourages exciton dissociation and oxygen adsorption to build bridges for the generation of reactive species, mainly attributed to their intrinsic push-pull electronic effects and polar nature [30,31]. Furthermore, the choice of semiconductor unit module is crucial for the photocatalytic performance, which is closely related to the performance of the photoresponse [32]. Among them, the pyrene monomer with a unique large π-conjugation system exhibits a wide light absorption range and effectively absorbs visible light, which has been proved to be an ideal type for the construction of COF photocatalysts [33]. At the same time, the chemical structure of pyrene is easy to modify for functionalization, which provides a solid foundation for the surface hydroxylation of COFs [34]. Therefore, the synergistic strategy combining hydroxylation modulation of the surface microenvironment with the expansion of the π-conjugated structure to simultaneously enhance the hydrophilicity, light-absorption capacity, and charge-carrier separation efficiency is crucial to achieving visible photocatalytic degradation of antibiotics.
Herein, imine-bonded COFs with pyrene units as the photosensitive active center (PyTTA-COF-H) were constructed and further hydroxylation modification of COFs (PyTTA-COF-OH) was achieved by surface microenvironment engineering, which was employed for the degradation of sulfadiazine (SDZ) model antibiotics under visible light conditions. Based on microscopic morphology and chemical structure characterization, PyTTA-COF-OH with good crystallinity and hydrophilicity connected by imine bonds was successfully constructed via the simple one-pot synthesis method, which effectively modulated the interfacial contact between PyTTA-COF-OH and antibiotics. Experimental photoelectric properties and theoretical calculations indicated that the surface microenvironmental hydroxylation of PyTTA-COF-OH effectively reduced the bandgap width and broadened the photoresponse range. In addition, the introduction of the hydroxylated microenvironment further widened the electrostatic potential (ESP) difference, thereby facilitating the dissociation of excitons on the pyrene units and the separation of photogenerated charge carriers within the molecular structure. The results of visible photocatalytic degradation experiments showed that the hydroxylated PyTTA-COF-OH not only achieved up to 99% SDZ degradation rate but also further enhanced the adsorption capacity of SDZ much better than PyTTA-COF-H. Mechanistic studies further demonstrated that the active species acting as the dominant role was e- and O2•- in the visible light-catalyzed degradation of SDZ. The hydroxyl–functionalized microenvironment significantly promoted the generation of O2•- by modulating the oxygen adsorption energy barriers and electron transfer processes.
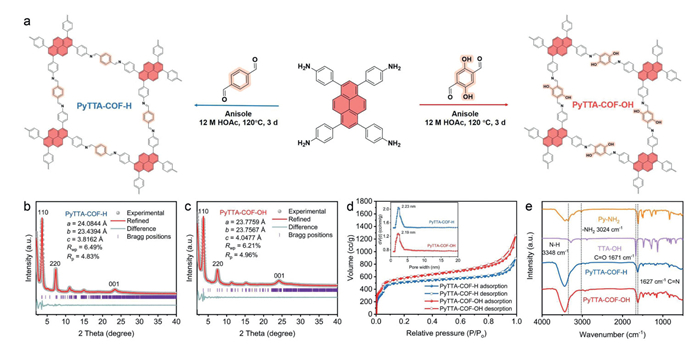
PyTTA-COF-H were constructed through a Schiff-base reaction condensation between 4,4′,4″,4′″-(pyrene-1,3,6,8-tetrayl)tetraaniline (Py-NH2) as the amino monomer and terephthalaldehyde (TTA-H) as the aldehyde monomer in anisole [35]. Then, the surface microenvironment of COFs was modulated by changing the functional groups on the aldehyde-based monomer, based on which the surface hydroxyl–rich PyTTA-COF-OH was synthesized via Py-NH2 and 2,5-dihydroxyterephthalaldehyde (TTA-OH) (Fig. 1a). Subsequently, the crystallinity of PyTTA-COF-H and PyTTA-COF-OH was confirmed by powder X-ray diffraction (PXRD) patterns, and the structural features of as-prepared COFs were further analyzed based on actual measured PXRD data and theoretical modeling (Figs. 1b and c). Notably, two sharp diffraction peaks at 2θ = ~3.7° in the