Figure 1.
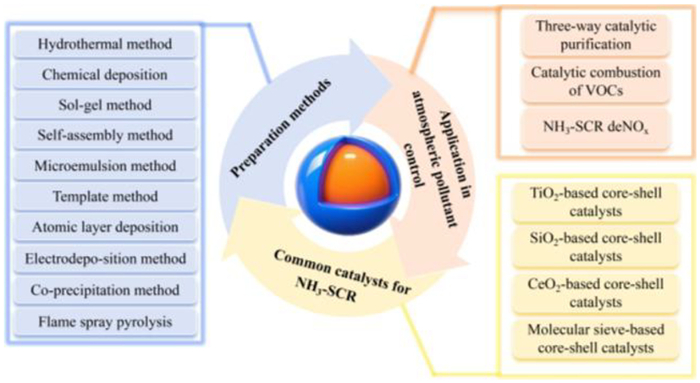
The main contents of this review.
Research progress of core-shell catalysts in the deNOx field
Tao He , Chunxue Wang , Yifei Liang , Tingjie Fu , Kunlin Li , Xin Sun , Kai Li , Ping Ning , Fei Wang

A core-shell structured material is usually a composite structure formed using a nanoparticle as a core and a thin film encapsulated on the surface of the core. Since the 1990s, core-shell materials have experienced rapid advancements in composition, structure, and preparation methods while witnessing a continuous expansion of their application domains. Many kinds of core-shell structural materials, according to their composition, can be classified into inorganic@inorganic, inorganic@organic, organic@inorganic, and organic@organic core-shell materials, etc. Based on morphology and structure, these materials can be categorized into spherical core-shell structure, polygonal core-shell structure (single-layer core-shell structure), multi-core-shell structure, multi-shell core-shell structure, and yolk-shell structure [1]. The composition and structure of core-shell materials can be designed and regulated on the nanoscale, which can effectively integrate the properties of the core and shell components and give full play to the interactions between the core and shell components [2,3]. Based on these characteristics, core-shell structured materials exhibit superior stability and catalytic activity in catalytic applications. Furthermore, core-shell materials can provide a relatively confined microenvironment, facilitating the enrichment and adsorption of reactants [4], thereby enabling widespread application in the catalytic degradation of atmospheric pollutants. This paper primarily reviews the application of core-shell catalysts in several fields, including three-way catalytic purification, catalytic combustion of VOCs, deNOx by the NH3-SCR method, and multi-pollutant synergistic control, with a special focus on deNOx by the NH3-SCR method. In terms of three-way catalytic purification, Li et al. [5] developed a yolk-shell Pd@void@CeO2 core-shell catalyst, which exhibited 80% NO conversion, 100% CO conversion, and 87% C3H8 conversion at 600 ℃. This catalyst is a structurally stable ternary material with high catalytic activity, demonstrating potential for practical application. For the catalytic combustion of VOCs, Li et al. [6] prepared a series of core-shell Mn2O3@δ-MnO2 catalysts with different degrees of orientation growth by a hydrothermal method, among which the Mn2O3@δ-MnO2–8h catalyst showed the most prominent methanol oxidation activity. Core-shell catalysts commonly used in deNOx include TiO2-based, SiO2-based, CeO2-based, and molecular sieve-based core-shell catalysts. For example, the nanocomposite catalyst TiO2@CeMnOx-NC [7] among TiO2-based core-shell catalysts, and the bifunctional core-shell structured catalyst Cu-SSZ-13@Ce0.75Zr0.25O2 (Cu-SSZ-13@CZO) [8] among molecular sieve-based core-shell catalysts, both exhibit excellent NH3-SCR catalytic performance. In addition to being widely used in the treatment of single pollutants, core-shell catalysts also demonstrate significant advantages in the collaborative control of multiple pollutants.
The “shell” of the core-shell material can also act as an effective barrier to protect the core layer. Based on this feature, many researchers have constructed deNOx catalysts with a core-shell structure to improve the stability and sulfur resistance of the catalysts. In the research of core-shell structured catalysts, several reviews have analyzed the application of core-shell structured catalysts in NH3-SCR from the aspects of classification, preparation, catalytic performance, structure-performance relationship [9,10], and anti-poisoning improvement [11,12]. However, no literature systematically summarizes the advantages and disadvantages of the preparation methods and the advantages of core-shell catalysts in deNOx. Our review comprehensively analyzes the advantages and disadvantages of various preparation methods from the perspectives of operation, cost, and particle size, thus facilitating the selection of suitable preparation processes. In addition, the review broadens the application areas by exploring core-shell catalysts in various application scenarios and emphasizes the significant advantages of core-shell catalysts for deNOx, including excellent poisoning resistance, structural regulation, and interfacial synergy. In view of the existing challenges, the review further proposes forward-looking directions such as technology integration, functional integration, and green preparation methods, aiming to promote technological innovation in this field. Our review is expected to provide a practical reference for advancing the application of core-shell catalysts in the field of air pollutants control. The main contents of this review are shown in Fig. 1.
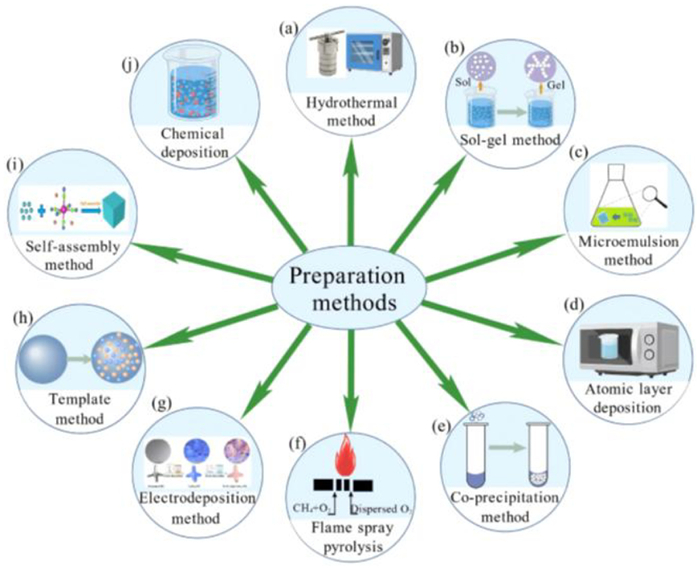
With the increasingly wide and in-depth application of core-shell structure catalysts, researchers have gradually found that the preparation method of core-shell structure catalysts significantly influences the activity and structural properties of the catalysts. In recent years, the preparation technology of core-shell structures has developed rapidly, and the preparation methods of core-shell catalysts applied in air pollution control can be broadly categorized into hydrothermal, chemical deposition, sol-gel, and other methods. As shown in Fig. 2, each technique will be discussed separately in the following sections [13–15].
Hydrothermal is the most used method, and it can control different crystal shapes and pore sizes by adjusting the hydrothermal temperature. Hydrothermal preparation of core-shell catalysts is often categorized into one-step hydrothermal and two-step hydrothermal methods.
Hydrothermal methods generally prepare the core particles first and then use thermal decomposition, hydrogen reduction, laser, or plasma arc radiation to encapsulate the nanoparticles as shells on the surface of the core particles [16]. The core-shell structure catalysts prepared by this method have higher sulfur and water resistance than the supported catalysts prepared by the one-step hydrothermal method. The hydrothermal method is mainly applied to preparing nanomaterials, synthesizing compounds, crystal growth, etc. It can also be used for preparation reactions in small doses. Gong et al. [17] designed and controllably synthesized a series of core-shell structured hydrodenitrogenation (HDN) catalyst supports with H-Beta zeolite nanoparticles as the core and tunable mesoporous materials SBA-16 thin film thickness as shell by hydrothermal method and prepared the corresponding Ni2P catalysts, then tested their catalytic performances for quinoline HDN. The results showed that the catalyst with suitable shell thickness (70 nm) contributed to forming tiny and uniformly sized (4.6 nm) Ni2P nanoparticles with more active sites, which improved the catalytic activity. Tan et al. [18] synthesized Mn@ZSM-5 by hydrothermal approach and then added nano-Ag on Mn@ZSM-5 to prepare Ag-Mn@ZSM-5 core-shell catalyst. Compared with the Ag-Mn/ZSM-5 catalyst prepared by the traditional impregnation method, the Ag-Mn@ZSM-5 core-shell catalyst exhibited superior catalytic performance, remarkable stability, and excellent water resistance.
Zhang et al. [19] synthesized a unique structure of nickel-cobalt (Ni-Co) nanoparticles@Ni0.19Co0.26P nanowires core/shell arrays on Ni foam as a cost-effective and pH-universal electrocatalyst for hydrogen evolution by a simple hydrothermal process and thermal reduction treatment, offering promising prospects for sustainable hydrogen production. Liu et al. [20] utilized a two-step hydrothermal process to first grow a layer of NiCoO4 with spinel structure in situ on nickel foam. Then, they encapsulated a layer of MnO2 nanoparticles and finally calcined the process to obtain the MnO2@NiCo2O4@Ni core-shell nanowire arrays with three-dimensional as an overall deNOx catalyst, which was synthesized as shown in Fig. S1 (Supporting information).
Chemical deposition methods generally prepare nuclear particles first, disperse them in solution, and control the reaction conditions of the system to make the precursor of the shell material react slowly or deposit, adsorb, and penetrate on the surface of the nuclear particles [21]. Li et al. [22] prepared MnOx@CeO2 catalysts with a core-shell structure by a chemical deposition method, in which the catalysts prepared by this method have strong Mn-Ce interactions, thus enhancing the redox cycle between the two components of the core-shell, which can effectively improve the low-temperature activity. The CeO2 shell can act as a protective layer, first reacting with SO2 to inhibit the formation of manganese sulfate. After the addition of SO2, the NOx conversion rate in the MnOx catalytic system decreased by 61%, while the MnOx@CeO2 only decreased by 25%, thus demonstrating a certain sulfur resistance.
The sol-gel method is a common material preparation method, especially for synthesizing metal oxide nanoparticles. The sol-gel method generally includes hydrolysis, polycondensation, drying, and sintering of precursor metal alkoxides. The method first disperses the core particles uniformly into the shell layer precursor solution and then triggers the gelation reaction under certain conditions. The gel is dried, sintered, and solidified, forming a coating layer on the surface of the particles to obtain the core-shell structure catalyst. In the early research, the primary precursor used in the sol-gel method was the organometallic alcohol solution. With the continuous deepening of the study of this method and the broadening of the application range, the selection of precursors is also expanding. For example, some inorganic salts can be used. Chung et al. [23] used the sol-gel method to encapsulate a SnO2 nanoscale film on the surface of the Au substrate, which was precisely adhered to the Au surface with uniformity and integrity. Tariq J. Al-Musawi et al. [24] synthesized a novel core-shell photocatalyst denoted as Fe3O4@SiO2@PAEDTC-doped MIL-101(Fe) by sol-gel method and applied it for the degradation of bisphenol A (BPA) and pyrocatechol (PCT). The catalyst achieved the highest degradation efficiencies of 94.2% for BPA and 100% for PCT. Yao et al. [25] synthesized a series of CeO2@Ce-O-P “multi-core@shell” catalysts by sol-gel method and used them for deNOx. The experimental results showed that the core-shell catalyst CeO2@Ce-O-P increased the activity by about 30% compared to the pure Ce-O-P catalyst in the presence of sulfur. However, the sol-gel method also has some problems: First, the cost of raw materials currently used is relatively high, some of which are toxic and harmful polymer organics; second, there are a large number of micropores in the gel. During the drying process, a lot of gases and organic substances escape and contract, which is easy to cause shell cracks.
