Citation:
Yi Zhang, Xin Yao, Jiaxin Yu, Haili Qin, Huaiping Cong. Gradient crosslinking of anisotropic hydrogels for programmable shape morphing and actuation[J]. Chinese Chemical Letters,
2026, 37(6): 112041.
doi:
10.1016/j.cclet.2025.112041
Gradient crosslinking of anisotropic hydrogels for programmable shape morphing and actuation
English
Gradient crosslinking of anisotropic hydrogels for programmable shape morphing and actuation
Anhui Province Engineering Research Center of Flexible and Intelligent Materials, School of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei 230009, China
Received Date:
26 July 2025 Accepted Date:
30 October 2025 Revised Date:
23 October 2025 Available Online:
15 June 2026
Abstract:
Biological systems exploit their sophisticated hierarchical anisotropic architectures to achieve complex shape morphing under external stimuli. However, artificial soft intelligent actuators often suffer from limited response speeds and poor programmability of deformation, primarily due to densely crosslinked network designs and insufficient anisotropic resolution. Here, we report a synergistic method combining directional freezing-induced self-assembly and dynamic metal coordination crosslinked mechanical alignment to fabricate anisotropic hydrogels with fast multiple responsiveness and programmable three-dimensional (3D) deformation. Benefiting from an interconnected lamellar network and open-oriented mass transport channels, the hydrogel exhibits rapid anisotropic stimuli-responsive deformations with a shrinkage along the lamellar direction that is 1.9 times greater than that in the perpendicular direction in 5 s of thermal stimulation, and a complete recovery within 4 s upon cooling. By developing a spatially modulated coordination photodissociation strategy, a gradient crosslinking network is further constructed within the hydrogel, enabling diverse and programmable 3D deformations in response to external stimuli, which facilitate complex actuation behaviors such as object grasping, biomimetic gestures, and light-driven lifting. Thus, the hydrogel with hierarchically anisotropic structure and porous dynamic crosslinked network is potential for intelligent soft robotics requiring flexible controllable deformation.
Biological organisms with intrinsic hierarchical anisotropic architectures are exceptionally capable of complex and fascinating three-dimensional (3D) shape deformations. These dynamic shape-shifting abilities provide enduring inspiration for the design of bioinspired soft actuators [1-4]. Among various smart materials, hydrogels have emerged as key candidates for applications particularly in soft robotics, biomimetic actuators, and intelligent medical devices owing to their tissue-like structures, multifaceted responsiveness to multiple stimuli, and excellent biocompatibility [5-7]. However, a key limitation of conventional hydrogels stems from their densely crosslinked networks, which severely restrict solvent mobility and rendered macroscopic deformation dominated by slow, diffusion-limited water transport. Consequently, their intrinsically slow response dynamics significantly hinders practical applications [8-10].
Recently, the construction of ordered and interconnected channels within hydrogel networks via ice-templating has markedly enhanced the transport of water molecules, offering a promising strategy to overcome the longstanding limitation of slow actuation in conventional hydrogels [11-14]. Compared with conventional pore-forming techniques such as chemical foaming or leaching [15,16], ice-templating offers distinct advantages in multiscale structural control, as the freezing parameters can be precisely tuned to regulate pore size and architecture. Besides, the formation of continuous, low-tortuosity channels facilitates water transport and accelerates the response of hydrogel [17-19]. However, the structural homogeneity of pore networks formed by unidirectional freezing results in isotropic macroscopic deformation, which limits the realization of anisotropic and programmable shape transformations commonly found in biological systems [20,21].
Inspired by the highly ordered architectures of natural organisms, extensive efforts have been made to construct anisotropic hydrogels with orientated structure by introducing highly aligned nanoscale building blocks into polymer networks through the application of external fields such as electric, magnetic, or acoustic fields, as well as shear forces [22-26]. Among these approaches, mechanical stretching has emerged as a particularly attractive strategy for inducing anisotropic structures in hydrogels due to its simplicity, efficiency, and versatility [27,28]. Unfortunately, anisotropic hydrogels induced by mechanical pre-stretching can only undergo in-plane contraction and recovery under external stimuli due to their intrinsically planar alignment and the absence of spatial heterogeneity [29,30]. Therefore, it is highly desired to precisely program internal structures to enable accurate regulation of complex deformation behaviors in hydrogels that simultaneously exhibit rapid anisotropic responsiveness but remains challenging [22,23].
Here, a hierarchical orientation assembly combined with dynamic crosslinking strategy for the fabrication of rapidly anisotropic responsive hydrogels with long-range ordered layered structure and directional open honeycomb-like network by unidirectional freezing-induced assembly cooperated with mechanical stretching-enhanced orientation and Fe3+-COO− coordination. Benefiting from directionally aligned lamellar networks with open interconnected porous structure, the hydrogels achieve fast anisotropic deformation under light/heat stimuli, outperforming existing actuators in responsive and recovery speeds. Furthermore, a dynamic and controllable photodissociation is employed to spatially modulate coordination crosslinking, enabling the formation of a gradient network for complex programmable 3D shape transformation. Finally, the spatially programmed hydrogels demonstrate a variety of biomimetic motions, including object gripping, submersible lifting, and hand-like movements. This work establishes a new structural paradigm for next-generation soft actuators, offering significant potential for applications in minimally invasive surgery and adaptive biomedical systems.