PXRD patterns of PyTTA-COF-H and PyTTA-COF-OH, respectively, both corresponding to the (110) crystal plane [36]. The differences in the positions and intensities of the diffraction peaks were not significant in the PXRD patterns, indicating that the introduction of hydroxyl units had a negligible effect on the crystal structure of COFs. Furthermore, Pawley refinement was performed to fit the experimental data after geometric optimization of the COFs by the Materials Studio, and then the consequences suggested that the obtained experimental results match well with the constructed simulation model of the eclipsed (AA) stacking mode [37]. Accordingly, the high crystallinity and AA stacking of PyTTA-COF-R provided better electron transfer channels and promoted charge carrier migration, which benefited the enhanced photo-oxidation activity [34]. Meanwhile, the specific surface area and permanent porosity of PyTTA-COF-R were determined by nitrogen (N2) adsorption-desorption isotherm measurements. As shown in Fig. 1d, PyTTA-COF-H and PyTTA-COF-OH displayed typical type-Ⅳ isotherms, and the presence of significant hysteresis loops in the adsorption-desorption isotherms indicated the presence of mesopores in the PyTTA-COF-R. The Brunauer–Emmett–Teller (BET) specific surface areas of PyTTA-COF-H and PyTTA-COF-OH were found to be 1639 and 1878 m2/g, respectively. Furthermore, the nonlocal density functional theory (NLDFT) model revealed that the actual pore size of the PyTTA-COF-H and PyTTA-COF-OH was 2.23 and 2.19 nm, respectively, in good accord with the predicted pore diameters for the eclipsed AA geometries (Fig. S1 in Supporting information). As illustrated in Fig. 1e, accompanied by the disappearance of -N-H (~3348 cm-1) and -NH2 (~3024 cm-1) in Py-NH2 and the -C=O (~1671 cm-1) in TTA-OH, and then the peaks at ~1627 cm-1 were present in the Fourier transform infrared spectroscopy (FT-IR) spectra of both COFs, which belonged to the imine bond (-C=N-). This phenomenon suggested the successful formation of imine linkers through the Schiff-base reaction. Based on the above studies, PyTTA-COF-OH with the larger surface area was beneficial to enhance the full contact of organic pollutants with the surface of PyTTA-COF-OH in aqueous environments, while a smaller pore size reduced the scattering and compounding of charge carriers during the transfer process, which further improved the photocatalytic performance [17,38].
Figure 1
Figure 1.
(a) The schematic representation of PyTTA-COF-R synthesis. XRD images of PyTTA-COF-H (b) and PyTTA-COF-OH (c). (d) N2 adsorption and desorption isotherms of PyTTA-COF-H and PyTTA-COF-OH (The inset of the pore size distributions for the two COFs). (e) FT-IR spectra of monomers and COFs.
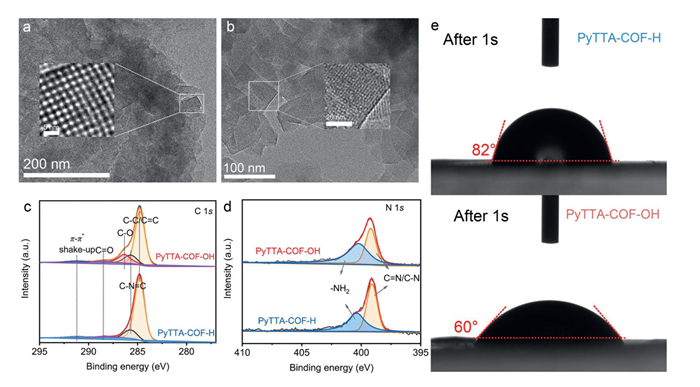
Meanwhile, the microstructure of PyTTA-COF-H and PyTTA-COF-OH was characterized by scanning electron microscopy (SEM). As shown in Fig. S2 (Supporting information), PyTTA-COF-H exhibited a typical micro-morphological structure of uniformly sized spherical stacks. In contrast, PyTTA-COF-OH appeared to have a morphological structure of stacked particles with smaller particle sizes after the introduction of hydroxyl surface sites, which was closely related to the increased specific surface area of PyTTA-COF-OH (Fig. S3 in Supporting information). The changes in the microscopic morphology were mainly attributed to the enhancement of molecular hydrogen bonding interactions based on hydroxyl–rich surface microenvironments [39]. In addition, transmission electron microscopy (TEM) and the high-resolution TEM (HR-TEM) images of PyTTA-COF-H and PyTTA-COF-OH exhibited a similar irregular layered stacked structure and