The self-assembly method uses the chemical bond or electrostatic force between molecules to self-assemble nanoparticles to the surface of core components in an orderly manner. Cu-SSZ-13@meso-CeO2 core-shell catalysts with microporous and mesoporous structures were successfully synthesized by Xu et al. [26] using a self-assembly technique. Firstly, Cu-SSZ-13 samples were prepared using a one-step hydrothermal preparation technique. Then, the in-situ cladding of mesoporous CeO2 cells was accomplished by using a self-assembly technique to coat the CeO2 shell on the Cu-SSZ-13 surface. Finally, the product Cu-SSZ-13@meso-CeO2 core-shell catalyst can be obtained by drying and calcination. The self-assembly technique can effectively synthesize the materials with microporous and mesoporous structures Cu-SSZ-13@meso-CeO2 core-shell catalyst, and the introduction of CeO2 shell can significantly improve the low-temperature catalytic efficiency, hydrothermal stability and sulfur resistance of the catalyst. Cai et al. [15] constructed MnFeOx@TiO2 double-walled nanocages with a hollow porous structure by annealing the core-shell structure of Mn3[Fe(CN)6]2·nH2O@Ti(OH)4 using an in situ self-assembly method. It has been confirmed by a series of characterizations that the MnFeOx@TiO2 catalyst has a large specific surface area, abundant structural defects and oxygen vacancies, and a strong interaction between MnOx, FeOx, and TiO2 shells, resulting in better catalytic activity. The NO removal rate can reach 80% at 145–260 ℃. Moreover, the MnFeOx@TiO2 catalyst has good thermal stability and water resistance. Xia et al. [27] synthesized 3D Cs3Bi2Br9/FeS2 (CBB/FS) heterojunctions by electrostatic self-assembly strategy, where the CBB/FS Z-scheme heterojunction exhibited exceptional stability and outstanding photothermal catalytic activity.
In recent years, with the increasing requirements on the performance and preparation technology of catalysts with core-shell structure, researchers have developed the layer-by-layer self-assembly (LbL) method based on the self-assembly method to synthesize core-shell catalysts with more complex structures and diversified functions. The principle of LbL is to utilize electrostatic force for layer-by-layer alternating adsorption of polymerized dielectrics or charged particles on the surface of the core layer. Compared with other preparation methods, LbL has more advantages, such as no special requirements on the core layer material’s composition and size, and the shell layer’s thickness can be adjusted by controlling the concentration of polymerized dielectrics or the number of adsorption times. Zhang et al. [28] first functionalized the surface of Fe-ZSM-5 molecular sieve with polycation agent poly(diallyl dimethyl ammonium chloride) (PDDA) to adsorb the cationic surfactant cetyltrimethyl ammonium bromide (CTAB), and then self-assembled the negatively charged SiO2 on the surface of Fe-ZSM-5 through electrostatic attraction to form a mesoporous SiO2 shell. Fe-ZSM-5@SiO2 core-shell catalyst was obtained after calcination and removal of organic matter.
The microemulsion method uses two kinds of insoluble solvents to form microemulsion under the action of surfactant, which then prepares nanoparticles through nucleation, coalescence, and agglomeration. Firstly, it is necessary to select a suitable surfactant, co-surfactant, oil phase, and water phase, and mix them according to a specific ratio to form a stable microemulsion system, and then introduce the reactants; after finishing the reaction, carry out the product separation and treatment to get the final desired product. Min et al. [29] prepared Fe3O4@SiO2 nanoparticles with uniform spherical core-shell structure by the reverse microemulsion method and then fixed Co(Ⅲ)-Salen complexes on Fe3O4@SiO2 nanoparticles through covalent bonding to synthesize Fe3O4@SiO2@Co(Ⅲ)-Salen nanocomposites. Catalytic performance studies showed that the catalyst exhibited high yields of 99% and 100% selectivity for the ring-opening addition reaction of CO2 and epoxides under solvent-free reaction conditions. Zhu et al. [30] prepared highly dispersed Ni@SiO2 catalysts with a unique core-shell structure by a one-pot microemulsion method, which synthesized catalysts with not only spatial limitation but also strong metal-support interactions, and the catalytic activity and stability of the synthesized Ni@SiO2 catalysts were better than those synthesized by other methods. Characterization results showed that the dispersion and sintering resistance of Ni@SiO2 catalysts were significantly improved compared with those prepared by conventional methods.
The template method is a common catalyst preparation method. Its basic principle is to use template materials with specific structures to make the catalyst precursor react on the pore or surface of the template through physical, chemical, or biological methods to prepare catalysts with particular morphology and structure.
The process of preparing core particles and covering their surfaces with the desired shell material is called “bottom-up” preparation. It involves both hard and soft template methods. The most attractive of the “top-down” preparation methods, starting with forming the shell and then introducing the core material, is the self-templating method, in which substances’ inward or outward migration plays an important role [31]. Boyjoo et al. [32] prepared a MnO2 capping layer on the surface of SiO2 using SiO2 nanospheres as a hard template. They obtained SiO2@MnO2 by low-temperature redox precipitation method using the redox reaction between KMnO4 and Mn(NO3)2 at 30 ℃. SiO2@MnO2 core-shell spheres are calcined and the SiO2 template is removed by NaOH etching to obtain MnO2 hollow spheres. Yuan et al. [33] synthesized ZIF-67 hollow spheres in situ by the one-step soft template method, including four steps: Adsorption, nucleation, in-situ growth, and template removal. The ZIF-67 hollow spheres were air-calcined to obtain Co3O4 hollow spheres (Co3O4–HS). Then, HAuCl4 solution and sodium citrate solution were added to the aminated Co3O4–HS; after stirring and centrifugation, Au/Co3O4–HS was obtained, and finally, SiO2 was deposited as a shell layer, successfully preparing the core-shell structured gold-based catalyst mSiO2/Au/Co3O4–HS. Due to the core-shell structure, the mSiO2/Au/Co3O4–HS catalyst maintained excellent reusability and stability over five consecutive cycles.
In practical applications, direct preparation without additional templates is desirable, which can significantly reduce the cost of preparation. The self-template method generally consists of two steps: Synthesizing the template nanomaterial and converting the template into a hollow structure. Self-templating methods have been developed based on different principles, including surface-protected etching, Ostwald ripening, the Kirkendall effect, galvanic replacement, and so on [34]. Li et al. [35] synthesized TiO2@NM/Ni10% hollow double-shell layer heterostructures in situ using tannic acid as a protecting agent. Usually, NM/Ni10% is unstable in water and will be hydrolytically etched into TiO2 particles. Researchers utilized the instability and selected tannic acid as a protective agent for the NM/Ni10% outer layer. The formation of Ti4+/Ti3+ double-cycle electron transfer channels between NM/Ni10% and TiO2, as well as the strong built-in electric field established by the type-Ⅱ heterojunction accelerated the separation and transport of photogenerated supports, which greatly improved the catalytic efficiency.
In addition to the above six widely used methods for preparing core-shell catalysts, there are other preparation methods, such as atomic layer deposition, electrodeposition, co-precipitation, flame spray pyrolysis. The atomic layer deposition method enables the precise regulation of the shell layer thickness of core-shell catalysts down to the atomic scale. Zhang et al. [36] combined the deposition-precipitation (DP) method with the atomic layer deposition (ALD) method to prepare a large number of Au@Pd/SiO2 catalysts on SiO2 carriers by precisely regulating the Au core size and Pd shell thickness. Au@Pd catalysts with shell thicknesses of 2–3 layers obtained record-high activities in the benzyl alcohol oxidation reaction. Zhao et al. [37] first prepared PtCu3/C nanoparticles by wet chemical method, then deposited Pt shells on PtCu3/C by atomic layer deposition (ALD) technology and successfully synthesized a series of PtCu3@Pt/C core-shell catalysts. Changing the number of ALD cycles between 1 and 6 to precisely adjust the Pt-shell thickness revealed that four ALD cycles can deposit about one layer of Pt atoms on the surface of PtCu3 nanoparticles. The Pt-shell thickness of the PtCu3@PtALD-4/C catalyst fully met the requirements of the ideal core-shell structure, showing the best catalytic performance of all catalysts. Wen et al. [14] prepared OER electrocatalysts Fe-Ni-S@CoSe2/NF with Fe-Ni-S nanoparticles encapsulated on the surface of CoSe2 ultrathin nanosheets using nickel foam (NF) as a substrate by a two-step electrodeposition method. The incorporation of Fe-Ni-S significantly improved the OER and water electrolysis performance of Fe-Ni-S@CoSe2/NF composites. Zhang et al. [38] synthesized trimetallic heterogeneous core-shell nanoboxes based on Prussian blue analogues (Ni-Co@Fe-Co PBA) with by iterative co-precipitation method. Ni-Co PBA truncated nanoboxes were prepared using potassium hexacyanocobaltate(Ⅲ) as an organic linker and nickel ions as metal nodes. Then, under the same reaction conditions, Fe-Co PBA shells were synthesized with identical ligands and ferrous ion nodes. Finally, the highly active Ni-Co PBA core was wrapped in the strong Fe-Co PBA shell to obtain Ni-Co@Fe-Co PBA nanoboxes with core-shell structure. FSP, as an innovative and advanced method for synthesizing nanomaterials, plays a crucial role in producing various metal oxide nanoparticles with customized morphologies and compositions. By skillfully tuning the operating parameters—such as precursor solution concentration, solvent type, flame temperature, oxygen-fuel ratio, and residence time of the particles in the flame zone—the researchers can efficiently control the nanoparticle size, distribution, and phase composition [39]. Zhou et al. [40] prepared core-shell catalysts CuOx@SiO2 by flame spray pyrolysis (FSP), the FSP system is shown in Fig. S2 (Supporting information). The precursor solutions were fed into the nozzle via a syringe pump to form a spray. CuOx nanoparticles were formed in the spray flame through pyrolysis, nucleation, surface growth, coagulation, and coalescence. Then, the precursor vapors of the shells were introduced into the spray flame to form the SiO2 coatings. Finally, the CuOx@SiO2 particles were collected on a glass filter above the combustor by a vacuum pump. FSP is a one-step and sequential method to synthesize nanomaterials with high thermal stability, high crystallinity and high purity.
We have summarized the advantages and disadvantages of different methods (Table S1 in Supporting information), which vary in terms of controllability, efficiency, cost and product quality. High-precision preparations (e.g., atomic layer deposition, self-assembly methods) are often accompanied by high cost or material limitations; however, the low-cost methods (e.g., chemical deposition, electrodeposition) are susceptible to the influence of reaction conditions or contamination issues; meanwhile, the preparation methods under mild-conditions (e.g., sol-gel, microemulsion methods) face challenges such as cracking or residues. Overall, method selection must be combined with the precision requirements of the target material, production scale and environmental friendliness.