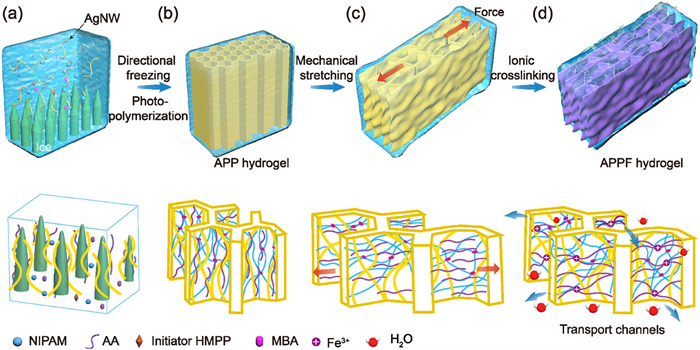
As shown in Fig. 1, a directional freezing-induced self-assembly combined with metal coordination crosslinked mechanical alignment method was developed to fabricate anisotropic photo- and thermo-responsive hydrogels with highly ordered oriented architectures and open, low-tortuosity mass transport channels. Firstly, intrinsic geometrically anisotropic silver nanowires (AgNWs) (diameter: 50–120 nm, length: 12–16 µm) with a high aspect ratio were selected to obtain the polymerizable scaffold through directional freezing assembly of a precursor solution of N-isopropylacrylamide (NIPAM) and acrylic acid (AA) (Fig. S1 in Supporting information). During the freezing, adjacent ice dendrites expelled solutes into interstitial regions, where water diffusion was confined by hydrogen bonding between NIPAM and AA, shortened ice crystal growth paths, reduced repulsion of AgNWs, and ultimately formed a refined honeycomb-like porous structure [31,32]. Through cryo-photopolymerization under ultraviolet (UV) light irradiation at −10 ℃, a primary AgNWs/PNIPAM/PAA (APP) hydrogel featuring an interconnected 3D honeycomb network with an average pore size of ~30 µm was fabricated (Fig. 1b and Fig. S2 in Supporting information). Subsequently, a mechanical stretch-induced orientation method was applied to the primary APP hydrogel, with the stretching direction perpendicular to the freezing direction, to obtain a hierarchically oriented structure (Fig. 1c). Finally, the AgNWs/PNIPAM/PAA/Fe3+ (APPF) hydrogel was fabricated by immersing the pre-stretched hydrogel in an FeCl3 solution, during which the temporarily oriented network was permanently fixed through secondary coordination crosslinking between the exposed carboxyl groups of the PAA chains and Fe3+ ions (Fig. 1d). The tensile stress-strain curves showed great increment in both tensile strength and elongation at break of APPF hydrogel compared with those of APP hydrogel, indicating that coordination crosslinking significantly enhanced the mechanical properties (Fig. S3 in Supporting information).
Figure 1
Figure 1.
Schematic illustration of the fabrication of the APPF hydrogel by a consecutive directional freezing-induced self-assembly and metal coordination crosslinked mechanical alignment process. (a) A precursor solution containing NIPAM, AA, photoinitiator 2-hydroxy-2-methyl-1-phenyl-1-propanone (HMPP), crosslinker N,N’-methylenebisacrylamide (MBA), Fe3+ and AgNWs was assembled into an aligned scaffold via directional freezing. (b) The APP hydrogel with a 3D ordered honeycomb network structure was synthesized through cryo-photopolymerization. (c) Mechanical stretching was applied to the APP hydrogel to induce anisotropic alignment of the polymer network. (d) The hierarchically anisotropic APPF hydrogel integrated with open transport channels was obtained via ionic crosslinking.