all presented well-defined lattice fringes, which further corroborated the observation that PyTTA-COF-R had excellent crystallinity in the PXRD patterns (Figs. 2a and b). The superficial composition and chemical state of PyTTA-COF-R were characterized by X-ray photoelectron spectroscopy (XPS). Additionally, the wide-scan XPS spectra indicated that the major elemental compositions for the surfaces of the two COFs were C, N, and O (Fig. S4 in Supporting information). Compared with PyTTA-COF-H, the peak intensity for O 1s in PyTTA-COF-OH was obviously enhanced, which was mainly attributed to the effective introduction of hydroxyl groups on the surface microenvironment of PyTTA-COF-OH. The C 1s spectrum of PyTTA-COF-H was divided into four peaks corresponding to C—C/C=C at 284.8 eV, C—N=C at 285.7 eV, C=O at 288.1 eV, and π-π* shake-up at 291.2 eV. Compared with PyTTA-COF-OH, the C 1s spectrum were further displayed a new peak at 286.3 eV corresponding to C—O, which attributed to the successful introduction of hydroxyl units (Fig. 2c). The N 1s spectra of two COFs were deconvoluted into two components attributed to C=N/C—N (399.1 eV) and -NH2 (400.4 eV), which represented the N in imine bonds and the unreacted amine groups, respectively (Fig. 2d) [40]. Simultaneously, the surface hydrophilicity of PyTTA-COF-R was investigated by the determination of the water contact angle. As indicated in Fig. 2e, the water contact angle of the PyTTA-COF-OH (~60°) was smaller than that of PyTTA-COF-H (~82°), which was closely associated with the intramolecular polarity [41]. This signified that the introduction of hydroxyl groups activates a more hydrophilic surface microenvironment, facilitating not only the entry of water molecules into the interface of PyTTA-COF-OH but also the utilization of dissolved oxygen [42,43]. The above finding provided a theoretical basis for PyTTA-COF-OH to produce more reactive oxygen species during visible photocatalysis.
Figure 2
Figure 2.
The TEM and HR-TEM images of PyTTA-COF-H (a) and PyTTA-COF-OH (b). C 1s XPS spectra (c) and N 1s XPS spectra (d) of PyTTA-COF-H and PyTTA-COF-OH. (e) Images of water contact angle on PyTTA-COF-H and PyTTA-COF-OH.
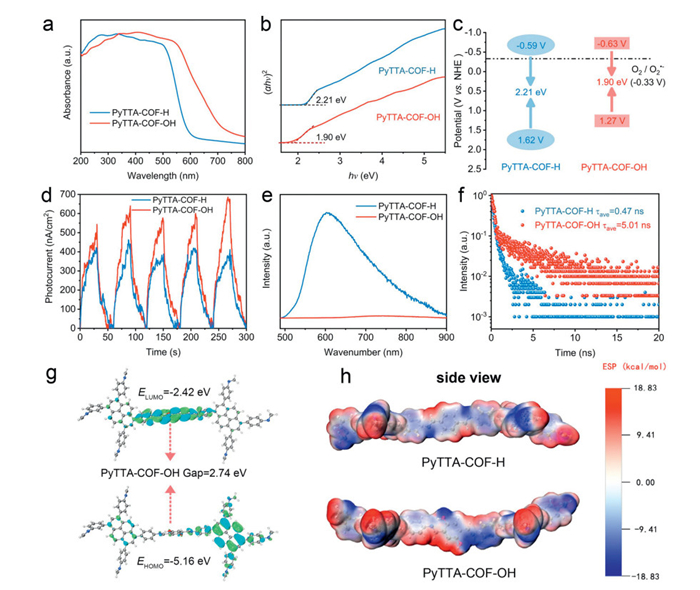
Afterwards, the energy band structures of the two COFs were determined via the UV–visible (UV–vis) diffuse reflectance spectra (DRS) and XPS valence-band spectra. As shown in Fig. 3a, the spectral coverage of PyTTA-COF-OH was remarkably extended from 600 nm to 750 nm compared to PyTTA-COF-H due to the insertion of hydroxyl units, which revealed that PyTTA-COF-OH had stronger visible-light trapping properties. The corresponding band gap energies of PyTTA-COF-H and PyTTA-COF-OH were calculated to be 2.21 and 1.90 eV via the Kubelka–Munk function equation, respectively (Fig. 3b). The narrower band gap of PyTTA-COF-OH indicated that the energy required for photo-induced excitation and generated charge carriers was lower, which favored visible-light-activated photocatalysis [44]. Moreover, Fig. S5 (Supporting information) estimates the valence band positions (EVB) of PyTTA-COF-H and PyTTA-COF-OH at 1.62 and 1.27 V vs. NHE, respectively. Therefore, the direct production of •OH (•OH/H2O, + 2.38 V vs. NHE) and 1O2 (1O2/O2•-, + 2.53 V vs. NHE) was quite challenging under the photocatalytic setting via the two COFs [11]. As indicated in Fig. 3c, the conduction band positions (ECB) of PyTTA-COF-H and PyTTA-COF-OH were deduced to be −0.59 and −0.63 V vs. NHE, respectively, which displayed that photogenerated electrons at the valence band position of PyTTA-COF-OH had a higher reduction potential beneficial for the generation of O2•- (O2/O2•-, −0.33 V vs. NHE) [7].