Based on the above characteristics, we can briefly analyze the applicable scenarios of each preparation method: Chemical deposition and electrodeposition can be used for large-scale production; atomic layer deposition is suitable for high-precision small-scale preparation; self-assembly or atomic layer deposition can be chosen for those with special requirements on the thickness of the shell layer, and the template or sol-gel method is suitable for the porous structure; the catalytic system that needs to regulate the structure of the pores can be chosen. The hydrothermal method can control the mesoporous structure of the shell layer by adjusting the hydrothermal temperature and time. To address the challenges of high cost and complex processes associated with core-shell catalysts, the improvement efforts should focus on optimizing low-cost preparation methods (e.g., electrodeposition, co-precipitation), reducing by-products (e.g., adopting green reagents or biodegradable templates), and synergizing with composite methods (e.g., combining sol-gel and self-assembly). Meanwhile, the efficient, environmentally friendly, and economical preparation of core-shell catalysts can be achieved through scale-up adaptation (continuous flow reactors) and the precise application of high-cost technologies (e.g., atomic layer deposition is used only for critical steps).
Due to their unique structure, the core-shell catalysts can effectively enhance the interaction between the core and shell components and show better catalytic activity and stability than the traditional supported catalysts. Meanwhile, due to the significant mobility and relatively small concentration of atmospheric pollutants, the relatively closed micro-environment inside the core-shell catalysts is conducive to the enrichment and adsorption of the atmospheric pollutants and thus has received extensive attention from many scholars. At present, core-shell catalyst has some application research in the field of atmospheric pollutant control, especially in the three-way catalytic purification, catalytic combustion of VOCs, and NH3-SCR, as shown in Fig. S3 (Supporting information). The following will review the three aspects, especially the research progress of core-shell catalysts in deNOx.
The primary pollutants in gasoline vehicle exhaust are nitrogen oxides (NOx), carbon monoxide (CO), and hydrocarbons (HC), which can cause environmental problems such as acid rain and photochemical smog when discharged into the atmosphere. At present, the three-way catalytic technology is one of the most effective technologies in the gasoline vehicle engine external purification technology, and the three-way catalyst (TWCs) has been proven to be the most effective material to reduce the automobile exhaust emissions because of its high selectivity of CO, HC, and NOx in the automobile exhaust. The noble metals platinum (Pt), rhodium (Rh) and palladium (Pd) have excellent activity in lowering the reaction temperature and are often used as the main active components of TWCs, but the noble metals have the disadvantages of scarce resources and high prices; On the other hand, Al2O3 with high specific surface area, CeO2 with high oxygen storage capacity, and ZrO2 with mechanical stability are widely used as supports for TWCs [41–43]. However, the poor heat resistance of these supports may lead to severe sintering and collapse of the catalyst at high temperatures [44,45]. In addition, weak interactions between the active centers and the supports cause the catalyst to migrate and aggregate, leading to catalyst deactivation. Therefore, developing catalysts with high catalytic efficiency, low cost, and good stability is of great research significance. Currently, the core-shell structure has been utilized to enhance the interaction between the active components of the noble metal and the support, improve the dispersion of the metal, and inhibit the migration and aggregation of the active components [46].
Ozawa et al. [47] proposed a new method using core-shell CeO2/ZrO2(csCZ) as a support to reduce the Pt content in a TWC. Analysis showed that 0.1 wt% Pt/csCZ had better activity than 0.1 wt% Pt/CeO2. The core-shell CeO2/ZrO2 support enhanced the catalytic activity of the low-content Pt catalyst. Due to the separation morphological characteristics of CeO2 nanoparticles on ZrO2, the agglomeration of Pt was inhibited, and the csCZ support plays an essential role in the reduction of Pt. Wei et al. [48] intercalated well-dispersed Pd nanoparticles into halloysite nanotubes (HNTs) to form Pd@HNTs. The results showed that Pd@HNTs have good dispersion and stability due to the strong interaction between Pd and HNTs with Schiff base ligands and the high thermal resistance of HNTs as a sintering barrier. Then, a CeO2 film was coated on the Pd@HNTs to form a Ce@Pd@HNTs catalyst, and CeO2 acted as a promoter and a protector to provide more oxygen vacancies for promoting NO reduction and CO and C3H8 oxidation, thus further improving the catalytic activity. The performance of more core-shell catalysts in three-way catalytic studies is listed in Table S2 (Supporting information) [49–55].
Volatile organic compounds (VOCs) are a kind of pollutant that can cause serious harm to the atmospheric environment, mainly from transportation, industrial production processes, and life activities, etc. VOCs can contribute to the formation of photochemical smog and destroy the ozone layer. The catalytic combustion method is one of the most promising methods for dealing with the above problems because of its apparent advantages, such as low ignition temperature, less energy consumption, high treatment efficiency, and no secondary pollution [56]. Precious metal catalysts are the industry’s most widely used VOC catalysts, with advantages like superior low-temperature activity, long service life, and good selectivity. The activity of precious metal catalysts is usually better than non-precious metal oxide catalysts. However, the disadvantage is that it is easy to be poisoned by carbon, chloride, or water vapor on the catalyst surface. The catalytic properties of precious metal catalysts are related to their content, dispersion, oxidation state, and interaction with the support [57,58]. Constructing a core-shell structure can improve the dispersion of the precious metal catalyst. In addition, enhancing the interaction between the precious metal and the support can improve the low-temperature catalytic activity.
Peng et al. [59] prepared a linear core-shell catalyst (Pt-CeO2NW@SiO2) by reverse-micellar emulsion. Due to the high dispersion of platinum, strong Pt-CeO2 interaction, and the protective effect of porous silicon shell on Pt nanoparticles, Pt-CeO2NW@SiO2 had a high low-temperature catalytic efficiency in toluene catalytic activity experiments (1000 ppm, GHSV = 20,000 mL g−1 h−1), and the conversion of toluene can reach 90% at 170 ℃. The unique core-shell structure also retained excellent sintering resistance up to 700 ℃ for 100 h. Ren et al. [60] proposed a bottom-down design to prepare ZIF-derived Co3O4, denoted as α-MnO2@Co3O4, grown in situ on a one-dimensional (1D) α-MnO2 material. The obtained α-MnO2@Co3O4 catalyst required a temperature of about 229 ℃ to reach 90% toluene conversion, which exhibited excellent catalytic activity compared with the pure MnO2 and Co3O4 catalysts, and the synergistic effect derived from the coupled interface established between α-MnO2 and Co3O4 was the main reason for the improved catalytic activity. The increased number of adsorbed oxygen species on the surface accelerated oxygen transport and enhanced the redox pairs of Mn4+/Mn3+ and Co2+/Co3+. In addition, the α-MnO2@Co3O4 catalyst exhibited excellent stability and water resistance to the toluene oxidation reaction. He et al. [61] used a facile and eco-friendly method to prepare Al2O3@CoMn2O4 core-shell microspheres with a hollow hierarchical structure. Introducing hollow Al2O3 led to a well-defined hollow layered structure, resulting in a more accessible active site and enhancing the interaction between CMO nanosheets and Al2O3 nanospheres. In addition, adding Pd NPs further improved the catalyst’s reduction capacity, oxygen mobility, and storage capacity, so the 1% Pd/h-Al@4CMO catalyst was successfully synthesized. The catalytic activity test for VOCs was carried out, and the results showed that toluene, benzene, and ethyl acetate could achieve 100% conversion at 165, 160, and 155 ℃, respectively. And 1% Pd/h-Al@4CMO has excellent catalytic activity for the three kinds of VOCs, which is a promising catalyst for VOC combustion.
Other core-shell structures, such as Pt-Ni(O)@SiO2 [62] and Al2O3@Pd-CoAlO [63], also exhibited significant catalytic performance in the low-temperature combustion of toluene. The performance of more core-shell catalysts in catalytic combustion of VOCs studies is listed in Table S2 [64–76].
Nitrogen oxides (NOx) from fossil fuel combustion and diesel exhaust emissions cause environmental problems such as photochemical smog, acid rain, and the hole in the ozone layer, as well as increasing threats to human life and health. Ammonia selective catalytic reduction (NH3-SCR) is one of the most efficient and widely used deNOx technologies.
Wang et al. [77] synthesized MnCeOx@PrCeOx core-shell deNOx catalysts with hollow structures by chemical precipitation method. The large specific surface area and well-dispersed active sites of the MnCeOx@PrCeOx catalyst, together with the strong interactions among Ce, Mn, and Pr, resulted in the catalyst exhibiting remarkable NH3-SCR activity, with >90% NO conversion and 100% N2 selectivity at 90–270 ℃. Bai et al. [78] prepared core-shell structure catalyst Co3O4@CoMn2O4 through the decomposition of ZIF-67 and the introduction of manganese and used it for the selective catalytic reduction of nitrogen oxides (NH3-SCR) with ammonia. Cobalt and manganese oxides showed good catalytic activity in the SCR reaction at low temperatures, but N2O was easily generated when the temperature was higher than 200 ℃. Core-shell structure catalyst Co3O4@CoMn2O4 exhibited excellent NOX conversion at 90–270 ℃, and the addition of SO2 and H2O inhibited the formation of N2O, and the selectivity of N2 was significantly improved. Xu et al. [26] prepared Cu-SSZ-13@meso-CeO2 core-shell catalysts with microporous and mesoporous structures by self-assembly technique. Due to introducing the CeO2 shell layer, the synergistic effect between the reducing substances increased the number of acidic sites and Cu2+−2Z substances. It enhanced the redox capacity, which resulted in the excellent deNOx activity of Cu-SSZ-13@meso-CeO2 catalysts, with the NO conversion rate of >90% at 175–470 ℃. Core-shell catalysts have a wide range of applications, and the performance of many core-shell catalysts in the study of NH3-SCR is listed in Table S2 [79–94]. To highlight the advantages of core-shell catalysts, we have listed some deNOx performance data of conventional catalysts in Table S2 [95–100], it can be observed that core-shell catalysts achieve higher deNOx efficiency at lower temperatures.
In addition to its wide application in single pollutants purification, the core-shell catalysts also demonstrate significant advantages in multi-pollutant synergistic control, with efficient and stable catalytic performance. For example, Li et al. [101] synthesized a core-shell structured catalyst, Cu-SSZ-13@Mn2Cu1Al1Ox, by an in situ growth method, which achieved simultaneous 100% removal of VOCs and NOx at 300 ℃ through charge transfer and multiple active sites in the core-shell structure, while exhibiting excellent water resistance. Similarly, Pan et al. [102] designed a MnOx@CeO2@MgO core-shell catalyst applied for the simultaneous removal of NOx and chlorobenzene (CB). The protective effect of the MgO shell was utilized to significantly enhance the synergistic removal efficiency of CB and NOx in SO2-containing environments. The sulfur resistance was attributed to the preferential reaction of MgO with SO2 to form sulfate, protecting the core active component and increasing the proportion of lattice oxygen. These studies show that the core-shell structure can not only optimize the redox properties and distribution of acidic sites in the catalyst but also inhibit the competitive adsorption of pollutants through the synergistic effect between components, thus providing an innovative solution for the efficient synergistic control of multiple pollutants in complex industrial exhaust gases.