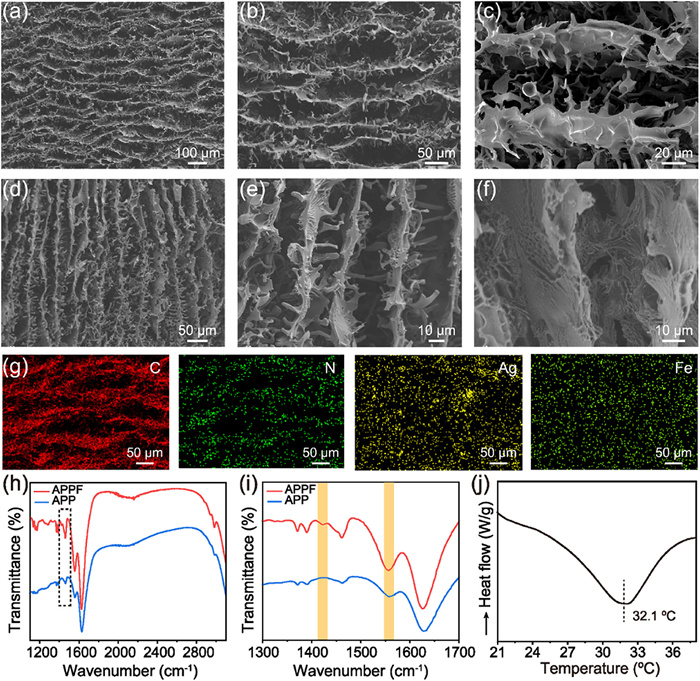
Top-view scanning electron microscope (SEM) images revealed that the APPF hydrogel exhibited a long-range ordered lamellar structure with an interlamellar spacing of approximately 50 µm (Fig. 2a). High-magnification SEM images showed that the adjacent lamellae were bridged by slender and ramiform polymer structures, resulting in a highly interconnected and 3D ordered network (Figs. 2b and c). Parallel to the growth direction of ice crystals, a uniform 30-µm-spaced lamellar structure was observed where the neighboring lamellae were interconnected and formed a secondary network (Figs. 2d and e). Enlarged SEM image demonstrated that the surface of the polymer framework possessed a rich microporous structure (Fig. 2f). Uniform distributions of C, N, Ag and Fe elements confirmed the incorporation of AgNWs into the PNIPAM/PAA network, as well as the formation of metal-coordination crosslinking (Fig. 2g). Furthermore, spatial enrichment of C and N elements along the stretching direction was observed, consistent with the oriented lamellar network, which confirmed the structural anisotropy and polymer network orientation induced by the stretching process. In the Fourier transform infrared spectroscopy (FT-IR) spectra, compared with the APP without coordination crosslinking, a peak at 1420 cm−1, corresponding to the symmetric stretching vibration of carboxylate groups, was appeared in the APPF (Figs. 2h and i). In addition, the peak at 1557 cm−1, assigned to the asymmetric stretching vibration of carboxylate groups, showed marked enhancement, strongly proving metal-carboxylate coordination within the APPF [33-35]. As detected from a distinct endothermic transition peak in differential scanning calorimetry (DSC) curve, the lower critical solution temperature (LCST) of the APPF hydrogel was determined at 32.1 ℃ (Fig. 2j) [36,37].
Figure 2
Figure 2.
SEM images with different magnifications of the APPF hydrogel: Top view (a-c) and side view (d-f). (g) Elemental mappings of C, N, Ag, and Fe on the APPF hydrogel surface. (h) FT-IR spectra of APPF and APP hydrogels. (i) The magnification of FT-IR spectra in the squared area. (j) Temperature- modulated DSC curve of the APPF hydrogel.
Distinct from previously reported porous hydrogels fabricated by unidirectional freezing, which exhibited isotropic deformation upon stimulation, the APPF hydrogel prepared through the integrated assembly strategy combining unidirectional freezing-induced self-assembly and mechanical stretching-induced orientation, possessed a hierarchically anisotropic network with four distinct levels: The geometrically anisotropic AgNWs were assembled into compartmental walls at the nanoscale (first level), which were crosslinked into a cellular network by cryo-photopolymerization (second level). Then, an aligned lamellar framework with enlarged porous structure were generated via mechanically induced orientation (third level). Through the crosslinking by Fe3+ coordination, an interconnected, long-range ordered anisotropic hydrogel network was finally constructed (fourth level). For comparison, two control hydrogels were prepared via similar polymerization process but without directional freezing assembly (denoted as S-APPF hydrogel), and directional freezing without mechanical stretching (denoted as D-APPF hydrogel), respectively. The S-APPF hydrogel showed a densely packed network that inhibited the formation of continuously connected porous channels (Figs. S4a-c in Supporting information), whereas the D-APPF hydrogel retained open channels but randomized network architectures, which intrinsically constrained anisotropic responsiveness and directional mass transport (Figs. S4d-f in Supporting information).
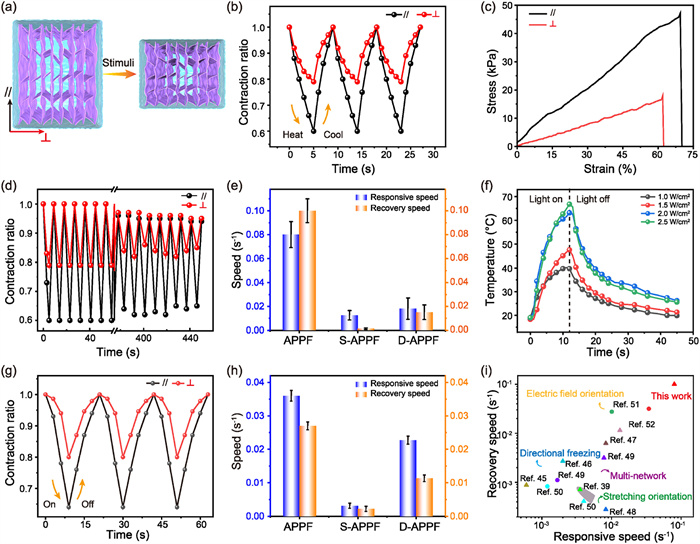
Benefiting from the hierarchical anisotropic structure and open low-tortuosity transport channels, the APPF hydrogel exhibited rapid anisotropic deformation in response to external stimuli (Fig. 3a). When immersed in a 50 ℃ water bath, which exceeded the LCST of PNIPAM, the APPF hydrogel contracted rapidly to 0.60 along the stretching-induced direction (//) within 5 s. In contrast, a contraction ratio of 0.79 was observed in the perpendicular direction (┴), resulting in an anisotropic factor α of 1.9. Once transferred into a water bath at 25 ℃, the shrunk hydrogel was immediately expanded to the original shape within 4 s (Fig. 3b and Fig. S5 in Supporting information). Such pronounced anisotropic deformation was attributed to the oriented lamellar network of the APPF hydrogel, which enabled a greater strain response along the stretching direction under thermal stimulation (Fig. 3c and Fig. S6 in Supporting information) [14,38,39]. The rapid actuation originated from the ordered AgNWs assembly enhancing mechanical strength and elasticity, the interconnected porous channels accelerating water transport, and the oriented lamellar polymer network enabling efficient stress transfer and rapid directional deformation under external stimulation. Impressively, no apparent fatigue was observed after 50 deformation-recovery cycles (Fig. 3d). Even after being immersed in 45 ℃ water for 24 h, the hydrogel rapidly recovered within 2 min after cooling (Fig. S7 in Supporting information).