Figure 3
Figure 3.
UV–vis DRS spectra (a), the corresponding Tauc plots (b), and electronic band structure (c) of PyTTA-COF-H and PyTTA-COF-OH. Transient photocurrent responses (d), PL spectra (e), and time-resolved PL decay curves (f) of PyTTA-COF-H and PyTTA-COF-OH. (g) Frontier molecular orbitals of PyTTA-COF-OH. (h) Side view of electrostatic potential distribution for two COFs.
The photoelectric properties and charge carrier separation efficiency were further investigated in the two COFs by transient photocurrent response, electronic impedance spectroscopy (EIS), and steady-state photoluminescence (PL). As shown in Fig. 3d, the transient photocurrent intensity of PyTTA-COF-OH was higher than that of PyTTA-COF-H, suggesting that the modulation strategy for hydroxylation of the surface microenvironment enhanced the charge separation efficiency [45]. The introduction of hydroxyl substituents resulted in a smaller arc radius compared to the pristine COFs, indicating that PyTTA-COF-OH had a lower charge transfer barrier (Fig. S6 in Supporting information) [46]. The PL intensity of PyTTA-COF-OH was significantly lower than PyTTA-COF-H, which demonstrated the reduced e- and h+ recombination rate in PyTTA-COF-OH (Fig. 3e). Meanwhile, the average fluorescence lifetime (τave) of PyTTA-COF-OH (5.01 ns) was much higher than that of PyTTA-COF-H (0.47 ns, about 10.65-fold) according to fluorescence decay fitting, suggesting that PyTTA-COF-OH exhibited a higher nonradiative attenuation transition and a stronger charge transfer ability under photoexcitation (Fig. 3f) [47]. The above findings were not sufficient to fully explain that the insertion of hydroxyl sites modulated the surface microenvironment, thereby mediating the efficient separation of photogenerated charge carriers in the internal structure of PyTTA-COF-OH. Obviously, internal electron transfer mechanisms needed to be studied in depth.
To deeply elucidate the photocatalytic activity differences in electron transfer, the density functional theory (DFT) theoretical calculations were utilized, aiming to investigate the Frontier molecular orbital theory (FMO) and charge carrier separation in the structures of PyTTA-COF-R series. Theoretical results showed that the highest occupied molecular orbital (HOMO) for PyTTA-COF-R was predominantly located on the Py-NH2 units, while the lowest unoccupied molecular orbital (LUMO) for PyTTA-COF-R was mainly distributed centrally on the imine bonds and imine-bonded benzene ring units (Fig. 3g and Fig. S7 in Supporting information). Based on this, the photogenerated e- tended to shift in the direction of TTA-H or TTA-OH units, while the photogenerated h+ was mainly concentrated in the Py-NH2 units when the charge carriers were generated by PyTTA-COF-R under photoexcitation. The transfer paths of charge carriers within the molecular structure were further clarified to provide support for the subsequent investigation into the generation mechanism of reactive oxygen species. Moreover, the forbidden bandwidth of PyTTA-COF-OH was lower than that of PyTTA-COF-H after the introduction of hydroxyl units, in agreement with the trend of the actual test data, which was beneficial for the separation and transfer of charge carriers. Furthermore, the driving force of charge carrier separation within the molecule was closely related to the electrostatic potential (ESP) distribution of COFs. For this reason, the ESP distribution of COFs was investigated using a typical section of PyTTA-COF-R as a theoretical calculation model [30]. As shown in Fig. 3h and Fig. S8 (Supporting information), the difference in ESP around the TTA-OH units was significantly enlarged after the introduction of the hydroxyl units, which was mainly attributed to the high electronegativity of the oxygen atoms that caused electrons to be adsorbed around the amino atoms, leading to a localized asymmetric distribution of charge in this region. Consequently, the TTA-OH units exhibited more electron build-up, which benefited in providing richer reaction sites and generated a higher charge density in the localized region to facilitate charge carrier separation. Based on the analysis of the above results, the hydroxylation strategy of the surface microenvironment was modulated by PyTTA-COF-OH, which facilitated the separation of photogenerated charge carriers and provided a reaction platform for the generation of O2•-.