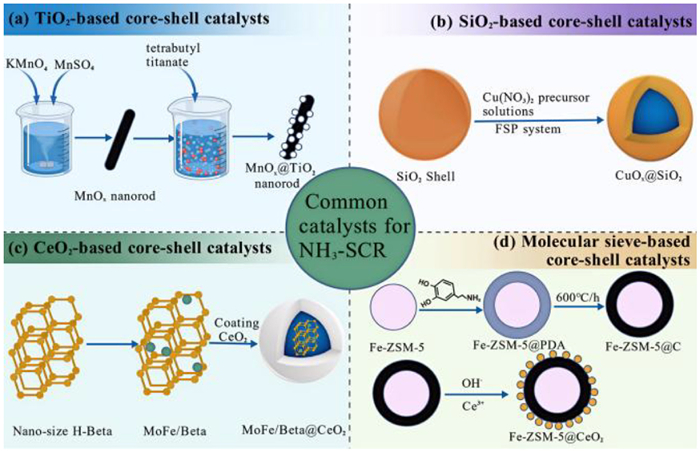
Researchers need to develop catalysts with excellent low-temperature activity, strong poisoning resistance, and good hydrothermal stability to solve the challenges faced by current deNOx catalysts. The composition of core particles and shell mainly determines the performance of the core-shell catalyst. In general, the active components are used as core particles. The shell layer components do not have catalytic activity. The shell layer mainly plays a protective role to avoid poisoning and inactivating the active components in contact with SO2, H2O, and other harmful components. A few researchers have prepared core-shell deNOx catalysts with active sites on the core and the shell simultaneously [20,86]. The NO in the gas stream is first oxidized to NO2 in the shell layer, which can realize the “fast SCR” reaction, and the strong interaction between the core and shell components accelerates the redox cycle to improve the low-temperature catalytic activity. As shown in Fig. 3, the commonly used components of core-shell catalysts for deNOx mainly include TiO2 [88], SiO2 [40], CeO2 [103], and molecular sieve [95]. The paper will review these four types of core-shell catalysts for deNOx based on the properties and functions of the above commonly used components.
TiO2 is widely used in deNOx as the support of supported catalysts, especially vanadium-based catalysts with TiO2 as the support have become commercial deNOx catalysts. Although many researchers have been involved in the preparation of TiO2-based core-shell catalysts, the TiO2-based core-shell catalysts used in NH3-SCR reactions reported in most works of literature are mainly materials with metal oxides as the core and TiO2 as the shell. Most of the TiO2-based core-shell deNOx catalysts previously reported used titanium nanotubes as shells [88,104]. Granular metal oxide@TiO2 core-shell catalysts have been gradually synthesized for NH3-SCR reaction [84,97]. The mesoporous structure and abundant acid sites of the TiO2 shell prevent the formation of ammonium sulfate species and act as an effective barrier to inhibit the sulfation of the active center, resulting in high SO2 resistance. Meanwhile, TiO2 shell coated on the outer surface of metal particles can also act as an effective barrier to prevent the aggregation of metal particles, thus improving the thermal stability of the catalyst [104].
Jiang et al. [105] developed a new low-temperature SCR catalyst, Cu-HPMo/TiO2, with a core-shell structure. The test results showed that the deNOx efficiency of Cu(3)-HPMo/TiO2 at 200 ℃ was close to 99%, and the Cu(3)-HPMo/TiO2 had a firm surface acidity, which inhibited the adsorption of SO2 and showed a good anti-SO2 performance at the SO2 concentration of <400 ppm. Huang et al. [7] elaborated and prepared the nanocage-confined composite catalyst TiO2@CeMnOx-NC by directional hydrolytic etching of MIL-125. MnOx was encapsulated in the cavities of TiO2 nanocages, and CeO2 was mainly anchored on its outer surface. Characterization results revealed that amorphous TiO2 formed after annealing, and CeO2 with fluorite cubic structure inhibited the crystallization of MnOx clusters, which led to strong metal loading interactions between Ce/Mn oxides and TiO2. Therefore, the catalyst exhibited excellent NH3-SCR catalytic performance over a wide temperature range. In addition, thanks to the double protection of TiO2 nanocages and CeO2 on the outer surface, the MnOx encapsulated in the cavity could be effectively protected from water vapor and SO2 in the flue gas, thus exhibiting good water and sulfur resistance. As shown in Fig. S4 (Supporting information), TiO2@CeMnOx-NC has better catalytic activity, water resistance, and sulfur resistance compared to other catalysts.
Nano-SiO2 has potential application value in catalyst and catalyst supports due to its advantages of large specific surface area, high porosity, and abundant acidic sites [106,107], so it is one of the commonly used catalyst supports. Zhou et al. [40] selected SiO2 as shell material and CuOx as core material to synthesize core-shell catalyst MnOx@SiO2 with high thermal stability, crystallinity, and purity by flame spray pyrolysis (FSP). The results of the activity tests showed that CuOx@SiO2 possessed a higher N2 selectivity and better thermal stability than CuOx. SEM and TEM results showed that the core-shell structure significantly improved the sintering resistance of CuOx@SiO2.
Meso-SiO2 shells are easily obtained by colloidal chemical methods [108]. Therefore, the main focus is preparing SiO2-based core-shell catalysts applied in NH3-SCR reactions using Meso-SiO2 as the shell material. Zhang et al. [109] synthesized a catalyst SiO2@FeCeOx/CNTs with a core-shell structure, as shown in Fig. S5 (Supporting information), which improved the stability and SO2 resistance of the catalyst by establishing an effective barrier in the SiO2 shell layer, hindered the generation of FeSO4 and inhibited the reduction of chemisorbed oxygen as compared to FeCeOx/CNTs without SiO2 encapsulation. Although the SiO2 shell layer was beneficial in improving the catalytic performance and sulfur resistance, excessive SiO2 rather decreased the catalytic activity because the SiO2 shell layer improved the catalytic performance of the catalyst by increasing the catalyst’s specific surface area and chemisorbed oxygen content, and excessive SiO2 could lead to an increase in the catalyst’s acidic sites, and the higher acid strength adsorbs too much NH3, which could not be efficiently used for the conversion of NO to N2 and thus inhibited the catalytic activity. Therefore, when SiO2-based core-shell catalysts are applied to deNOx, the amount of SiO2 added, that is, the thickness of the Meso-SiO2 shell layer, should be fully considered.
CeO2 has robust oxygen storage and release capacity. It is often added as an additive to the catalyst system to improve the redox properties and low-temperature activity [110]. Nkinahamira et al. [111] synthesized a series of In/H-Beta@Ce core-shell catalysts using In/H-Beta as the core and CeO2 as the amorphous shell. They investigated the effect of challenging conditions, such as SO2 and H2O, on deNOx performance. The results showed that the amorphous CeO2 shell layer enhanced the SO2 and H2O tolerance by stabilizing the InO+ active site, promoting redox cycling, and reducing the formation of sulfate substances, significantly improving the catalyst’s performance. CeO2-based core-shell deNOx catalysts reported in many works of literature are divided into two categories. One is CeO2 as a core material, which acts as the main active component or additive to enhance the low-temperature activity of the catalyst [84]. The second is that CeO2 is used as the shell; the CeO2 shell primarily serves as a sacrificial site to preferentially contact and react with SO2, thereby protecting the active center from sulfation and enhancing the sulfur resistance of the catalyst [22].
Liu et al. [103] prepared a MoFe/Beta@CeO2 core-shell catalyst with a nanoscale Beta support MoFe bimetallic oxides as the core and a CeO2 film as the shell layer. The results of NH3-SCR tests on NO showed that the activity of the MoFe/Beta@CeO2 catalyst was significantly better than that of the catalyst without the CeO2 shell layer. In the presence of 100 ppm SO2, the catalytic performance of the MoFe/Beta@CeO2 catalyst was almost unaffected, and the NOx conversion was still maintained at about 98%, indicating that the MoFe/Beta@CeO2 catalyst has strong resistance to SO2 poisoning. The characterization showed that the CeO2 shell layer could effectively inhibit the formation of ammonium sulfate substances at low temperatures, preventing SO2 from poisoning the active components on the MoFe/Beta@CeO2 catalyst. The migration and aggregation of active metal oxides were prevented at high temperatures, which improved the stability of the catalyst. Chen et al. [95] prepared Fe-ZSM-5@CeO2 core-shell structure catalyst. The synergistic effect between Fe-ZSM-5 and CeO2 promoted the redox cycle between Ce3+/Ce4+ redox-couple and high-rate active oxygen species. Reactive oxygen species in the CeO2 shell promoted the conversion of NO to NO2, which was conducive to rapid SCR. The formed NO2 also activates Fe2+/Fe3+ redox-couple, providing an additional redox cycle promoting rapid SCR. CeO2 shell could also improve the sulfur and water resistance of Fe-ZSM-5. When Fe-ZSM-5@CeO2 was exposed to 10% H2O and 100 ppm SO2, the NOx conversion rate decreases by 8%, showing excellent sulfur and water resistance.
Molecular sieves are widely used in synthesizing catalytic materials because of their hydrophobicity, large specific surface area, abundant acidic sites, and good structural stability. Currently, the application of ion-exchanged molecular sieves in NH3-SCR is becoming increasingly attractive, among which Fe or Cu ion-exchanged molecular sieves are more widely studied and applied. The main reasons hindering their practical applications are the poor low-temperature activity, hydrothermal stability, and sulfur resistance of ion-exchanged molecular sieve catalysts. Constructing a core-shell structure is conducive to improving the performance of ion-exchanged molecular sieve catalysts. In preparing core-shell deNOx catalysts, ion-exchanged molecular sieves can be used as both the core material and the shell layer.
Jia et al. [8] designed and prepared a bifunctional core-shell catalyst Cu-SSZ-13@Ce0.75Zr0.25O2 (Cu-SSZ-13@CZO) with Cu-SSZ-13 as core and dispersed CZO as a shell by using hydrothermal induced self-assembly method. They systematically studied the activity of Cu-SSZ-13@CZO for the selective reduction of nitrogen oxides (NOx) with ammonia. The results showed that the NOx conversion rate of Cu-SSZ-13 was almost 100% in the temperature window of 275–475 ℃, compared with the original Cu-SSZ-13 at 350–450 ℃. In addition, the CZO shell could serve as the sacrifice site of the Cu-SSZ-13 active core, protecting the Cu-SSZ-13 active core from SO2 poisoning formed by Ce2(SO4)3. When the molecular sieve is used as the shell material, the adsorption activation process of NH3 and NO is significantly affected by the shell composition, pore structure, and thickness. Microporous molecular sieves such as SAPO and SSZ-13 are unsuitable as shell materials because the diffusion resistance limits the reactant’s accessibility to the active site on the core of the core-shell catalyst [112].
Chi et al. [113] prepared a CeO2@Fe-ZSM-5 (CeO2 as core and Fe-ZSM-5 as shell layer) hollow-structured catalyst with significantly better H2O/SO2 tolerance than CeO2 and Fe-ZSM-5 catalysts. The Fe-ZSM-5 shell layer could oxidize NO to NO2, preventing the agglomeration of nano-CeO2 and avoiding the direct contact between CeO2 and SO2. The hollow structure reduced the ineffective region of the catalyst and improved its internal efficiency.
The performance of core-shell catalysts for deNOx applications, which require a balance of low-temperature activity, resistance to poisoning, and hydrothermal stability, depends primarily on the selection of core-shell components. TiO2-based core-shell catalysts exhibit excellent low-temperature activity and sulfur resistance but are less stable at high temperatures than other systems, making them suitable for low and medium-temperature flue gases; SiO2-based core-shell catalysts are ideal for high sulfur and water vapor environments under high hydrothermal stability and the inert protective shell layer, though they rely on the core layer for active sites; CeO2-based core-shell catalysts are excellent in low-temperature deNOx due to their outstanding oxygen storage capacity and redox properties, but need to be optimized for sulfur resistance; molecular sieve-based core-shell catalysts have regular pores and firm acidity, and have excellent high-temperature stability, but are costly and sensitive to sulfur. Future research directions can further enhance the comprehensive performance of core-shell catalysts through the design of composite shell layers, such as a TiO2-SiO2 hybrid shell layer that exhibits both high activity and stability. Acid modification strategies, including the grafting of acidic sites onto inert shell layers (e.g., SiO2), represent a viable approach to further optimize the comprehensive performance of core-shell catalysts. This modification method enables the catalysts to better adapt to diverse industrial application scenarios and fulfill specific functional requirements.