Figure 3
Figure 3.
(a) Schematic illustration of anisotropic response of the APPF hydrogel. (b) Contraction ratios of the APPF parallel and perpendicular to the stretching direction under thermal stimulus. (c) Tensile stress-strain curves of the APPF parallel and perpendicular to stretching direction. (d) Cycling stability of the thermo-responsive performance of the APPF over 50 cycles. (e) Response and recovery speeds of the APPF, S-APPF, and D-APPF under thermal stimulus. (f) Temperature evolution of the APPF irradiated with NIR light at different intensities. (g) Contraction ratio of the APPF parallel and perpendicular to the stretching direction under NIR light stimulus. (h) Response and recovery speeds of the APPF, S-APPF, and D-APPF under light stimulus. (i) Thermal-responsive (triangles) and photo-responsive (circles) speeds of the APPF and reported hydrogels. The statistical data in (e) and (h) were presented as mean ± standard deviation (SD) (n = 3).
Furthermore, the influences of the mechanical stretching, unidirectional freezing temperature, and the coordination crosslinking on the responsiveness of APPF hydrogels were explored. As the pre-stretch increased from 25% to 75%, the anisotropic factor α was increased continuously (Fig. S8 in Supporting information). This was attributed to the stretching-induced alignment of polymer chains and the formation of oriented pores, which enhanced contraction deformation of the hydrogel network along the stretching direction upon stimulation [14,40]. A decrease in the unidirectional freezing temperature from −10 ℃ to −30 ℃ resulted in a marked reduction in the response speed of the hydrogel (Fig. S9 in Supporting information), primarily due to the accelerated formation of finer ice crystals and a densified gel network, which impeded efficient heat and mass transfer during thermal stimulation [11,12,17]. Besides, as the Fe3+ ion concentration or soaking time increased, the α of the APPF hydrogel initially increased and then decreased (Figs. S10 and S11 in Supporting information). This behavior was arising from that moderate crosslinking promoted the mobility of polymer chains through the dynamic orientation of coordination bonds, thereby enhancing the α; whereas an overhigh crosslinking density induced network rigidification, suppressing the anisotropic response [9,41].
For comparison, the thermal responsiveness of control samples with different structures were investigated. Although the S-APPF hydrogel exhibited anisotropic responsive behavior, the response process was remarkably slow, with a contraction time up to 40 s and a recovery time extending to several minutes. This sluggish response was attributed to its dense and compact microstructure, which significantly hindered water transport and internal structural reconfiguration (Fig. S12a in Supporting information). Meanwhile, isotropic contraction and recovery were observed for the D-APPF hydrogel due to the absence of stretch-induced alignment (Fig. S12b in Supporting information). Furthermore, to investigate the role of AgNWs, the thermal-responsive properties of a PNIPAM/PAM/Fe3+ (PPF) hydrogel without AgNWs was illustrated in Fig. S12c (Supporting information). The PPF hydrogel implemented a shrinkage of 0.68 along the direction parallel to stretching and 0.80 along the perpendicular direction within 8 s under hot water stimulation, and fully recovered to its original shape within 6 s upon immersion in cold water. Although the PPF hydrogel exhibited anisotropic deformation, its anisotropy factor was only 1.6, lower than that of the APPF hydrogel (1.9). Fig. 3e and Fig. S12d in Supporting information clearly concluded the markedly enhanced anisotropic actuation and recovery performances of the APPF hydrogel compared to the control structures.