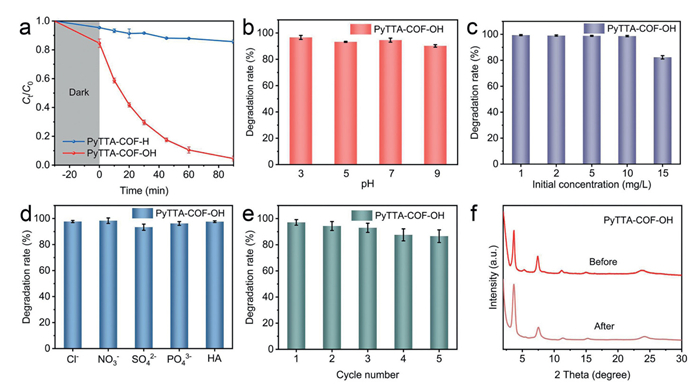
The photo-oxidation activity of PyTTA-COF-R under visible-light conditions was evaluated using sulfadiazine (SDZ) as an oxidizing model pollutant in the aqueous environment. In the absence of added photocatalysts, 10 mg/L SDZ can hardly be degraded under visible-light irradiation. Subsequently, the SDZ adsorption reached equilibrium at 30 min by the two photocatalysts under dark conditions before undergoing visible-light irradiation. As shown in Fig. 4a, PyTTA-COF-OH (15.4%) had a stronger adsorption capacity compared to PyTT-COF-H (4.6%), which was attributed to the excellent hydrophilicity of PyTTA-COF-OH and the hydrogen-bond interaction between the hydroxyl group on the surface of PyTTA-COF-OH and the amino and sulfanilamide groups in SDZ [48]. Subsequently, the removal of SDZ reached only 14% in 90 min via PyTTA-COH—H under visible-light irradiation. Compared with PyTTA-COF-H, PyTTA-COF-OH enhanced the photo-oxidation activity for SDZ with a degradation rate as high as 99% within 90 min under visible-light irradiation, which indicated that the presence of hydroxyl groups effectively gained the photocatalytic activity of COFs in line with the above experimental findings. By quantitative analysis, the first-order kinetic rate constant (kobs) of PyTTA-COF-OH for the SDZ degradation under visible-light conditions was calculated to be 0.033 min-1, which was 33 times higher than that of PyTTA-COF-H (0.001 min-1), suggesting that the hydroxyl units of PyTTA-COF-OH had a crucial role in the efficient activation of photocatalytic function (Fig. S9 in Supporting information). Based on the degradation rate constants, PyTTA-COF-OH had excellent photocatalytic activity under low dose conditions compared to other photocatalytic systems (Table S1 in Supporting information). Next, considering the complexity of the actual water environment, the effect on SDZ degradation was further evaluated in the presence of pH differences and different initial concentrations of antibiotics by PyTTA-COF-OH. As illustrated in Fig. 4b, the removal rates of SDZ all reached > 90% via the COFs at the pH values of 3–9, indicating that PyTTA-COF-OH still had good application prospects in the environmental system. At the same time, PyTTA-COF-OH displayed remarkable photocatalytic activity with degradation rates above 99% for initial SDZ concentrations ranging from 1 mg/L to 10 mg/L. When the SDZ concentration solution was as high as 15 mg/L, the SDZ removal rate of PyTTA-COF-OH was still up to 82% (Fig. 4c).
Figure 4
Figure 4.
(a) Photocatalytic performance for SDZ degradation via two COFs under visible light conditions. SDZ degradation rate of PyTTA-COF-OH at different pH (b) and initial concentrations (c). (d) SDZ removal rate under the coexistence of interfering anions and humic acids via PyTTA-COF-OH. (e) SDZ removal ratios by PyTTA-COF-OH over the course of five successive reaction rounds. (f) XRD patterns of PyTTA-COF-OH before and after the reaction.
Furthermore, humic acids (HA) and a variety of ions were present in wastewater systems containing organic matter, and their presence tends to interfere with the degradation of organic matter [49]. As shown in Fig. 4d and Fig. S10 (Supporting information), the SDZ degradation activity of PyTTA-COF-OH was almost unperturbed by HA or various coexisting ions, demonstrating that the material had a strong anti-interference ability in aqueous matrices. Meanwhile, the photocatalytic degradation performance of PyTTA-COF-OH on the other five antibiotics was further investigated (Fig. S11 in Supporting information). The results showed that the degradation rates were all greater than 95%, which indicated that the catalyst had a good general applicability in the field of degrading antibiotics. Based on the favorable properties exhibited via PyTTA-COF-OH, the reproducibility of PyTTA-COF-OH was further evaluated by cycling experiments and PXRD characterization. After five consecutive cycles, the SDZ photocatalytic degradation performance of PyTTA-COF-OH was only slightly attenuated. The PXRD pattern remained virtually unchanged, confirming the preservation of crystallinity (Figs. 4e and f). Meanwhile, no new characteristic peaks appeared in the FT-IR spectra of PyTTA-COF-OH after reaction, indicating that the PyTTA-COF-OH had good stability in the system of visible-light catalytic SDZ degradation (Fig. S12 in Supporting information). In addition, the specific surface area of the PyTTA-COF-OH did not show significant differences for the pre-reaction period. In particular, the decrease in the pore size of PyTTA-COF-OH was mainly attributed to the clogging of the pores by small molecules produced by degradation (Fig. S13 in Supporting information). The above analysis undoubtedly verified that PyTTA-COF-OH had great potential for application in the field of purifying antibiotic wastewater.