Core-shell catalysts are widely used in air pollutant control, especially in removing nitrogen oxides with relatively more applications. In actual operation, fossil fuel combustion and diesel exhaust contain not only a large amount of NOx but also SO2, H2O, and some acid gases such as HCl, which can cause problems such as agglomeration of active species on the catalyst, structural collapse, and poisoning deactivation. Protecting the active sites from poisoning inactivation and enhancing the catalyst’s hydrothermal stability is necessary to meet various complex operating conditions. The unique structure and excellent performance of core-shell catalysts enable their broad application in addressing the limitations of existing deNOx catalysts. The paper will review the advantages of core-shell catalysts in the selective catalytic reduction of nitrogen oxides by ammonia from the following three aspects.
The core-shell structure exhibits excellent corrosion resistance and enhanced stability, while the newly-formed active heterointerface demonstrates favorable synergistic effect [114]. Because of its unique structure, the core-shell catalyst can maximize the interface area of the interaction between the “core” and “shell” components and fully play the synergistic effect between different elements. At the same time, it can effectively improve the dispersion of each component and reduce the amount of expensive metals. Wang et al. [115] prepared a core-shell deNOx catalyst Fe3O4@TiO2 with Fe3O4 as core and TiO2 as shell and investigated the catalytic efficiency of Fe3O4@TiO2 core-shell catalyst and Fe3O4/TiO2 supported catalyst in desulfurization and deNOx process. The results showed that the core-shell catalysts were superior to those supported in physicochemical properties, removal efficiency, and stability. The characterization results yielded that the core-shell catalysts with very favorable pore structure and large specific surface area contribute to the enhancement of low-temperature catalytic activity through the synergistic interaction between the core and shell components, which promotes redox pairing cycling, maintains high Fe2+ concentration and accelerates H2O2 activation for NO oxidation. Zhang et al. [28] prepared a Fe-ZSM-5@Ce/Meso-SiO2 core-shell catalyst. The characterization results showed that Ce was mainly distributed in the pores of the Meso-SiO2 shell layer rather than on the outer surface of the shell layer. The Meso-SiO2 shell layer had abundant pores and a large specific surface area, which was mainly used to increase the catalyst’s specific surface area and improve the dispersion of the active component Ce. Thus, part of the NO in the gas stream is oxidized to NO2 by Ce on the shell layer and then, together with the remaining NO gas, undergoes a rapid SCR reaction on the Fe-ZSM-5 core layer, which was efficacious in improving the catalyst’s low-temperature activity. Jiang et al. [116] synthesized the core-shell Cu/Beta@CeO2 catalyst using Cu/Beta as the core and CeO2 as the shell by the self-assembly method for simultaneous removal of NO and toluene. The test results showed that the CeO2 shell increased the number of Brønsted acid sites and promoted the adsorption of NH3 and toluene. The intense Cu-Ce core-shell interfacial interaction generated Ce3+ and isolated Cu2+ in large amounts, which played an important role in improving catalytic performance. Compared with a series of copper-doped Cu/Beta@CeO2 catalysts, 1.5% Cu/Beta@CeO2 showed the most substantial core-shell interaction and better catalytic performance. NO removal efficiency exceeded 80% in the range of 250–400 ℃, and T50 and T90 of toluene were 157 ℃ and 298 ℃, respectively. Furthermore, 1.5%Cu/Beta@CeO2 showed strong resistance to SO2.
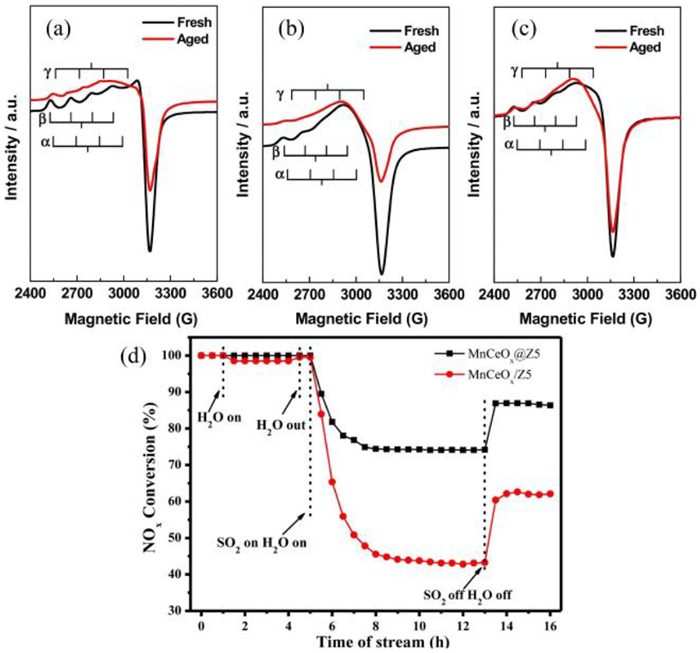
The shell components of core-shell catalysts often have a protective effect, which can effectively reduce the exposure of the active site to SO2, H2O, and other harmful components, prevent the sulfation of the active components and the formation of byproducts such as ammonium bisulfate to a certain extent, and inhibit H2O and some acidic gases leading to catalyst poisoning inactivation. At the same time, the shell can be used as an effective barrier to prevent the agglomeration of nanoparticles at high temperatures and enhance the thermal stability of the catalyst. Zhang et al. [117] introduced a novel core-shell composite material, Cu/SSZ-13@CeO2. Characterization results indicated that encapsulating Cu/SSZ-13 with a CeO2 shell markedly enhances hydrothermal stability by maintaining the functionality of [Cu(OH)]+ active sites and averting their deactivation. Yang et al. [118] developed a novel Fe3O4@TiO2-Ce core-shell catalyst for removal by the heterogeneous Fenton method. The characterization results showed that the number of TiO2 particles encapsulating the Fe3O4 core increased significantly after Ce doping, indicating that Ce doping affected the hydrolysis of tetrabutyl titanate (TBOT) and promoted the deposition of TiO2 on the surface of Fe3O4. The shell layer of TiO2 had a protective effect on the core of Fe3O4, and the doping of Ce could enhance the water resistance of the catalysts by preventing the entry of H2O into the interface of the two phases and inhibiting the formation of hydroxyl groups at the active sites. Zhang et al. [119] prepared the core-shell structured catalyst meso-Cu-SSZ-13@MAS (mesoporous aluminosilicate) using a combination of alkaline desilication and self-assembly. The hydrothermal aging Cu-SSZ-13 and meso-Cu-SSZ-13 catalysts lost the NH3-SCR activity, and the core-shell structured meso-Cu-SSZ-13@MAS catalysts still showed better activity. The EPR results showed that hydrothermal treatment had significantly different effects on the active sites of the three catalysts. As can be seen from Figs. 4a-c, two major Cu2+ species (labeled as α and β) were present in the three fresh samples. After hydrothermal aging, the α and β species in Cu-SSZ-13 and meso-Cu-SSZ-13 decreased, and new Cu2+ species (labeled as γ) appeared, indicating the collapse of the zeolite structure in both samples. In contrast, both α and β species on meso-Cu-SSZ-13@MAS had minimal changes, and almost no γ species were formed, suggesting the high hydrothermal stability of meso-Cu-SSZ-13@MAS. In addition, propene caused coke deposition on the active component, reducing catalytic performance. In contrast, the meso-Cu-SSZ-13@MAS catalyst had fewer active sites for propylene oxidation reaction, so the catalyst also had a high resistance to propylene poisoning. Yan et al. [96] mainly adopted the idea of the synergistic effect of shielding and protective effects with acid redox. Using the dry gel transformation method, they synthesized the core-shell structured catalyst MnCeOx@Z5 with a mesoporous zeolite (ZSM-5) as the shell and Mn-Ce composite oxides as the core. The shell layer of the MnCeOx@Z5 catalyst reduced the active component’s contact with SO2, which could inhibit the sulfation of active metal oxides to a certain extent. At the same time, the mesoporous ZSM-5 shell layer could also effectively inhibit the reaction between adsorbed SO2 and NH3 species, reducing the formation of sulfate and avoiding the clogging of shell pores by sulfate species. In addition, the ZSM-5 shell layer in the MnCeOx@Z5 catalyst was inherently hydrophobic and exhibited a certain degree of resistance to poisoning even in the atmosphere containing water and sulfur. As shown in Fig. 4d, when 100 ppm of SO2 and 5 vol% H2O were jointly introduced at 300 ℃, the NO conversion decreased from 100% to 75% for the MnCeOx@Z5 catalyst. Whereas the NO conversion of the supported catalyst, MnCeOx/Z5, rapidly decreased by 50% under the same conditions, indicating that the formation of a core-shell structure is beneficial for improving the sulfur resistance of the catalyst.
In addition to SO2 and H2O, the catalyst can be poisoned by other flue gas impurities, including alkali and alkaline earth metals such as K, Na, Mg, and Ca [11]. Liu et al. [120] designed and constructed Fe/Beta@meso-CeO2 core-shell catalysts with abundant mesoporous by template-assisted self-assembly. Through the coating of mesoporous CeO2 shells, K was prevented from entering the interior of the catalyst, and the exchange of K with isolated iron ions was effectively prevented, avoiding the formation of Fe2Ox clusters. The CeO2 shells also increased the chemisorbed oxygen species and promoted the formation of reactive NO2 and cis-N2O2− species, further improving the catalysts’ anti-potassium poisoning performance. After potassium poisoning, the NOx conversion of the Fe/Beta@meso-CeO2 catalyst was significantly higher than that of the Fe/Beta catalyst with an uncoated CeO2 shell, especially in the high-temperature region (>375 ℃). Reasonable design of core-shell structure, protection of acidic sites, optimization of pore structure, and introduction of alkali metal-resistant additives can effectively improve catalysts’ alkali metal poisoning resistance.
The structure of the core-shell catalyst can be optimized in a targeted manner by modulating the preparation method and preparation conditions. For example, in the hydrothermal process, the thickness of the shell layer can be synergistically regulated between 10 nm and 100 nm within a temperature range of 100–220 ℃ and a reaction time of 12–72 h. Additionally, the sol-gel method can be used to control the precursor ratios. The number of deposition cycles can be adjusted by the atomic layer deposition method to optimize the elemental distribution at the core-shell interface. The pH or reduction temperature can be regulated by the precipitation method to improve properties such as surface oxygen vacancy concentration and crystal orientation. These strategies, based on the quantitative correlation between process parameters and structure and properties, can precisely optimize the exposure efficiency of active sites, the mass transfer pathways, and sintering resistance. The key to deNOx catalysts using NH3-SCR technology lies in the collaboration of redox and acidic sites [121,122]. Wang et al. [123] obtained a Pt-based catalyst with a core-shell structure by a simple strategy that precisely controlled the distance between the acidic sites and Pt, thereby improving the N2 selectivity in the selective catalytic reduction of NOx. Ma et al. [124] prepared two kinds of yolk-shell structured CeO2-TiO2 catalysts with different cavity sizes using carbon and hybrid Ce-C spheres as templates. The sizes of the large-cavity catalysts (CeTi-L) and small-cavity catalysts (CeTi-S) were 5 µm and 500 nm, respectively, and the TEM results of the samples are shown in Figs. S6a and b (Supporting information). The CeTi-L catalyst exhibited higher deNOx activity than CeTi-S. In a 40-h stability test in the presence of SO2, the NO conversion of CeTi-L was maintained above 80% (4.7% loss), while the NO conversion of CeTi-S dropped sharply to 20% (72.6% loss). Ammonium bisulfate (ABS) can be deposited on the catalyst surface through physical or chemical effects, blocking the active sites and hindering the NH3-SCR process. The thermogravimetric curves (Fig. S6c in Supporting information) from the three ABS-deposited samples clearly show that the decomposition temperature of ABS on SiO2 hollow spheres decreases with the increase of the cavity size. This result suggests that larger cavity sizes may accelerate the decomposition of ABS by altering the radius of curvature of the surface and affecting the vapor pressure of ABS. Therefore, deNOx activity as well as sulfur resistance can be improved by adjusting the cavity size of the core-shell catalyst for NOx purification under sulfur-containing conditions.