In addition to the thermal responsiveness, the APPF hydrogel exhibited a rapid response to light irradiation, based on the notable photothermal conversion of the incorporated AgNWs. Specifically, the aligned AgNWs acted as photon-absorbing units, whose localized surface plasmon resonance under near-infrared (NIR) light irradiation enabled remote activation of the hydrogel, thereby promoting efficient heat conduction and enhancing overall photothermal conversion efficiency [42-44]. The photothermal conversion efficiency of the hydrogel was correlated with the content of AgNWs and power intensity (Fig. 3f and Fig. S13 in Supporting information). Under NIR light stimulation at an intensity of 2.5 W/cm2, the APPF hydrogel exhibited anisotropic deformation, contracting by 0.36 along the stretching direction, while the contraction in the perpendicular direction was 0.2, resulting in an α of 1.80. Once the NIR light was turned off, the hydrogel recovered rapidly to its original shape within 12 s (Fig. 3g, Fig. S14 and Video S1 in Supporting information). It was found that the light-induced deformation speed of the APPF hydrogel was slower than that induced by temperature. This was because the hot water bath could uniformly and rapidly increase the temperature of the whole hydrogel, whereas the light-to-heat conversion of AgNWs was localized and limited by the filler distribution and the depth of light penetration. Furthermore, no obvious deterioration in responsiveness was detected after 50 deformation cycles (Fig. S15 in Supporting information). Impressively, the APPF hydrogel achieved a deformation rate of 0.036 s−1 and a recovery rate of 0.027 s−1, which were approximately ten times higher than 0.003 s−1 and 0.002 s−1 observed for S-APPF, respectively, and also superior to those of D-APPF, which underwent isotropic deformation under stimulation (Fig. 3h and Fig. S16 in Supporting information). These results collectively demonstrated that the APPF hydrogel exhibited rapid anisotropic responsiveness under thermal and NIR light stimulation. Moreover, we systematically summarized and compared the key performance parameters of various high-performance hydrogel actuators. The results revealed that the anisotropic stimuli responsiveness of the APPF hydrogel surpassed that of previously reported hydrogels with alternative structural designs (Fig. 3i and Table S1 in Supporting information) [39,45-52].
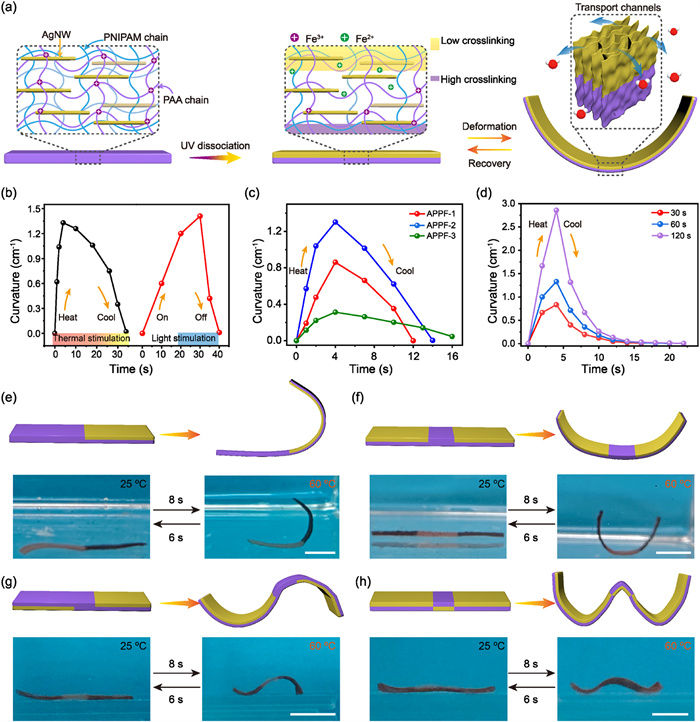
Inspired by biological systems that achieve locomotion, predation, and environmental adaptation, hydrogel actuators with 3D deformation capabilities have emerged as essential components for mimicking natural intelligent multi-degree-of-freedom functions [53,54]. In fact, shape transformations typically originated from non-uniform internal stresses caused by differential swelling or shrinkage, which were induced by the spatial distribution of functional components within the hydrogel matrix. To enable the transition from 2D planar contraction to complex 3D shape-shifting, a programmable deformation system was developed by exploiting the UV-induced dynamic dissociation between Fe3+ ions and PAA. As shown in Fig. 4a, through spatiotemporally controlled UV light irradiation, Fe3+ ions within the illuminated region were reduced to Fe2+ ions, leading to a significant decrease in the coordination crosslinking formed between Fe3+ ions and the carboxyl groups of PAA. Consequently, a crosslinking density gradient was established along the thickness direction of the APPF hydrogel, resulting in the formation of a low-crosslinking (LC) region and a high-crosslinking (HC) region [55,56]. The spatially heterogeneous coordination crosslinking network enabled preferential deformation of LC side under thermal or light stimulation, as the relatively looser network structure and lower contraction resistance on this side allowed faster contraction compared to the HC region. Under thermal stimulation in 60 ℃ water, the hydrogel achieved a bending curvature of 1.33 cm−1 within 5 s and fully recovered to its initial state within 30 s (Fig. 4b and Fig. S17a in Supporting information). Moreover, under NIR stimulation at 2.5 W/cm2, a maximum curvature of 1.41 cm−1 was reached in approximately 30 s, with complete recovery occurring within 10 s (Fig. 4b and Fig. S17b in Supporting information). Impressively, no apparent fatigue was observed after 50 deformation-recovery cycles (Fig. S18 in Supporting information). These results demonstrated that the UV-induced spatial dissociation strategy imparted the APPF hydrogel with remarkable light-responsive 3D deformation capability, highlighting its strong potential for advanced soft actuators and bioinspired systems.