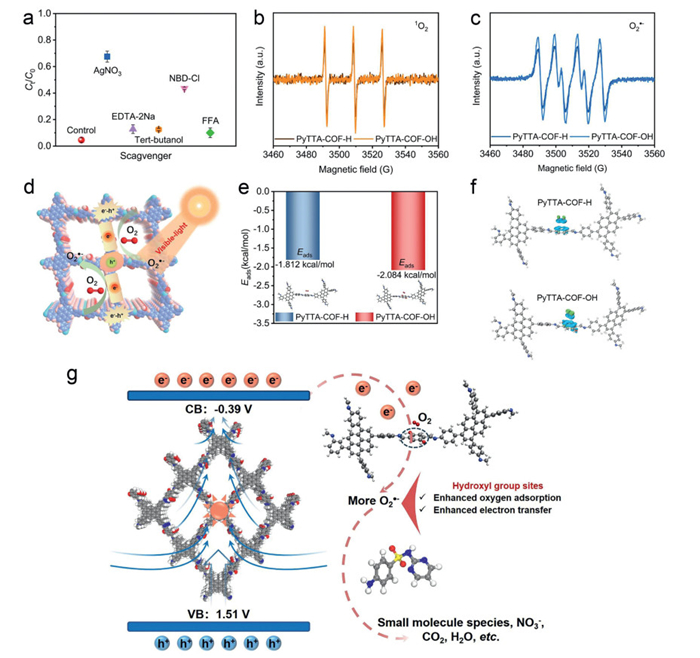
To reveal the reason for SDZ being degraded under visible-light conditions via PyTTA-COF-OH, the contributing behavior of active species in the photocatalytic systems was explored based on a series of quenching experiments (AgNO3 as the scavenger of e-, EDTA-2Na as the scavenger of h+, tert–butanol as the scavenger of •OH, NBD-Cl as the scavenger of O2•-, FFA as the scavenger of 1O2). As shown in Fig. 5a, AgNO3 and NBD-Cl exhibited a strong inhibitory effect on SDZ degradation in the photocatalytic reaction system through PyTTA-COF-OH, while tert–butanol, EDTA-2Na, and FFA did not have a significant effect on SDZ degradation, illustrating that e- and O2•- were the reactive species in the dominant photo-oxidation process. In addition, the electron paramagnetic resonance (EPR) test was utilized to measure the reactive oxygen species (ROSs) generated during the photocatalytic process. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) were used as the trapping agents for the free-radical ROSs (•OH and O2•-) and 1O2, respectively. As illustrated in Fig. S14 (Supporting information), neither PyTTA-COF-H nor PyTTA-COF-OH showed significant signal peaks of DMPO-•OH adducts in the EPR spectra, and the experimental results were strongly supported by quenching experiments as well as energy band structure parameters of the two COFs. Astonishingly, the typical EPR signals of TEMP-1O2 adducts were found for both COFs, this phenomenon contradicted the above experimental findings (Fig. 5b). Based on this, spiked micropollutants (metronidazole (MDE)) and singlet oxygen sensor green (SOSG) were used as monoclinic oxygen probes, respectively, to further monitor the exposure of 1O2via PyTTA-COF-OH under visible-light catalysis [50,51]. Fig. S15 (Supporting information) demonstrates the photocatalytic reaction time of 90 min with < 1% removal in 100 µmol/L MDE solution through PyTTA-COF-OH. Meanwhile, no obvious fluorescence signal at ~525 nm for PyTTA-COF-OH with the increase of irradiation time, the above experimental results sufficiently proved the absence of unilinear oxygen in this system, and the appearance of the typical EPR signal peaks for the TEMP-1O2 adducts was mainly ascribed to the light-induced electron transferred from TEMP to the photosensitizer resulted in the formation of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) with EPR activity and incorrectly indicated the presence of 1O2 (Fig. S16 in Supporting information) [52]. Moreover, the strong signals of DMPO—O2•- adducts appeared in the EPR spectra of PyTTA-COF-OH under the light compared to PyTTA-COF-H, indicating the production from a large amount of O2•- (Fig. 5c). To further confirm the presence of superoxide radicals in the photocatalytic system by PyTTA-COF-OH. The concentration of O2•- was analyzed semi-quantitatively in the system by nitro blue tetrazolium (NBT) reduction experiments over time based on the principle of the mechanism that NBT was solely reduced by O2•-. As shown in Fig. S17 (Supporting information), the absorbance intensity of NBT at 260 nm was significantly reduced with the increase of light exposure time, indicating the effective generation of O2•-. The above experimental conclusions were supported by the energy band structure parameters of PyTTA-COF-OH, which effectively reacted the excited state electrons with adsorbed oxygen (O2) to generate O2•-.