Different core-shell catalysts exhibit distinct reaction mechanisms in deNOx, and the common reaction mechanisms are illustrated below with specific examples. The TiO2@CeMnOx-NC catalysts mainly follow the Eley-Rideal (E-R) reaction mechanism in the NH3-SCR process: The adsorbed NH3 is activated by subtracting a hydrogen atom to -NH2, and -NH2 intermediate further reacts with NO (NH2 + NO → NH2NO → N2 + H2O) to promote the NH3-SCR reaction [7]. The MnFe@CeOx-60 catalysts follow the L-H mechanism in the NH3-SCR reaction: NH3 is first adsorbed on the surface of the catalyst to form weakly adsorbed NH3, Lewis-acid-liganded NH3, and Brønsted-acid-bound NH4+; subsequently, NO + O2 adsorption generates a variety of nitrate species, both of which react at the core-shell interface in the presence of reactive species (Mn4+, Fe3+, Ce3+, and Oads species) [80].
Cai et al. [80] synthesized a series of core-shell MnFe@CeOx-60 nanocage catalysts. Due to the core-shell interfacial diffusion effect, some active species can diffuse with each other, which significantly increases the oxygen vacancy defect sites and surface acid sites. Meanwhile, this interfacial diffusion effect can effectively enhance the electronic interactions and significantly increase the proportion of active species. The role of constructing the CeO2 shell layer is shown in Fig. 5a. The MnFe@CeOx-60 catalyst followed the L-H mechanism in the low-temperature NH3-SCR reaction: NH3 was first adsorbed on the catalyst surface to form weakly adsorbed NH3, Lewis-acid-liganded NH3, and Brønsted-acid-bound NH4+, followed by the adsorption of NO+O2 to generate a variety of nitrate species, and the two were reacted by the active species at the core-shell interface (Mn4+, Fe3+, Ce3+, and Oads species). The core MnFeOx provides active sites and facilitates electron transfer. In contrast, the CeO2 shell layer introduces defects and acidic sites through interfacial diffusion effects, protecting the core from water vapor and regulating the redox capacity to prevent the overoxidation of NH3.
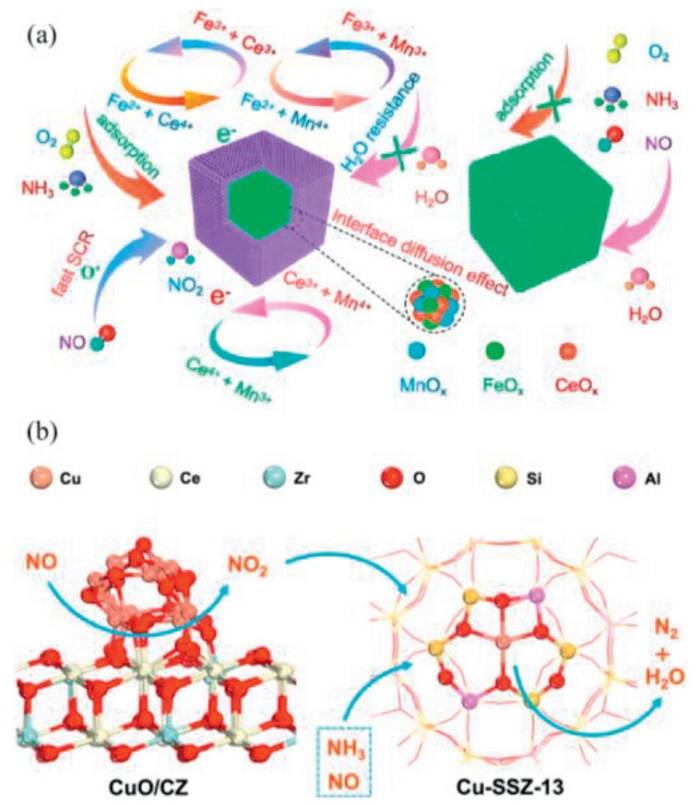
Jia et al. [125] prepared a Pt-CZO/Cu-SSZ-13 coupling catalysts by a facile grinding strategy. Pt species were distributed on the CZO surface in the form of single atoms and nanoclusters, which significantly enhanced the reactivity of lattice oxygen and the number of Brønsted acid sites, thus improving the low-temperature SCR activity. However, this coupling approach provides weak control over the interfacial structure and limited dispersion and stability of Pt species. In comparison, Jia et al. [126] also prepared a Cu-SSZ-13@CZ catalysts through a sophisticated core-shell structure design, which induced the migration of Cu species to the surface of the CZ shell layer during hydrothermal aging, forming a highly controllable CuO/CZ interface. This interface not only protected the Cu-SSZ-13 core layer by sacrificially adsorbing SO2 but also promoted a fast SCR pathway by enhancing NO oxidation, thereby achieving a wider activity temperature window (200–425 ℃) and excellent aging resistance. The contribution of the CuO/CZ interface to the NH3-SCR performance is shown in Fig. 5b. The comparison of the two catalysts shows that the interface effect in the core-shell structure is more capable of achieving precise regulation of active sites and optimization of reaction paths than simple physical coupling, providing a better strategy for designing efficient and stable SCR catalysts.
This paper reviews the primary preparation methods of core-shell catalysts, their applications in controlling air pollutants and, in particular, the research progress on core-shell catalysts in the field of deNOx. It helps researchers in this field to understand the common preparation methods of core-shell catalysts and their advantages and disadvantages and to understand the progress of the application of core-shell catalysts in three-way catalytic purification, catalytic combustion of VOCs, deNOx and application of core-shell catalysts in atmospheric pollutant control, especially the commonly used core-shell deNOx catalysts as well as the advantages they have in the field of deNOx, which can provide ideas for the development of efficient core-shell catalysts. Although significant efforts have been made to develop core-shell catalysts with high activity, anti-poisoning, and high hydrothermal stability, many challenges remain.
The following are some of the challenges that still exist in the area of deNOx for core-shell catalysts:
(1) The performance of core-shell catalysts is highly dependent on their preparation methods and conditions. Although various preparation methods can successfully synthesize catalysts with core-shell structures, each method has its advantages and disadvantages. For example, the hydrothermal method is highly controllable but prone to lead to defects in the shell layer; the chemical deposition method is low-cost but sensitive to the preparation conditions, and the template method can precisely regulate the morphology but is difficult to remove.
(2) Supported catalysts are the mainstream of commercial catalysts and dominate the deNOx field (>95%), which are widely used in mobile pollution sources (e.g., automobile exhaust) and stationary pollution sources (e.g., power plants and industrial boilers). Still, supported catalysts have the drawbacks of poor anti-poisoning performance, easy to sinter at high temperatures, and poor hydrothermal stability. Meanwhile, core-shell catalysts are widely studied and applied in the field of deNOx due to their high catalytic activity, resistance to poisoning, and high-temperature hydrothermal stability. We compared the main characteristics of supported catalysts and core-shell catalysts (Fig. S7 in Supporting information), and found that the core-shell catalysts have significant advantages over the supported catalysts. However, core-shell catalysts have the drawback of high preparation costs, including the expensive price of certain raw materials and the high cost of processing time. This problem limits the large-scale industrial production and application of core-shell catalysts, which is one of the problems that need to be solved urgently;
(3) Although core-shell catalysts show excellent performance under laboratory conditions, the long-term stability problem is more prominent in practical applications. During catalytic reactions, especially at high temperatures, in high humidity, or complex chemical environments, the core-shell structure may be destroyed due to high-temperature sintering, exposing the active components and reducing the activity and selectivity of the catalyst. In addition, prolonged operation may also trigger changes in the interfacial structure between the core and shell, weakening the synergistic effect between the two and thereby affecting the durability of the catalytic performance.
To develop more promising core-shell catalyst catalytic systems for the practical industrial applications, future research can be carried out from the following aspects:
(1) For the drawbacks of different preparation methods, the methods can be optimized and integrated. For example, the hydrothermal method tends to cause shell defects, which can be reduced by precisely controlling the reaction temperature, time, and solution concentration, as well as adding surfactants to lower the surface tension. Additionally, by combining the hydrothermal method with the chemical deposition method, a stable core structure is first formed using the hydrothermal method. Then, a uniform shell layer is grown via chemical deposition. In this way, the advantages of both methods can be leveraged while avoiding the limitations of either alone.
(2) To address the high preparation cost of core-shell catalysts, improvements can be made by optimizing low-cost preparation methods (e.g., electrodeposition, co-precipitation). It is also feasible to achieve high-efficiency mass production for cost reduction by scaling up suitable continuous-flow reactors and precisely applying high-cost technologies, such as atomic layer deposition, to key processes like active site construction, thereby balancing performance and cost. Meanwhile, exploring inexpensive and widely available raw materials to replace expensive ones as active components or carriers is also essential.
(3) In practical industrial applications, the long-term stability of core-shell catalysts needs to be synergistically optimized in terms of material structure, preparation processes, and operating conditions. Currently, the commonly used components of core-shell deNOx catalysts are mainly concentrated in four categories: TiO2, SiO2, CeO2, and molecular sieve. Among them, molecular sieve-based core-shell catalysts are often used in mobile pollution sources, such as marine exhaust and diesel vehicle exhaust deNOx. TiO2-based, CeO2-based, and SiO2-based core-shell catalysts are commonly employed in stationary pollution sources, including waste incineration, power plants, and flue gas deNOx of industrial boilers. It is crucial to select the appropriate catalyst type based on the specific requirements of the application. As shown in Fig. S8 (Supporting information), core-shell catalysts can be classified based on their core-shell composition and application scope. Advanced technologies, such as atomic layer deposition, are used to precisely control the growth of the shell layer. At the same time, preparation processes, including hydrothermal preparation with gradient heating, are optimized to minimize defects. At the same time, intelligent monitoring systems are used to regulate reaction temperature, humidity, and other parameters in real time, thereby enhancing the structural stability and durability of the catalysts under complex industrial conditions through whole-process control.