Figure 4
Figure 4.
(a) Schematic illustration of the spatially modulated photodissociation of APPF hydrogel for the stimulus-responsive 3D deformation. Spatially controlled UV irradiation reduced Fe3+ to Fe2+ within the exposed regions, generating a gradient network dissociation along the light path. Upon thermal or light stimulation, the hydrogel rapidly bent toward the low-crosslinking region with the help of open transport channels. (b) Curvature changes over time under thermal (left) and light (right) stimulus. (c) Curvature changes over time for systems with different amounts of AA. (d) Effects of different UV light durations on the curvature change of APPF hydrogels. (e-h) Schematic diagrams and corresponding optical images showing thermally induced shape transformations of APPF hydrogels with region-selective UV irradiation. All deformations were induced in 60 ℃ water and recovered at 25 ℃. Scale bar: 1 cm.
To investigate the influence of the crosslinking network on the 3D deformation of UV-programmed APPF hydrogels, three hydrogel formulations (APPF-1 to APPF-3, with AA/NIPAM molar ratios of 1:7, 1:5 and 1:4, respectively) were prepared, and their curvature evolution under thermal stimulation at 60 ℃ was recorded after 1 min of UV exposure (Fig. 4c). The APPF-1 hydrogel with the lowest AA content, exhibited a maximum bending curvature of 0.86 cm−1, whereas the APPF-2 hydrogel reached 1.3 cm−1 within 4 s. However, further increasing the AA content resulted in a marked decrease in curvature to 0.31 cm−1 (Fig. S19a in Supporting information). This non-monotonic behavior could be attributed to the underlying coordination and network structure. At low AA content, the limited number of Fe3+-COO− coordination sites led to insufficient crosslinking density gradients, thereby weakening the deformation capability [19,57,58]. In contrast, excessive AA content reduced the free volume fraction within the PNIPAM network, restricting thermal contraction and diminishing actuation efficiency. The influence of UV irradiation duration was further investigated. As shown in Fig. 4d, APPF hydrogels irradiated for 30, 60, and 120 s achieved linearly increased deformation magnitude under temperature stimulus, reaching maximum curvatures of 0.83, 1.31 and 2.86 cm−1, respectively, within 4 s. Furthermore, the maximum bending deformation increased stepwise with temperature (Fig. S19b in Supporting information). Similarly, as the power increased from 1.0 W/cm2 to 3.0 W/cm2, the maximum curvature rose from 0.21 cm−1 to 0.47 cm−1, while the response time decreased from 4 s to 2 s (Fig. S20 in Supporting information). Compared to the APPF hydrogel with deformation and recovery velocities of 0.33 and 0.13 cm−1 s−1 respectively, the D-APPF hydrogel displayed bending deformation velocities of merely 0.24 and 0.094 cm−1 s−1. Meanwhile, despite rapid bending deformation upon stimulation being measured for the S-APPF sample, it was difficult to recover its initial shape upon cooling (Fig. S21 in Supporting information). This remarkable performance was attributed to unique anisotropic architecture of the APPF hydrogel, whose hierarchical network structure and open transport channels collectively enhanced its actuation and recovery capabilities compared to control structures.
To enhance the programmability and spatial control of hydrogel deformation, a photomask-assisted region-selective UV irradiation strategy was employed. By customizing photomask patterns, the spatial dissociation of Fe3+-COO− coordination bonds were regulated, thereby enabling the construction of an intelligent hydrogel capable of multimodal biomimetic deformation. As illustrated in Figs. 4e-h, the synergistic regulation of photomask design and irradiation parameters enabled the precise programming of multimodal biomimetic deformations. Specifically, when UV-induced dissociation was localized to the right half of a rectangular hydrogel (40 mm × 10 mm × 0.8 mm), a characteristic L-shaped curvature was observed upon thermal stimulation at 60 ℃. Utilizing a composite photomask to shield the central region while irradiating the upper bilateral zones resulted in a dual-curvature U-shaped configuration. Furthermore, asymmetric dissociation targeting the upper-left and lower-right regions facilitated the self-morphing of the hydrogel into an S-shaped structure. Finally, gradient programming achieved by applying gradient dissociation to both upper sides in combination with localized dissociation in the central lower region induced a triple-curvature wave-like deformation resembling a W-shape, demonstrating the system’s high spatial programmability and shape-morphing versatility (Video S2 in Supporting information).
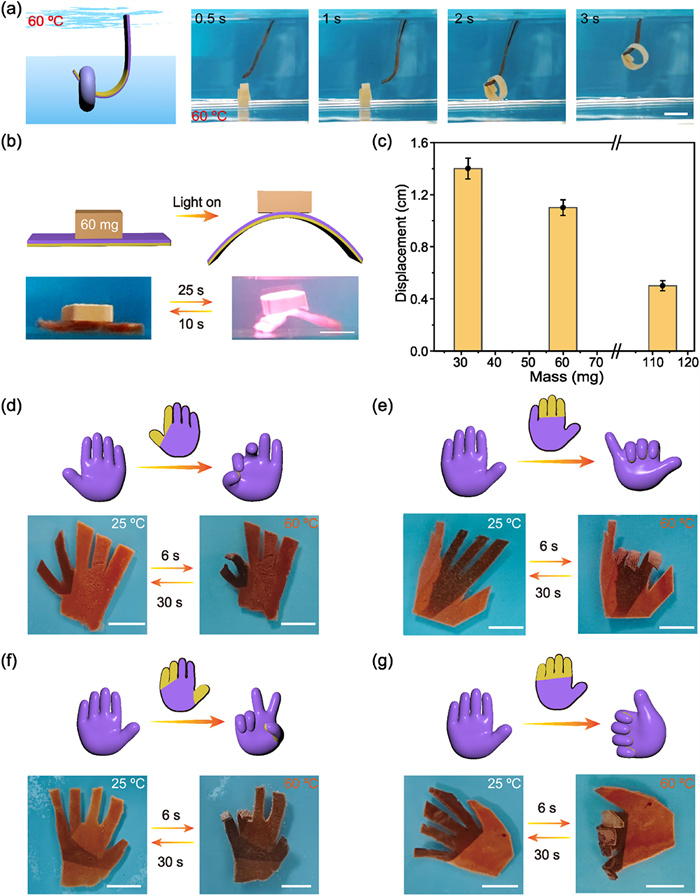
To further explore the practical utility and multifunctionality of the APPF hydrogels, a series of actuation demonstrations were conducted under various stimuli and application scenarios. As shown in Fig. 5a, the UV-irradiated APPF hydrogel (dimensions: 40 mm × 10 mm × 0.8 mm) exhibited ultrafast actuation under thermal stimulation at 60 ℃, reaching a curvature of 0.62 cm−1 within 3 s. This rapid deformation enabled efficient object manipulation, with a load-bearing capacity up to 200% of its own weight (Video S3 in Supporting information). In addition, a spatially programmed hydrogel jack (length: 40 mm, width: 8 mm, mass: 30 mg) was designed (Fig. 5b). Upon NIR irradiation (2.5 W/cm2), the APPF hydrogel exhibited rapid bending actuation within 25 s, successfully lifting a 60 mg load, which returned to its original state within 10 s. An effective displacement was generated by the actuator under loads of up to 3.8 times its self-weight, demonstrating its good load adaptability and load-bearing capacity. Specifically, under 30 mg load (1× self-weight), a displacement of 1.4 cm was achieved. When the load increased to 113 mg (3.8× self-weight), the actuator maintained a displacement of 0.5 cm (Fig. 5c, Fig. S22 and Video S4 in Supporting information). These results confirmed that the UV light-spatially programmed hydrogel actuator possessed multifunctionality, along with notable load-bearing ability with rapid responsiveness, indicating its potential for application in minimally invasive surgical manipulators and microscale industrial robotics.
Figure 5
Figure 5.
(a) Schematic illustration and time-sequenced optical images of a hydrogel actuator rapidly grabbing an object in 60 ℃ hot water. (b) Schematic diagram and corresponding optical images showing the bending of the hydrogel actuator lifting a 60 mg object under NIR light irradiation. (c) Bar graph showing the lifting height of the hydrogel actuator under various loads ranging from 30 mg to 113 mg. (d-g) Schematic diagrams and optical images of bioinspired hand-like actuators performing different finger-pinching motions by spatially programmed UV light irradiation. Scale bar: 1 cm. The statistical data in (c) were presented as mean ± SD (n = 3).
Impressively, using a spatial coordination dissociation strategy by the photomask-assisted region-selective UV irradiation, programmable and multimodal biomimetic hand-like deformations were achieved. By spatially encoding the irradiation patterns, distinct regions in finger were selectively activated, enabling a series of heat-triggered gesture transformations at 60 ℃. For example, in the two-finger actuation mode, gradient irradiation was applied to the index finger and thumb, inducing synchronized bending that brought them into contact and resulted in a biomimetic “OK” gesture (Fig. 5d). In the three-finger actuation mode, two distinct gestures were obtained depending on the masking design: for the “digital six” gesture, the thumb and little finger were masked while the middle three fingers bent cooperatively (Fig. 5e); for the “yeah” gesture, only the index and middle fingers were masked, while the thumb, ring, and little fingers were irradiated, leading to asymmetric deformation of the exposed fingers (Fig. 5f). In the four-finger actuation mode, the thumb was masked and the remaining fingers responded independently to irradiation, with the index fingertip reaching a curvature of 1.21 cm−1, ultimately forming a recognizable “thumbs-up” gesture (Fig. 5g). Although the recovery time of the hand-shaped hydrogel was prolonged, owing to the increased geometric complexity and the difficulty in achieving coordinated deformation among multiple units, the results still highlighted the versatility and spatial precision of UV-programmed hydrogel actuators in generating complex, gesture-specific deformations, thereby offering promising potential for applications in soft robotics, biomimetic grippers, and intelligent human-machine interfaces.
In conclusion, we have reported a synergistic fabrication approach that integrated directional freezing-induced self-assembly with mechanical stretching-induced alignment crosslinked by the metal coordination to construct anisotropic hydrogel actuators featuring rapid responsiveness and programmable 3D deformation. The resulting hydrogel, endowed with a hierarchically aligned lamellar network and open mass transport channels, demonstrated pronounced anisotropic contraction under thermal and NIR stimulation, with shrinkage ratios reaching 0.60 parallel and 0.79 perpendicular to the lamellar direction under the thermal activation. These anisotropic actuation behaviors were rapid and highly reversible, surpassing the response and recovery speeds of the reported hydrogel actuators. Importantly, by leveraging spatially dynamic modulated coordination photodissociation, a gradient crosslinking architecture were introduced within the hydrogel matrix, enabling complex, localized deformations. This capability facilitated diverse biomimetic actuation modes, including shape reconfiguration, object grasping, and programmable finger gestures. Overall, this work addresses the limitations in hydrogel-based actuators related to slow response and poor programmability, offering a generalizable strategy for constructing next-generation intelligent soft materials. The combination of structural anisotropy, dynamic programmability, and multifunctional responsiveness positions this system as a promising platform for applications in soft robotics, minimally invasive medical tools, and adaptive optomechanical systems.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Yi Zhang: Writing – original draft, Validation, Methodology, Data curation. Xin Yao: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Conceptualization. Jiaxin Yu: Validation, Formal analysis. Haili Qin: Methodology, Formal analysis. Huaiping Cong: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (Nos. 22471052, 22171066), the Anhui Provincial Natural Science Foundation (No. 2308085MB46), and the Fundamental Research Funds for the Central Universities (No. JZ2023YQTD0074).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112041.
Figure 1
Schematic illustration of the fabrication of the APPF hydrogel by a consecutive directional freezing-induced self-assembly and metal coordination crosslinked mechanical alignment process. (a) A precursor solution containing NIPAM, AA, photoinitiator 2-hydroxy-2-methyl-1-phenyl-1-propanone (HMPP), crosslinker N,N’-methylenebisacrylamide (MBA), Fe3+ and AgNWs was assembled into an aligned scaffold via directional freezing. (b) The APP hydrogel with a 3D ordered honeycomb network structure was synthesized through cryo-photopolymerization. (c) Mechanical stretching was applied to the APP hydrogel to induce anisotropic alignment of the polymer network. (d) The hierarchically anisotropic APPF hydrogel integrated with open transport channels was obtained via ionic crosslinking.
Figure 2
SEM images with different magnifications of the APPF hydrogel: Top view (a-c) and side view (d-f). (g) Elemental mappings of C, N, Ag, and Fe on the APPF hydrogel surface. (h) FT-IR spectra of APPF and APP hydrogels. (i) The magnification of FT-IR spectra in the squared area. (j) Temperature- modulated DSC curve of the APPF hydrogel.
Figure 3
(a) Schematic illustration of anisotropic response of the APPF hydrogel. (b) Contraction ratios of the APPF parallel and perpendicular to the stretching direction under thermal stimulus. (c) Tensile stress-strain curves of the APPF parallel and perpendicular to stretching direction. (d) Cycling stability of the thermo-responsive performance of the APPF over 50 cycles. (e) Response and recovery speeds of the APPF, S-APPF, and D-APPF under thermal stimulus. (f) Temperature evolution of the APPF irradiated with NIR light at different intensities. (g) Contraction ratio of the APPF parallel and perpendicular to the stretching direction under NIR light stimulus. (h) Response and recovery speeds of the APPF, S-APPF, and D-APPF under light stimulus. (i) Thermal-responsive (triangles) and photo-responsive (circles) speeds of the APPF and reported hydrogels. The statistical data in (e) and (h) were presented as mean ± standard deviation (SD) (n = 3).
Figure 4
(a) Schematic illustration of the spatially modulated photodissociation of APPF hydrogel for the stimulus-responsive 3D deformation. Spatially controlled UV irradiation reduced Fe3+ to Fe2+ within the exposed regions, generating a gradient network dissociation along the light path. Upon thermal or light stimulation, the hydrogel rapidly bent toward the low-crosslinking region with the help of open transport channels. (b) Curvature changes over time under thermal (left) and light (right) stimulus. (c) Curvature changes over time for systems with different amounts of AA. (d) Effects of different UV light durations on the curvature change of APPF hydrogels. (e-h) Schematic diagrams and corresponding optical images showing thermally induced shape transformations of APPF hydrogels with region-selective UV irradiation. All deformations were induced in 60 ℃ water and recovered at 25 ℃. Scale bar: 1 cm.
Figure 5
(a) Schematic illustration and time-sequenced optical images of a hydrogel actuator rapidly grabbing an object in 60 ℃ hot water. (b) Schematic diagram and corresponding optical images showing the bending of the hydrogel actuator lifting a 60 mg object under NIR light irradiation. (c) Bar graph showing the lifting height of the hydrogel actuator under various loads ranging from 30 mg to 113 mg. (d-g) Schematic diagrams and optical images of bioinspired hand-like actuators performing different finger-pinching motions by spatially programmed UV light irradiation. Scale bar: 1 cm. The statistical data in (c) were presented as mean ± SD (n = 3).

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