Figure 5
Figure 5.
(a) Contribution of different active species to SDZ degradation during photocatalysis. EPR spectra of 1O2 radicals (b) and O2•- radicals (c) via PyTTA-COF-R. (d) Mechanism of photocatalytic molecular oxygen activation via PyTTA-COF-OH. (e) Oxygen adsorption energy and activation molecular modelling of two COFs on TTA-H and TTA-OH units. (f) Calculated charge density difference for TTA-H units of PyTTA-COF-H and TTA-OH units of PyTTA-COF-OH in the presence of adsorbed oxygen (Green and blue colors represent charge accumulation and charge depletion, respectively). (g) The mechanism for antibiotics degradation via PyTTA-COF-OH under visible light irradiation.
In addition to factors such as energy band structure and intensity of the photoresponse, the adsorption behavior of O2 and the charge transfer pathway on the catalyst surface were particularly important for the generation of superoxide radicals. Based on the results of quenching experiments, the generation of O2•- was reasonably assumed to be an electron transfer process. The pyrene unit acts as a photosensitive active center that generated excitons upon photoexcitation and dissociated into charge carriers due to the energetic disorders within the structure of PyTTA-COF-OH (Fig. 5d) [53]. And then, the electrons tended to transfer to TTA-H or TTA-OH units, and the path of electron transfer within the molecule of COFs was supported by calculations from FMO. Remarkably, the above processes had been extensively studied in semiconductor-based photocatalytic systems [54]. Therefore, oxygen adsorption was critical for the electron transfer process to generate O2•- before and after the introduction of the hydroxyl sites. Based on the above knowledge, the adsorption energy between oxygen and the photocatalyst can be utilized to assess the probability of the electron transfer process. For this, further DFT theoretical calculations were introduced, aiming to evaluate the adsorption energy of O2 on the TTA-H and TTA-OH units of PyTTA-COF-R. The results demonstrated that the binding ability of PyTTA-COF-OH (as shown in the adsorption free energy) to O2 was stronger than that of PyTTA-COF-H based on the surface microenvironmental modulation of hydroxyl sites, which reflected that the insertion of hydroxyl groups effectively enhanced the adsorption and electron transfer process of oxygen on the surface of PyTTA-COF-OH (Fig. 5e). This was one of the reasons why PyTTA-COF-OH was able to produce more O2•-. To gain insight into the above process, the charge density difference between TTA-OH units of PyTTA-COF-H and TTA-H units of PyTTA-COF-OH in the presence of oxygen was further evaluated by theoretical calculations. The results showed that the electron consumption in TTA-OH units was greater than that in TTA-H units, suggesting that hydroxylation of the surface microenvironment readily promoted the transfer of electrons to adsorbed oxygen, resulting in the generation of O2•-, the finding that was mutually consistent with the above observations (Fig. 5f) [55].
Based on the above experimental results, the proposed mechanism of enhanced visible light catalytic efficacy for antibiotics degradation via PyTTA-COF-OH is displayed in Fig. 5g. Firstly, surface hydroxyl functionalization of PyTTA-COF-OH not only reduced the band gap width and thus the reaction barrier but also enhanced the light absorption in the visible band. Meanwhile, quenching and radical trapping experiments further demonstrated that the catalyst system produced more O2•- by PyTTA-COF-OH. In addition, DFT theoretical calculations further demonstrated that the presence of hydroxyl sites induced an asymmetric distribution of localized charges within the molecular structure, which contributed to exciton dissociation and charge carrier separation. More importantly, the insertion of surface hydroxyl sites enhanced the adsorption of oxygen and facilitated the transfer of electrons to the adsorbed oxygen, which built a reaction bridge for the generation of abundant O2•-. Thus, the efficient degradation of antibiotics was realized by PyTTA-COF-OH under visible light irradiation.