In addition, we will further investigate how the composition and morphology of core-shell catalysts influence their physicochemical properties and performance, explore the formation mechanism of the core-shell structure, analyze the interfacial effect between the shell layer and the core particles, and optimize the preparation methods and conditions to achieve the precise control of the core-shell catalysts at the microscopic level. To optimize the diffusion paths of reactants and products and enhance catalytic efficiency, we will investigate the catalytic mechanism and reaction sequence of core-shell catalysts, optimizing the distribution of active and acidic sites. This will guide the design and development of practical core-shell catalysts in the field of deNOx. The challenges and future development prospects of core-shell catalysts in deNOx are shown in Fig. 6.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 52370113, 52460012), National Key Research and Development Program of China (No. 2024YFC3714400), Yunnan Fundamental Research Projects (No. 202401AW070015), Yunnan Key Laboratory of Phosphorous Gypsum Resource Recycling and Ecological Utilization (No. 202449CE340028), Yunnan International Joint R&D Center for Waste Resource Recovery in Metallurgical and Chemical Industries (No. 202403AP140008).
Supplementary material associated with this article can be found, in the online version, at doi:
R.G. Chaudhuri, S. Paria, Chem. Rev. 112 (2011) 2373–2433.
R. Wang, X. Zi, L. Liu, H. Dai, H. He, Prog. Chem. 22 (2010) 358–366.
N. Zhang, M. Qin, J. Zhu, et al., Chin. Chem. Lett. 36 (2025) 110177. doi: 10.1016/j.cclet.2024.110177
Y. Zhang, G. Zeng, L. Tang, et al., Biosens. Bioelectron. 22 (2007) 2121–2126. doi: 10.1016/j.bios.2006.09.030
T. Li, G. Lin, W. Li, M. Wey, J. Environ. Chem. Eng. 13 (2025) 115305. doi: 10.1016/j.jece.2024.115305
W. Li, X. Wen, X. Wang, et al., Mol. Catal. 514 (2021) 111847.
X. Huang, F. Dong, G. Zhang, Z. Tang, Chem. Eng. J. 432 (2022) 134236. doi: 10.1016/j.cej.2021.134236
L. Jia, J. Liu, D. Huang, et al., ACS Catal. 12 (2022) 11281–11293. doi: 10.1021/acscatal.2c03048
H. Li, L. Schill, R. Fehrmann, A. Riisager, Inorg. Chem. Front. 10 (2023) 727–755. doi: 10.1039/d2qi02106d
H. Zhou, W. Su, Y. Xing, et al., J. Environ. Chem. Eng. 12 (2024) 113941. doi: 10.1016/j.jece.2024.113941
Q. Cai, F. Wang, Y. Hou, et al., Fuel Process. Technol. 243 (2023) 107675. doi: 10.1016/j.fuproc.2023.107675
L. Jia, J. Liu, H. Cheng, Z. Zhao, J. Liu, J. Environ. Sci. 150 (2025) 451–465. doi: 10.1016/j.jes.2023.10.012
H. Ullah, N. Batisse, K. Guerin, G. Rogez, P. Bonnet, Langmuir 36 (2020) 8461–8475. doi: 10.1021/acs.langmuir.0c00889
T. Wen, W. Liu, L. Wang, Y. Gong, J. Alloys. Compd. 966 (2023) 171550. doi: 10.1016/j.jallcom.2023.171550
Z. Cai, G. Zhang, Z. Tang, J. Zhang, ACS Appl. Nano Mater. 4 (2021) 6201–6211. doi: 10.1021/acsanm.1c00979
C. Zhang, Q. Guo, M. Xu, et al., Acta Chim. Sin. 70 (2012) 1327–1331. doi: 10.6023/A1202222
H. Gong, Y. Zhuang, X. Zhang, J. Liu, S. Li, Appl. Catal. B 330 (2023) 122574. doi: 10.1016/j.apcatb.2023.122574
Y. Tan, X. Liu, Y. Zhang, et al., Fuel 388 (2025) 134475. doi: 10.1016/j.fuel.2025.134475
L. Zhang, K. Chen, L. Li, et al., Chin. Chem. Lett. 37 (2026) 111002. doi: 10.1016/j.cclet.2025.111002
Y. Liu, J. Xu, H. Li, et al., J. Mater. Chem. A 3 (2015) 11543–11553. doi: 10.1039/C5TA01212K
X. Ning, Y. Zhang, W. Wang, Chem. Bull. 72 (2009) 962–972.
S. Li, B. Huang, C. Yu, Catal. Commun. 98 (2017) 47–51. doi: 10.1016/j.catcom.2017.04.046
F. Chung, R. Wu, F. Cheng, Sens. Actuators B 190 (2014) 1–7.
T.J. Almusawi, N. Mengelizadeh, R. Rahimpoor, D. Balarak, Appl. Organomet. Chem. 39 (2025) e7961. doi: 10.1002/aoc.7961
W. Yao, Y. Liu, Z. Wu, Appl. Surf. Sci. 442 (2018) 156–163. doi: 10.1016/j.apsusc.2018.02.066
T. Xu, G. Li, X. Zhu, et al., Fuel 353 (2023) 129292. doi: 10.1016/j.fuel.2023.129292
Y. Xia, Z. He, G. He, et al., Chin. Chem. Lett. 36 (2025) 111521. doi: 10.1016/j.cclet.2025.111521
L. Zhang, T. Du, H. Qu, B. Chi, Q. Zhong, Chem. Eng. J. 313 (2017) 702–710. doi: 10.1016/j.cej.2016.12.108
J. Min, W. Song, T. Hu, et al., Ceram. Int. 47 (2021) 35320–35332. doi: 10.1016/j.ceramint.2021.09.075
X. Zhu, Y. Wang, L. Lv, et al., Ind. Eng. Chem. Res. 63 (2024) 15038–15050. doi: 10.1021/acs.iecr.4c01680
T. Wu, R. Guo, C. Li, Y. You, W. Pan, Chemosphere 333 (2023) 138942. doi: 10.1016/j.chemosphere.2023.138942
Y. Boyjoo, G. Rochard, J. Giraudon, J. Liu, J. Lamonier, Sustainable Mater. Technol. 20 (2019) e00091. doi: 10.1016/j.susmat.2018.e00091
S. Yuan, X. Lv, Y. Zhang, et al., J. Taiwan. Inst. Chem. Eng. 103 (2019) 138–148. doi: 10.1016/j.jtice.2019.06.021
X. Wang, J. Feng, Y. Bai, Q. Zhang, Y. Yin, Chem. Rev. 116 (2016) 10983–11060. doi: 10.1021/acs.chemrev.5b00731
J. Li, H. Chang, S. Feng, et al., Chem. Eng. J. 498 (2024) 155542. doi: 10.1016/j.cej.2024.155542
X. Zhang, Z. Sun, R. Jin, et al., Nat. Commun. 14 (2023) 530. doi: 10.1038/s41467-023-36147-2
X. Zhao, Z. Ma, X. Li, Y. Guo, P. Li, Mater. Chem. Phys. 329 (2025) 130145. doi: 10.1016/j.matchemphys.2024.130145
H. Zhang, J. Diao, M. Ouyang, et al., ACS Catal. 13 (2023) 1349–1358. doi: 10.1021/acscatal.2c05433
C. Dimitriou, P. Psathas, M. Solakidou, Y. Deligiannakis, Nanomaterials 13 (2023) 3006. doi: 10.3390/nano13233006
G. Zhou, Y. Zhang, X. Zhao, et al., Fuel 334 (2023) 126824. doi: 10.1016/j.fuel.2022.126824
L. Lan, S. Chen, H. Li, et al., J. Ind. Eng. Chem. 58 (2018) 246–257. doi: 10.1016/j.jiec.2017.09.034
S. Song, X. Wang, H. Zhang, NPG. Asia Mater. 7 (2015) e179. doi: 10.1038/am.2015.27
J. Wang, H. Chen, Z. Hu, M. Yao, Y. Li, Catal. Rev. 57 (2014) 79–144.
A. Uzunoglu, D.A. Kose, L.A. Stanciu, Int. Nano Lett. 7 (2017) 187–193. doi: 10.1007/s40089-017-0213-3
Z. Zhou, J. Ouyang, H. Yang, A. Tang, Appl. Clay. Sci. 121-122 (2016) 63–70. doi: 10.1016/j.clay.2015.12.017
D. Ma, H. Liu, Y. Chen, et al., Appl. Chem. Ind. 48 (2019) 1156–1162. doi: 10.1109/icme.2019.00202
M. Ozawa, M. Misaki, M. Iwakawa, et al., Catal. Today 332 (2019) 251–258. doi: 10.1016/j.cattod.2018.08.015
W. Li, M. Wey, Sci. Total. Environ. 675 (2019) 397–407. doi: 10.1016/j.scitotenv.2019.04.243
L. Li, N. Zhang, R. Wu, et al., ACS Appl. Mater. Interfaces 12 (2020) 10350–10358. doi: 10.1021/acsami.9b20734
L. Li, N. Zhang, X. Huang, et al., ACS Catal. 8 (2018) 3222–3231. doi: 10.1021/acscatal.8b00358
B. Liu, G. Zhang, H. He, et al., Chem. J. Chin. Univ. 34 (2013) 1936–1944.
W. Li, M. Wey, Appl. Catal. A 602 (2020) 117732. doi: 10.1016/j.apcata.2020.117732
W. Li, M. Wey, Sci. Total. Environ. 707 (2020) 136137. doi: 10.1016/j.scitotenv.2019.136137
S. Chen, J. Li, Q. Wu, et al., Catal. Lett. 145 (2015) 1420–1428. doi: 10.1007/s10562-015-1535-2
M. Ozawa, M. Takahashimorita, K. Kobayashi, M. Haneda, Catal. Today 281 (2017) 482–489. doi: 10.1016/j.cattod.2016.06.029