In summary, we successfully designed PyTTA-COF-OH with good crystallinity and hydrophilicity as a metal-free photocatalyst for the catalytic degradation of antibiotics under visible light. Antibiotic degradation experiments showed that PyTTA-COF-OH achieved SDZ removal rate of 99% within 90 min of reaction in 10 mg/L SDZ solution. Meanwhile, PyTTA-COF-OH exhibited excellent performance in both cycling and anti-interference experiments. The findings of the quenching and EPR experiments further confirmed that O2•- was the main reactive species involved in the oxidation process. More importantly, the insertion of surface hydroxyl sites not only optimized the molecular structure of PyTTA-COF-OH but also enhanced the oxygen adsorption and promoted the electron transfer process to the adsorbed oxygen. The synergistic strategy combining hydroxylation modulation of the surface microenvironment with the expansion of the π-conjugated structure simultaneously enhanced the interfacial contact between PyTTA-COF-OH and antibiotics, the light absorption capacity, the photogenerated charge-carrier separation efficiency, and the oxygen activation efficiency, which provided a new perspective for the construction of highly efficient COFs-based photocatalysis in the degradation of antibiotics under the real aqueous environment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Science and Technology Department of Sichuan Province (Nos. 2025HJRC0053 and 2024YFHZ0290), the China Education Association for International Exchange (No. 2023261), NSFC (No. 22308235), and Chengdu Science and Technology Bureau (No. 2024-YF06-00013-HZ). The authors would like to thank Shiyanjia Lab (http://www.shiyanjia.com) and Suzhou Deyo Bot Advanced Materials Co., Ltd. (http://www.szdybc.com) for providing support on material characterization.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112143.
X. Dong, F. Zhang, Y. Wang, et al., Appl. Catal. B: Environ. Energy 345 (2024) 123660.
[54]
P. Zhang, Y. Tong, Y. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 16209–16217. doi: 10.1002/anie.202006747
[55]
Y. Qian, D. Li, Y. Han, et al., J. Am. Chem. Soc. 142 (2020) 20763–20771. doi: 10.1021/jacs.0c09727
Figure 1
(a) The schematic representation of PyTTA-COF-R synthesis. XRD images of PyTTA-COF-H (b) and PyTTA-COF-OH (c). (d) N2 adsorption and desorption isotherms of PyTTA-COF-H and PyTTA-COF-OH (The inset of the pore size distributions for the two COFs). (e) FT-IR spectra of monomers and COFs.
Figure 2
The TEM and HR-TEM images of PyTTA-COF-H (a) and PyTTA-COF-OH (b). C 1s XPS spectra (c) and N 1s XPS spectra (d) of PyTTA-COF-H and PyTTA-COF-OH. (e) Images of water contact angle on PyTTA-COF-H and PyTTA-COF-OH.
Figure 3
UV–vis DRS spectra (a), the corresponding Tauc plots (b), and electronic band structure (c) of PyTTA-COF-H and PyTTA-COF-OH. Transient photocurrent responses (d), PL spectra (e), and time-resolved PL decay curves (f) of PyTTA-COF-H and PyTTA-COF-OH. (g) Frontier molecular orbitals of PyTTA-COF-OH. (h) Side view of electrostatic potential distribution for two COFs.
Figure 4
(a) Photocatalytic performance for SDZ degradation via two COFs under visible light conditions. SDZ degradation rate of PyTTA-COF-OH at different pH (b) and initial concentrations (c). (d) SDZ removal rate under the coexistence of interfering anions and humic acids via PyTTA-COF-OH. (e) SDZ removal ratios by PyTTA-COF-OH over the course of five successive reaction rounds. (f) XRD patterns of PyTTA-COF-OH before and after the reaction.
Figure 5
(a) Contribution of different active species to SDZ degradation during photocatalysis. EPR spectra of 1O2 radicals (b) and O2•- radicals (c) via PyTTA-COF-R. (d) Mechanism of photocatalytic molecular oxygen activation via PyTTA-COF-OH. (e) Oxygen adsorption energy and activation molecular modelling of two COFs on TTA-H and TTA-OH units. (f) Calculated charge density difference for TTA-H units of PyTTA-COF-H and TTA-OH units of PyTTA-COF-OH in the presence of adsorbed oxygen (Green and blue colors represent charge accumulation and charge depletion, respectively). (g) The mechanism for antibiotics degradation via PyTTA-COF-OH under visible light irradiation.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