T. Xu, W. Sun, X. Jin, et al., Adv. Fine Petrochem. 20 (2019) 53–57.
T.S. Ahmadi, Z.L. Wang, T.C. Green, A. Henglein, M.A. Elsayed, Science 272 (1996) 1924–1925. doi: 10.1126/science.272.5270.1924
A.T. Bell, Science 299 (2003) 1688–1691. doi: 10.1126/science.1083671
H. Peng, T. Dong, L. Zhang, et al., Appl. Catal. B 256 (2019) 117807. doi: 10.1016/j.apcatb.2019.117807
Q. Ren, S. Mo, J. Fan, et al., Chin. J. Catal. 41 (2020) 1873–1883. doi: 10.1016/S1872-2067(20)63641-5
J. He, F. Zheng, Y. Zhou, et al., J. Colloid. Interface Sci. 613 (2022) 155–167. doi: 10.1016/j.jcis.2022.01.023
W. Liu, L. Zhang, T. Dong, et al., ChemCatChem 10 (2018) 4134–4142. doi: 10.1002/cctc.201800925
S. Zhao, F. Hu, J. Li, ACS Catal. 6 (2016) 3433–3441. doi: 10.1021/acscatal.6b00144
Y. Zhang, Z. Tan, X. Wang, et al., Nanoscale 11 (2019) 4794–4802. doi: 10.1039/c8nr10523e
F. Dong, W. Han, W. Han, Z. Tang, Appl. Catal. B 315 (2022) 121524. doi: 10.1016/j.apcatb.2022.121524
X. Feng, H. Chen, Q. Xue, C. Su, Y. Zhou, Chem. Eng. J. 490 (2024) 151464. doi: 10.1016/j.cej.2024.151464
X. Du, F. Dong, Z. Tang, J. Zhang, Nanoscale 12 (2020) 12133–12145. doi: 10.1039/d0nr02334e
J. Jiang, X. Li, X. Liu, et al., Sep. Purif. Technol. 361 (2025) 131494. doi: 10.1016/j.seppur.2025.131494
A. Rokicińska, P. Łątka, B. Olszanski, et al., Chem. Eng. J. 480 (2024) 148173. doi: 10.1016/j.cej.2023.148173
B. Wang, Q. Yang, B. Li, et al., Appl. Catal. B 332 (2023) 122753. doi: 10.1016/j.apcatb.2023.122753
T. Dong, W. Liu, M. Ma, et al., Chem. Eng. J. 393 (2020) 124717. doi: 10.1016/j.cej.2020.124717
M. Wen, F. Dong, Z. Tang, J. Zhang, Microporous Mesoporous Mater. 322 (2021) 111156. doi: 10.1016/j.micromeso.2021.111156
Q. Zhao, Q. Liu, Y. Zheng, et al., Chemosphere 244 (2020) 125532. doi: 10.1016/j.chemosphere.2019.125532
W. Yang, Y. Peng, Y. Wang, et al., Appl. Catal. B 278 (2020) 119279. doi: 10.1016/j.apcatb.2020.119279
Q. Zhao, Y. Zheng, C. Song, et al., Appl. Catal. B 265 (2020) 118552. doi: 10.1016/j.apcatb.2019.118552
Y. Zheng, R. Han, L. Yang, et al., Chem. Eng. J. 465 (2023) 142807. doi: 10.1016/j.cej.2023.142807
F. Wang, Q. Cai, J. Gao, et al., Appl. Surf. Sci. 685 (2025) 162067. doi: 10.1016/j.apsusc.2024.162067
Y. Bai, Y. Hou, Y. Guo, et al., J. Colloid. Interface Sci. 616 (2022) 55–66. doi: 10.1016/j.jcis.2022.01.034
Z. Di, H. Wang, R. Zhang, et al., Appl. Catal. A 630 (2022) 118438. doi: 10.1016/j.apcata.2021.118438
Z. Cai, G. Zhang, Z. Tang, J. Zhang, ACS Appl. Nano Mater. 5 (2022) 3619–3631. doi: 10.1021/acsanm.1c04194
Z. Fu, G. Zhang, W. Han, Z. Tang, Chem. Eng. J. 426 (2021) 131334. doi: 10.1016/j.cej.2021.131334
Y. Li, Y. Hou, Y. Zhang, Y. Yang, Z. Huang, J. Colloid. Interface Sci. 608 (2022) 2224–2234. doi: 10.1016/j.jcis.2021.10.078
L. Xie, C. Liu, Y. Deng, F. Liu, W. Ruan, Ind. Eng. Chem. Res. 61 (2022) 8698–8707. doi: 10.1021/acs.iecr.2c00789
B. Huang, D. Yu, Z. Sheng, L. Yang, J. Environ. Sci. 55 (2017) 129–136. doi: 10.1016/j.jes.2016.05.038
R. Chen, S. Peng, Y. Wang, X. Qi, Z. Liu, J. Rare Earths 41 (2023) 959–964. doi: 10.1016/j.jre.2023.01.005
C. Fang, L. Shi, H. Hu, J. Zhang, D. Zhang, RSC Adv. 5 (2015) 11013–11022. doi: 10.1039/C4RA14735A
X. Chen, Q. Liu, Q. Wu, et al., Catal. Commun. 149 (2021) 106252. doi: 10.1016/j.catcom.2020.106252
Z. Sheng, D. Ma, D. Yu, et al., Chin. J. Catal. 39 (2018) 821–830. doi: 10.1016/S1872-2067(18)63059-1
Z. Cai, G. Zhang, Z. Tang, J. Zhang, Nanoscale 14 (2022) 12281–12296. doi: 10.1039/d2nr02255a
Z. Chen, L. Liu, H. Qu, et al., J. Catal. 392 (2020) 217–230. doi: 10.1016/j.jcat.2020.10.005
X. Huang, F. Dong, G. Zhang, Y. Guo, Z. Tang, Chem. Eng. J. 419 (2021) 129572. doi: 10.1016/j.cej.2021.129572
X. Huang, F. Dong, G. Zhang, Z. Tang, Nanoscale 12 (2020) 16366–16380. doi: 10.1039/d0nr04165c
S. Liu, J. Liu, Q. Lin, et al., ACS Sustainable Chem. Eng. 8 (2020) 13418–13429. doi: 10.1021/acssuschemeng.0c04243
S. Liu, H. Wang, Y. Wei, R. Zhang, Mol. Catal. 485 (2020) 110822.
L. Chen, X. Wang, Q. Cong, et al., Chem. Eng. J. 369 (2019) 957–967. doi: 10.1016/j.cej.2019.03.055
R. Yan, S. Lin, Y. Li, et al., J. Hazard. Mater. 396 (2020) 122592. doi: 10.1016/j.jhazmat.2020.122592
C. Huang, R. Guo, W. Pan, et al., J. Energy Inst. 92 (2019) 1364–1378. doi: 10.1016/j.joei.2018.09.005
C. Yu, D. Hou, B. Huang, et al., J. Hazard. Mater. 399 (2020) 123011. doi: 10.1016/j.jhazmat.2020.123011
S. Cheng, J. Shao, B. Huang, J. Guan, L. Zhou, RSC Adv. 10 (2020) 13855–13865. doi: 10.1039/d0ra00668h
G. Dai, Y. Peng, C. Yu, et al., J. Fuel Chem. Technol. 52 (2024) 1686–1695. doi: 10.1016/S1872-5813(24)60465-2
Z. Li, J. Xiao, Y. Gao, R. Gui, Q. Wang, Environ. Sci. Technol. 57 (2023) 20326–20338. doi: 10.1021/acs.est.3c04421
H. Pan, Z. Chen, X. Feng, et al., Appl. Catal. B: Environ 362 (2025) 124761. doi: 10.1016/j.apcatb.2024.124761
J. Liu, Y. Du, J. Liu, et al., Appl. Catal. B 203 (2017) 704–714. doi: 10.1016/j.apcatb.2016.10.039
L. Zhang, D. Zhang, J. Zhang, et al., Nanoscale 5 (2013) 9821–9829. doi: 10.1039/c3nr03150k
J. Jiang, R. Zheng, Y. Jia, et al., Mol. Catal. 493 (2020) 111044.
J.M. Kim, S.M. Chang, S. Kim, et al., Ceram. Int. 35 (2009) 1243–1247. doi: 10.1016/j.ceramint.2008.06.003
M. Ocaña, M. Andrésvergés, R. Pozas, C.J. Serna, J. Colloid. Interface Sci. 294 (2006) 355–361. doi: 10.1016/j.jcis.2005.07.020
K. An, Q. Zhang, S. Alayoglu, et al., Nano Lett. 14 (2014) 4907–4912. doi: 10.1021/nl502434m
H. Zhang, M. Zhang, L. Hao, et al., Fuel Process. Technol. 201 (2020) 106342. doi: 10.1016/j.fuproc.2020.106342
F. Wang, B. Shen, L. Gao, J. Yang, Fuel Process. Technol. 168 (2017) 131–139. doi: 10.1016/j.fuproc.2017.08.024
F. Nkinahamira, S. Sun, R. Zhu, J. Hazard. Mater. 487 (2025) 137204. doi: 10.1016/j.jhazmat.2025.137204
L. Han, S. Cai, M. Gao, et al., Chem. Rev. 119 (2019) 10916–10976. doi: 10.1021/acs.chemrev.9b00202
B. Chi, H. Qu, X. Xing, Q. Zhong, J. Alloys. Compd. 726 (2017) 906–912. doi: 10.1016/j.jallcom.2017.08.075
Y. Zhang, T. Liu, J. Yu, et al., Chin. Chem. Lett. 36 (2025) 110275. doi: 10.1016/j.cclet.2024.110275
B. Wang, Z. Song, F. Liu, et al., J. Cleaner Prod. 434 (2024) 140100. doi: 10.1016/j.jclepro.2023.140100
S. Jiang, L. Zhang, L. Zhao, J. Zhang, Y. Huang, Chem. Eng. J. 474 (2023) 145979. doi: 10.1016/j.cej.2023.145979
J. Zhang, Y. Zhang, G. Ma, et al., Chin. Chem. Lett. 36 (2025) 110516. doi: 10.1016/j.cclet.2024.110516
W. Yang, J. Du, L. Bei, et al., J. Energy Inst. 120 (2025) 101990. doi: 10.1016/j.joei.2025.101990
T. Zhang, F. Qiu, J. Li, Appl. Catal. B 195 (2016) 48–58. doi: 10.1016/j.apcatb.2016.04.058
J. Liu, J. Liu, Z. Zhao, et al., Catal. Surv. Asia 22 (2018) 181–194. doi: 10.1007/s10563-018-9251-8
S. Brandenberger, O. Kröcher, A. Tissler, R. Althoff, Catal. Rev. 50 (2008) 492–531. doi: 10.1080/01614940802480122
L. Wang, W. Li, S.J. Schmieg, D. Weng, J. Catal. 324 (2015) 98–106. doi: 10.1016/j.jcat.2015.01.011
H. Wang, M. Liu, Y. Ma, et al., ACS Catal. 8 (2018) 2796–2804. doi: 10.1021/acscatal.7b04327
K. Ma, K. Guo, L. Li, et al., Catal. Commun. 128 (2019) 105719. doi: 10.1016/j.catcom.2019.105719
L. Jia, Y. Shao, Z. Li, et al., ACS ES&T Eng 4 (2024) 1007–1015. doi: 10.1021/acsestengg.3c00530
L. Jia, L. Zhang, B. Liu, et al., Environ. Sci. Technol. 58 (2024) 15038–15051.

Figure 2 Review of preparation methods for core-shell catalysts. (a) Hydrothermal method. (b) Sol-gel method. (c) Microemulsion method. Reprinted with permission [13]. Copyright 2020, American Chemical Society. (d) Atomic layer deposition method. (e) Co-precipitation method. (f) Flame spray pyrolysis. (g) Electrodeposition method. Reprinted with permission [14]. Copyright 2023, Elsevier. (h) Template method. (i) Self-assembly method. Reprinted with permission [15]. Copyright 2021, American Chemical Society. (j) Chemical deposition method.

Figure 3 Core-shell catalysts commonly used in deNOx. (a) TiO2-based core-shell catalysts. Reprinted with permission [88]. Copyright 2018, Elsevier. (b) SiO2-based core-shell catalysts. Reprinted with permission [40]. Copyright 2023, Elsevier. (c) CeO2-based core-shell catalysts. Reprinted with permission [103]. Copyright 2017, Elsevier. (d) Molecular sieve-based core-shell catalysts. Reprinted with permission [95]. Copyright 2019, Elsevier.

Figure 4 EPR spectra of Cu-SSZ-13 (a), meso-Cu-SSZ-13 (b) and meso-Cu-SSZ-13@MAS (c) catalysts before and after hydrothermal aging. Reprinted with permission [119]. Copyright 2016, Elsevier. (d) The H2O/SO2 tolerance performances when used MnCeOx@Z5 and MnCeOx/Z5 at 300 ℃. Reprinted with permission [96]. Copyright 2020, Elsevier.

Figure 5 (a) Role of the CeO2 shell layer in the NH3-SCR reaction. Reprinted with permission [80]. Copyright 2022, American Chemical Society. (b) Schematic illustration of the promotional effect of the CuO/CZ interface on the NH3-SCR performance. Reprinted with permission [126]. Copyright 2024, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: