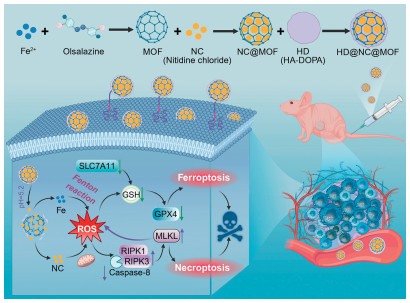
Scheme 1.
Schematic illustration of the fabrication of HD@NC@MOF and their synergistic therapeutic mechanisms for LUSC.
Targeted delivery of nitidine chloride inducing ferroptosis and necroptosis as a novel anti-lung squamous cell carcinoma strategy
Xiaoqin Pan , Changsheng Li , Xinyi Ai , Changqing Tao , Jingchuan He , Tingting Li , Zhihua Deng , Xiaocheng Mo , Ya Chen , Xiumei Qin , Dongmei Wang , Ronghua Jin , Jie Yang

Lung cancer remains the leading cause of cancer-related morbidity and mortality worldwide, with its burden particularly pronounced in China [1,2]. Lung squamous cell carcinoma (LUSC), accounting for approximately 20%–30% of all lung cancer cases [3], is often diagnosed at advanced stages, accompanied by extensive dissemination and metastasis, thereby precluding curative resection and relegating systemic chemotherapy to the frontline therapeutic modality [4]. Nevertheless, the efficacy of conventional cytotoxic regimens is significantly compromised by the emergence of drug resistance [5,6]. In recent years, bioactive constituents derived from traditional Chinese medicine (TCM) have emerged as promising agents that can reverse resistance and simultaneously target multiple pathways for therapy. These findings provide a complementary strategy to address the limitations of current chemotherapy and illuminate new avenues for LUSC treatment [7,8].
Active components constitute a significant reservoir for drug discovery, as evidenced by the fact that over one-third of drugs approved between 1981 and 2014 originated from TCM sources [9,10]. Nitidine chloride (NC), the principal bioactive alkaloid isolated from Zanthoxylum nitidum (Roxb.) DC., a medicinal herb indigenous to Guangxi, China, has demonstrated robust antitumor efficacy by inhibiting tumor proliferation, inducing reactive oxygen species (ROS) generation, and activating caspase 3/gasdermin E (GSDME)-dependent pyroptosis as we reported previously [11,12]. Notably, extracts of Zanthoxylum nitidum (Liang-Mian-Zhen), exemplified by the commercially available Liang-Mian-Zhen analgesic tablets, have been approved for the clinical management of cancer-associated pain in patients with hepatocellular carcinoma, highlighting the clinical application value of this Guangxi-specific botanical [13,14]. Nevertheless, the clinical translation of NC is severely hampered by its extremely low aqueous solubility (< 367 µg/mL) [15] and potential off-target toxicity [16,17]. Therefore, constructing novel delivery systems capable of achieving tumor-specific accumulation and sustained release has emerged as a pivotal strategy to enhance the chemotherapeutic index of NC.
Drug delivery systems (DDSs) represent a critical strategy to enhance the bioavailability of therapeutic molecules by improving solubility, prolonging circulation half-life, and enhancing targeting efficiency, thereby mitigating off-target toxicity [18-21]. Metal-organic framework (MOF), distinguished by its high specific surface area, tunable structure, and plentiful metal active sites, has emerged as prominent platform for drug delivery [22-26]. Leveraging the intrinsic advantages of MOF and the ability of NC to induce ferroptosis via ROS generation [27], we designed a mixed-valence iron-based MOF. Olsalazine, which has been used clinically since 1985 for the treatment of ulcerative colitis, demonstrated favorable efficacy and tolerability [28]. To overcome the biocompatibility limitations often associated with conventional MOF ligands, we developed an olsalazine-based MOF. Furthermore, given the pronounced overexpression of CD44 receptors on the membrane of LUSC [29], we functionalized nanoparticles with hyaluronic acid (HA) to exploit ligand-receptor-mediated active targeting. This approach enabled specific accumulation within LUSC lesions, thereby markedly enhancing therapeutic precision while minimizing off-target toxicity [19,30-33].
In this study, we designed and constructed a MOF (Fe-MOF) using olsalazine as the ligand and iron as the metal node, which was co-loaded with NC. The system was further functionalized with HA, serving as both a gating switch and a targeting molecule, to form the HD@NC@MOF nano-delivery platform (Scheme 1). This approach enables precise treatment of LUSC. Upon CD44-receptor-mediated endocytosis, the nanocomposites rapidly disassembled in the acidic intratumoral microenvironment, leading to synchronous release of NC and redox-active Fe ions. The released NC, coupled with Fe2+/Fe3+ cycling, triggered a Fenton-like reaction, resulting in ROS bursts that deplete glutathione (GSH) and promote lipid peroxidation, thereby inducing ferroptosis. Concomitantly, excessive ROS indirectly nucleated the receptor-interacting serine/threonine-protein kinase 1 (RIPK1)/RIPK3 necrosome, leading to mixed lineage kinase domain-like pseudokinase (MLKL) phosphorylation and necroptosis. Intriguingly, activated MLKL further stimulated nicotinamide adenine dinucleotide phosphate oxidase (NOX), amplifying ROS generation in a self-reinforcing positive-feedback loop [34-36]. The synergistic crosstalk between ferroptosis and necroptosis markedly potentiated the antitumor efficacy of HD@NC@MOF.
Emerging evidence suggested that ROS could trigger programmed cell death, positioning ROS-modulating strategies as promising anticancer avenues [37-39]. To synergistically amplify NC-mediated ROS generation, a Fe-MOF was synthesized and used as a carrier for NC encapsulation. To overcome the modification challenges caused by electrostatic repulsion, 3,4-dihydroxyphenylalanine (DOPA) functionalized HA (HA-DOPA, HD) was synthesized using the established protocol (Fig. 1A). This HD was then used to modify the MOF surface, endowing it with targeted recognition properties. As shown in Fig. S1 (Supporting information), the synthesis and preparation procedures were delineated in Fig. S1A. The peak observed at 1060 cm−1 was assigned to the stretching vibration of the glycosidic bond (C-O-C) in HA (Fig. S1B). Furthermore, the ultraviolet-visible (UV–vis) spectrum of HD showed a characteristic absorption peak at 280 nm, corresponding to DOPA (Fig. S1C). These results confirm the successful synthesis of HD.
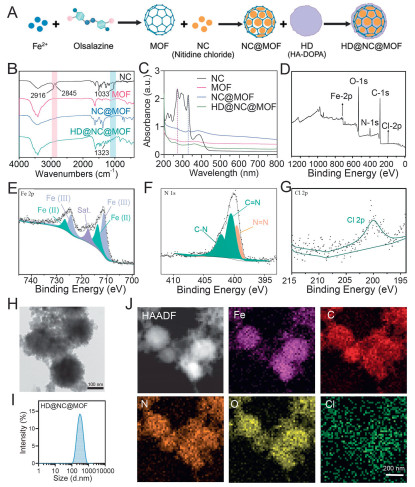
In the Fourier transform infrared spectroscopy (FTIR) spectrum of HD@NC@MOF (Fig. 1B), the peaks at 2845 and 2916 cm−1 were assignable to the symmetric and asymmetric C–H stretching vibrations of methylene groups. The peak at 1033 cm−1 corresponded to the stretching vibration of the ether bond (C-O-C) of NC, confirming successful NC encapsulation within the MOF framework. Additionally, the band at 1323 cm−1 was assigned to the N-H stretching vibration of the HA shell, corroborating surface HD functionalization. The UV–vis absorption spectra exhibited characteristic peaks of NC and DOPA (Fig. 1C), further confirming the successful loading of NC and the coating of HD onto the MOF surface.
To further elucidate the valence states and element composition of the HD@NC@MOF, XPS analysis was conducted. The survey spectrum in Fig. 1D confirmed the presence of C, N, O, Fe and Cl within the material. The Fe 2p spectra (Fig. 1E) exhibited peaks at 711.7 and 724.4 eV, which were characteristic of Fe(Ⅲ). Peaks at 713.8 and 726.8 eV were corresponded to Fe(Ⅱ), indicating a mixed-valence Fe species. The presence of mixed-valence metal centers provided abundant redox-active sites, significantly contributing to ROS generation in HD@NC@MOF. The N 1s spectrum showed the characteristic peak at 399.6 eV for the azo group, while the peaks at 400.5 and 402.2 eV corresponded to C-N and C=N bonds of NC, respectively (Fig. 1F). The distinctive peak at 200.5 eV in the Cl 2p spectrum further confirmed the successful loading of NC (Fig. 1G). As shown in Fig. S2 (Supporting information), the O 1s spectrum (Fig. S2A) exhibited two peaks at binding energies of 531.5 eV (O=C) and 533.4 eV (O-C). In the C 1s spectrum (Fig. S2B), three peaks were detected at 284.8, 286.5, and 288.4 eV, corresponding to C-C, C-O, and C=O bonds, respectively.
Further morphological characterization was performed by transmission electron microscopy (TEM). As shown in Fig. 1H, HD@NC@MOF presented rough-surfaced spherical particles' attributable to HD coating. Dynamic light scattering (DLS) revealed an increase in hydrodynamic diameter from 248.8 ± 10.2 nm (NC@MOF) to 281.3 ± 19.45 nm (HD@NC@MOF) (Fig. 1I, Fig. S3A in Supporting information), accompanied by a zeta potential shift from −16.07 ± 0.74 mV to −24.13 ± 1.30 mV (Fig. S3B in Supporting information). The concomitant increase in hydrodynamic diameter and the marked shift in zeta potential collectively corroborated the successful HD encapsulation. The elemental composition of the HD@NC@MOF was analyzed via elemental mapping (Fig. 1J), confirming the homogeneous distribution of C, N, O, Cl and Fe across the HD@NC@MOF. Furthermore, the detailed elemental composition data were summarized in Table S1 (Supporting information). Given that iron could generate ROS through the Fenton reaction [40], we further investigated the ROS-producing capacity of the carrier. As shown in Fig. S4 (Supporting information), the concentration-dependent decrease in UV absorption of methylene blue demonstrated the carrier's ability to generate ROS, thereby supporting its role in synergizing with NC to exert anti-tumor effects. The elemental composition correlated with a drug loading capacity of 16% (Fig. S5 in Supporting information), demonstrating consistency between compositional characterization and NC loading capacity.
The tumor microenvironment-responsive release mechanism spatiotemporally controls drug delivery at lesion sites, which enhances bioavailability while minimizing off-target toxicity. As illustrated in Fig. S6 (Supporting information), the HD@NC@MOF exhibited pronounced pH-responsive release behavior: At pH 5.2, ~70.5% of NC was released, approximately threefold higher than that at physiological pH 7.4 (~25.7%). The stability of HD@NC@MOF was evaluated across a variety of media. As demonstrated in Fig. S7 (Supporting information), both the hydrodynamic diameter (~280 nm) and ζ-potential (~−24 mV) remained virtually unchanged over a 48 h period in H2O, phosphate-buffered saline (PBS), and fetal bovine serum (FBS), confirming the high stability required for prolonged systemic circulation and efficient tumor accumulation.
The selective accumulation of nanomedicine at pathological sites critically governed its pharmacological efficacy, therapeutic duration, and ultimate treatment outcomes. Given the reported overexpression of CD44 receptor on LUSC membrane [29], CD44 expression across relevant cell lines was quantified. Western blot (WB) analysis (Fig. 2A and Fig. S8 in Supporting information) confirmed maximal CD44 expression in SK-MES-1 LUSC cells and minimal expression in normal HBE cells, establishing these as optimal models for subsequent studies. The tumor targetability of nanomedicine enabled discrimination between malignant and normal cells, thereby enhancing the selective cytotoxicity of nanomedicine. As illustrated in Fig. 2B, free NC exhibited higher toxicity toward normal cells than tumor cells, yielding a selectivity index (SI) of 0.54. Remarkably, HD@NC@MOF reversed this selectivity profile, demonstrating a 5-fold increased SI of 2.58 compared to free NC with enhanced tumor-selective cytotoxicity (Fig. 2C). This enhanced tumor-selective cytotoxicity was further validated by confocal laser scanning microscopy (CLSM) and flow cytometry (FCM). The concentration-dependent selective internalization was further investigated. As shown in Fig. S9 (Supporting information), the intracellular fluorescence intensity increased significantly with increasing concentration and reached a plateau at 50 µg/mL. As evidenced in Figs. 2D–F and Fig. S10 (Supporting information), SK-MES-1 cells exhibited 1.73-fold higher cellular uptake efficiency than HBE cells. This targeting effect was abolished by free HA competition, confirming HA-mediated receptor-specific endocytosis, which was consistent with the results presented in Fig. S11 (Supporting information).
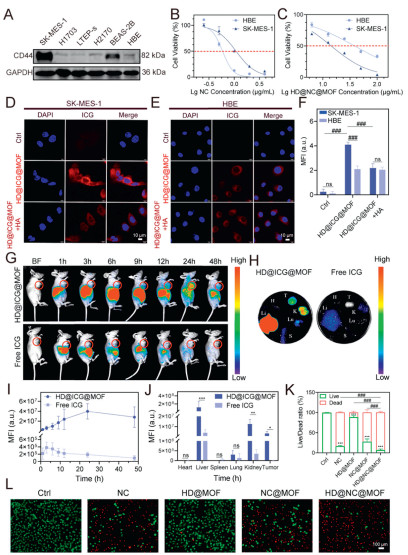
Enhanced intra-tumoral retention, as established in prior studies, was paramount for optimizing therapeutic efficacy in targeted DDSs. We encapsulated indocyanine green (ICG) into the system via the same procedure to prepare HD@ICG@MOF and observed its intratumoral retention. The animal model used was female BALB/c nude mice (5 weeks old), obtained at the Guangxi Experimental Animal Center, and the experimental protocol was approved by the Animal Welfare and Ethics Committee of Guangxi Medical University. Pharmacokinetic profiling revealed distinct biodistribution patterns of free ICG and HD@ICG@MOF. Free ICG exhibited rapid clearance, peaked at 12 h and cleared by 24 h. In contrast, HD@ICG@MOF showed a delayed peak concentration of 24 h and with 85% fluorescence intensity at 48 h (Figs. 2G and I). Ex vivo organ imaging (Figs. 2H and J) demonstrated predominant accumulation of HD@ICG@MOF in the liver and kidney compared to free ICG, with significantly enhanced tumor targeting. The superior selective cytotoxicity of HD@NC@MOF was further validated through cell counting kit-8 (CCK-8) assays and live/dead staining (Figs. 2K and L, Fig. S12 in Supporting information), demonstrating significantly enhanced SIs: HD@NC@MOF (2.58) vs. NC@MOF (0.44), bare MOF (1.07), and HD@MOF (2.51) (Table S2 in Supporting information). The cytotoxicity of the raw material, olsalazine, was also evaluated. As shown in Fig. S13 (Supporting information), even at a concentration as high as 200 µg/mL, the viability of SK-MES-1 cells remained around 75%, indicating minimal intrinsic cytotoxicity at this concentration. To explore the mechanism underlying the excellent anti-tumor performance, cell viability was assessed after treatment with 2 µg/mL NC or 25 µg/mL NPs for 12 h. Under these conditions, the viability of SK-MES-1 cells exceeded 80% in all groups, with no significant differences observed (Fig. S14 in Supporting information). Based on these results, this concentration was chosen for subsequent mechanistic studies evaluating effects on ROS levels, GSH, iron content, and malondialdehyde (MDA).
To elucidate the mechanism underlying HD@NC@MOF's selective cytotoxicity, intracellular ROS levels were quantitatively assessed using fluorescent microscopy and FCM in treated cells. As shown in Fig. 3A and Figs. S15A, C and S16 (Supporting information), free NC alone activated intrinsic ROS generation, and the HD@MOF itself also generated detectable ROS. Notably, HD@NC@MOF synergistically amplified ROS production, achieving 1.84-fold higher than HD@MOF and 1.33-fold greater than NC@MOF. Rescue experiments were conducted to validate whether ROS generation mediated the anti-tumor effect of NC. Pre-treatment with N-acetylcysteine (NAC) markedly reduced NC-induced cytotoxicity in SK-MES-1 cells (Fig. S17 in Supporting information), further supporting the critical role of ROS in the therapeutic mechanism of NC [41]. Given that mitochondria were the primary intracellular source of ROS [42], their role in this process required systematic investigation. As depicted in Fig. 3B, the colocalization studies demonstrated significant overlap between fluorescein isothiocyanate (FITC)-labeled HD@NC@MOF and mitochondria markers, providing evidence that HD@NC@MOF was likely perturbing mitochondrial function to enhance ROS production. Ultrastructural alterations were further analyzed by TEM. As shown in Fig. 3C, the untreated control (Ctrl) cells exhibited intact mitochondria (green arrows) and endoplasmic reticulum (ER, blue arrows). In contrast, HD@NC@MOF-treated cells displayed severe mitochondrial damage, characterized by cristae loss (red arrows), internal cytoplasmic vacuolization (pink arrows), and membrane blurring (orange arrows). Notably, these mitochondrial disruptions were partially restored in HD@NC@MOF + HA co-treated cells, confirming CD44-mediated endocytosis as the key pathway for mitochondrial targeting and subsequent antitumor efficacy.
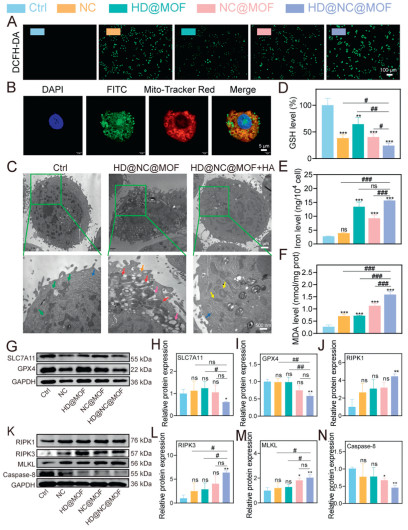
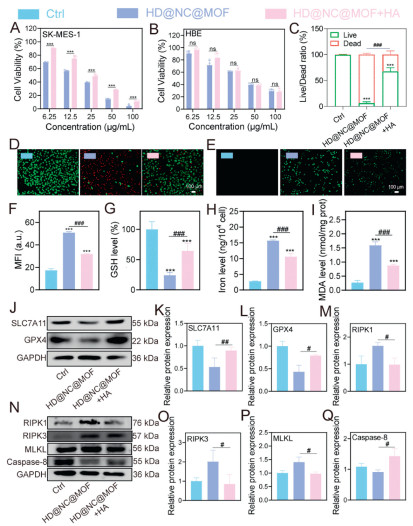
Cell death modalities were further quantified via 7-aminoactinomycin D (7-AAD)/Annexin V dual staining. As revealed in Fig. S18 (Supporting information), necrotic cell populations (20.40%) predominated over apoptotic cells (8.62%), demonstrating a 2.83-fold preference for a non-apoptotic cell death mechanism. This distinct death profile suggested that HD@NC@MOF treatment induced neroptosis rather than apoptosis. To further characterize the cell death modalities, pharmacological inhibition assays were conducted. As shown in Fig. S19 (Supporting information), significant cytoprotection was observed with the use of necroptosis inhibitor Nec-1, ferroptosis inhibitors (DFO, ferrostatin-1, liproxstatin-1), and ROS scavenger NAC. This rescue experiment supported that both ferroptosis and necroptosis pathways were involved in HD@NC@MOF-induced cell death. HD@NC@MOF was demonstrated to induce significant GSH depletion (Fig. 3D), elevated intracellular iron levels (Fig. 3E), and increased malondialdehyde (MDA), which was a lipid peroxidation biomarker (Fig. 3F). WB analysis further confirmed the downregulation of ferroptosis hallmark proteins, glutathione peroxidase 4 (GPX4) and solute carrier family 7 member 11 (SLC7A11), upregulation of necroptosis markers (RIPK1, RIPK3, and MLKL), and reduced caspase-8 expression (Figs. 3G–N) after HD@NC@MOF treatment. HD@NC@MOF activated the RIPK1/RIPK3/MLKL pathway through a system-specific dual mechanism: it not only induced high levels of ROS to promote RIPK1/RIPK3-MLKL activation but also inhibited caspase-8 to suppress apoptosis [34,43,44]. In contrast, conventional ROS-generating agents often failed to reliably induce necroptosis due to their lack of regulation over the apoptotic pathway. To further demonstrate whether all ROS inducers activate necroptosis, we treated SK-MES-1 cells with H2O2 (a widely used ROS inducer) and analyzed the expression of key necroptosis-related markers (RIPK1, RIPK3, MLKL) and caspase-8. As shown in Fig. S20 (Supporting information), neither 200 nor 400 µmol/L H2O2 treatment caused significant changes in the expression levels of these proteins compared to the control group, confirming that ROS generation alone was not sufficient to activate the necroptosis pathway. However, after 12 h of treatment with HD@NC@MOF, the expression of caspase-8 protein was significantly inhibited, while the expression of RIPK1, RIPK3, and MLKL proteins were significantly upregulated, thereby inducing necroptosis in cells.
The impact of free HA on antitumor efficacy was systematically validated through CCK-8 assays, live/dead staining, and WB. As illustrated in Figs. 4A and B, supplementation with free HA shifted the half maximum inhibitory concentration (IC50) from 14.36 ± 0.39 µg/mL to 39.00 ± 3.57 µg/mL with an approximately three-fold-reduction in tumor cell killing. Live/dead staining results (Figs. 4C and D) corroborated this observation of decreased cytotoxicity. Mechanistically, HA addition markedly attenuated intracellular ROS and restored GSH levels, while simultaneously diminishing labile iron and MDA accumulation (Figs. 4E–I, Figs. S15B and D in Supporting information). WB analysis further revealed a reversal of ferroptosis-relevant protein expression after free HA treatment, presenting as GPX4, SLC7A11, and cleaved caspase-8 were up-regulated, RIPK1, RIPK3, and MLKL were down-regulated (Figs. 4J–Q). Collectively, these data underscored the pivotal role of free HA in attenuating ferroptosis and enhancing tumor-selective cytotoxicity.
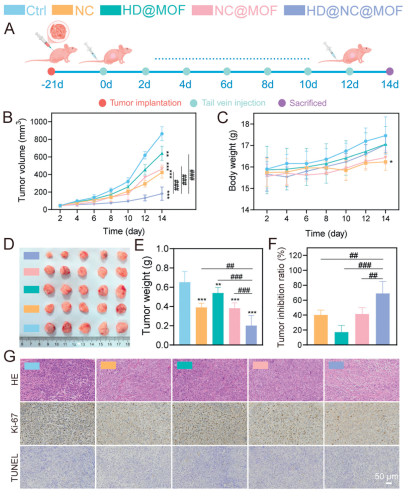
Inspired by promising in vitro therapeutic outcomes, we evaluated the in vivo antitumor efficacy of the engineered HD@NC@MOF. The process of the in vivo experiment is shown in Fig. 5A. As illustrated in Figs. 5B–F, HD@NC@MOF demonstrated superior antitumor efficacy with a 69.02% tumor inhibition rate, significantly outperforming both NC@MOF (41.41%) and free NC (40.18%). HD@NC@MOF-treated mice exhibited steady body weight gain, whereas free NC and NC@MOF groups showed significant weight loss attributed to off-target drug distribution. These results further demonstrated HD@NC@MOF's superior capacity to mitigate NC-induced systemic toxicity. To elucidate the in vivo antitumor mechanism, paraffin sections were subjected to hematoxylin-eosin (HE), Ki-67 and terminal-deoxynucleotidyl transferase mediated nick end labeling (TUNEL) staining. Consistent with the in vitro results, the HD@NC@MOF exhibited a pronounced reduction in Ki-67-positive proliferating cells, yet negligible TUNEL-positive apoptotic nuclei (Fig. 5G and Fig. S21 in Supporting information). Histopathological examination of major organs (Fig. S22 in Supporting information) revealed only mild injury in the heart, liver, and kidneys in the NC group, whereas no discernible lesions were observed in the HD@NC@MOF group. Hemocompatibility assays showed that HD@NC@MOF induced < 5% hemolysis even at a concentration of 400 µg/mL, demonstrating excellent blood compatibility (Fig. S23 in Supporting information). Furthermore, complete blood count analysis indicated no significant differences in key parameters, including red blood cell count (RBC), white blood cell count (WBC) and platelet count (PLT), between any treatment group and the control group (Table S3 in Supporting information). These results confirm that HD@NC@MOF did not induce significant adverse effects on the hematological system of tumor-bearing nude mice at the pharmacodynamic dose. Biochemical analyses further corroborated these findings: serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and serum creatinine (CREA) remained within the normal range for all treatment arms (Fig. S24 in Supporting information).
Zanthoxylum nitidum (Roxb.) DC., a characteristic medicinal herb in Guangxi, China, has a long history of application in TCM and is widely used in modern medicine. NC, as one of its main active components, has demonstrated significant potential as an anticancer agent [16,45]. However, its poor aqueous solubility and substantial toxicity towards normal cells severely limit its clinical utility [15,16]. Recent efforts have focused on employing nanomaterial-based delivery systems to overcome these limitations. For instance, Li et al. [46] utilized folic acid-modified d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS-FA) to deliver NC. Nevertheless, the lack of pH-responsive properties in TPGS-FA resulted in non-specific drug release in both normal and tumor tissues, failing to mitigate toxicity to healthy organs. Fe-MOF often exhibits intrinsic pH responsiveness and enables high drug loading capacities. Bi et al. [47] employed MIL-100(Fe) as a carrier for NC delivery. However, the inherent pharmacological activity of MIL-100(Fe) is relatively weak, and the absence of targeting modifications limits its antitumor efficacy and tumor cell selectivity.
This study designed and synthesized an Fe-MOF with features of high drug loading capacity, pH responsiveness, and intrinsic antitumor activity for NC encapsulation. This NC-loaded Fe-MOF (NC@MOF) was further coated with HA to enhance tumor-targeting ability. Furthermore, the HD@NC@MOF system enables the co-release of NC and ferric ions (Fe3+). NC stimulated mitochondrial dysfunction, and Fe3+ catalyzes the Fenton reaction, synergistically triggering a substantial burst of ROS. This ROS surge depleted GSH and promotes lipid peroxidation (LPO), ultimately inducing ferroptosis. Concurrently, ROS inflict damage on cellular membranes and DNA, indirectly activating Toll-like receptors (TLRs) or NOD-like receptors (NLRs). This activation initiates the assembly of the RIPK1/RIPK3 complex (necrosome), resulting in necroptosis. Notably, the necroptosis executor MLKL can further activate NOX, generating additional ROS and establishing a positive feedback loop. Therefore, the HA-functionalized MOF system not only addresses the inherent drawbacks of NC but also significantly enhances its antitumor efficacy through the orchestrated induction of ferroptosis and necroptosis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiaoqin Pan: Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation. Changsheng Li: Writing – review & editing, Visualization, Project administration, Investigation, Data curation. Xinyi Ai: Writing – review & editing, Validation, Supervision, Formal analysis, Conceptualization. Changqing Tao: Writing – original draft, Validation, Investigation, Data curation. Jingchuan He: Writing – review & editing, Visualization, Software, Methodology. Tingting Li: Writing – review & editing, Visualization, Methodology, Conceptualization. Zhihua Deng: Validation, Supervision, Formal analysis. Xiaocheng Mo: Supervision, Methodology, Conceptualization. Ya Chen: Validation, Methodology, Investigation. Xiumei Qin: Methodology, Formal analysis. Dongmei Wang: Writing – review & editing, Supervision, Resources, Methodology, Conceptualization. Ronghua Jin: Writing – review & editing, Supervision, Resources, Project administration, Data curation, Conceptualization. Jie Yang: Writing – review & editing, Supervision, Project administration, Methodology, Funding acquisition, Conceptualization.
This work is financially supported by Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation (No. 2023GXNSFDA026026) and the Distinguished Young Scholars Program of Guangxi Natural Science Foundation (No. 2025GXNSFFA069017), Science and Technology Planning Project of Nanning Qingxiu District (No. 2021013), the National Natural Science Foundation of China (Nos. 82374093, 82160698).
Supplementary material associated with this article can be found, in the online version, at doi:
F. Bray, M. Laversanne, H. Sung, et al., CA Cancer J. Clin. 74 (2024) 229–263. doi: 10.3322/caac.21834
H. Sung, J. Ferlay, R.L. Siegel, et al., CA Cancer J. Clin. 71 (2021) 209–249. doi: 10.3322/caac.21660
Y. Shen, J.Q. Chen, X.P. Li, Genes Dis. 12 (2025) 101374. doi: 10.1016/j.gendis.2024.101374
Z.Z. Wang, H. Tang, Acad. J. Naval Med. Univ. 46 (2025) 17–23.
N. Vasan, J. Baselga, D.M. Hyman, Nature 575 (2019) 299–309. doi: 10.1038/s41586-019-1730-1
S.Y. Sun, Cancer Lett. 604 (2024) 217203. doi: 10.1016/j.canlet.2024.217203
Y. Yang, B. Jiang, L. Shi, et al., J. Ethnopharmacol. 346 (2025) 119555. doi: 10.1016/j.jep.2025.119555
H. Zheng, Y. Chen, W. Luo, et al., J. Nanobiotechnol. 23 (2025) 357. doi: 10.1186/s12951-025-03378-y
C.L. Yao, J.Q. Zhang, J.Y. Li, et al., Nat. Prod. Rep. 38 (2021) 1618–1633. doi: 10.1039/d0np00057d
D.J. Newman, G.M. Cragg, J. Nat. Prod. 79 (2016) 629–661. doi: 10.1021/acs.jnatprod.5b01055
F. Yu, W. Tan, Z. Chen, et al., Chin. Med. 17 (2022) 115. doi: 10.1186/s13020-022-00671-y
F. Yu, Z. Luo, L. Li, et al., Chin. Pharmacol. Bull. 38 (2022) 1023–1031.
Y. Lin, K.F. Qiu, F.C. Qin, et al., Chin. J. Mod. Appl. Pharm. 29 (2012) 1032–1034.
F.H. Luo, Z.H. Chen, F.F. Zeng, et al., J. Ethnopharmacol. 337 (2025) 118783. doi: 10.1016/j.jep.2024.118783
Y.W. Chen, G.H. Liu, L. Yang, et al., Lishizhen Med. Mater. Med. Res. 21 (2010) 1121–1122.
Q. Lu, S. Luo, Z. Shi, et al., Front. Pharmacol. 13 (2022) 1046402. doi: 10.3389/fphar.2022.1046402
J. Bouquet, M. Rivaud, S. Chevalley, E. Deharo, V. Jullian, A. Valentin, Malar. J. 11 (2012) 67. doi: 10.1186/1475-2875-11-67
D.M. Anwar, M. El-Sayed, A. Reda, et al., Expert Opin. Drug Deliv. 18 (2021) 1609–1625. doi: 10.1080/17425247.2021.1955853
J. He, T. Li, X. Pan, et al., Biomaterials 323 (2025) 123407. doi: 10.1016/j.biomaterials.2025.123407
T. Briolay, T. Petithomme, M. Fouet, et al., Mol. Cancer 20 (2021) 55.
Z. Liu, S. Liu, B. Liu, et al., Chin. Chem. Lett. 35 (2024) 109626. doi: 10.1016/j.cclet.2024.109626
B. Niu, K. Liao, Y. Zhou, et al., Biomaterials 277 (2021) 121110.
X. Zhao, S. He, B. Li, et al., Nano Lett. 23 (2023) 863–871. doi: 10.1021/acs.nanolett.2c04042
M. Parsaei, K. Akhbari, Inorg. Chem. 61 (2022) 19354–19368. doi: 10.1021/acs.inorgchem.2c03138
G. Lin, J. Chen, X. Liu, et al., Chin. Chem. Lett. 36 (2025) 111018. doi: 10.1016/j.cclet.2025.111018
C. Yao, J. Tian, H. Wang, et al., Chin. Chem. Lett. 28 (2017) 893–899. doi: 10.1016/j.cclet.2017.01.005
Z. Yin, Y. Lv, L. Deng, et al., Free Radic. Biol. Med. 203 (2023) 86–101. doi: 10.1016/j.freeradbiomed.2023.04.003
J. Lu, Y. Li, X. Qin, et al., J. Colloid Interface Sci. 702 (2026) 138849. doi: 10.1016/j.jcis.2025.138849
Y.H. Ko, H.S. Won, E.K. Jeon, et al., BMC Cancer 11 (2011) 340. doi: 10.1186/1471-2407-11-340
H. Xu, M. Niu, X. Yuan, et al., Exp. Hematol. Oncol. 9 (2020) 36.
J. Chandra, N. Molugulu, S. Annadurai, et al., Environ. Res. 233 (2023) 116506.
A. Mansouri, N.K. Farsani, A. Javanmard, et al., Int. J. Biol. Macromol. 308 (2025) 142253.
X. Li, F. Xiong, X. Cao, et al., Chin. Chem. Lett. 37 (2026) 110970. doi: 10.1016/j.cclet.2025.110970
Y. Zhang, S.S. Su, S. Zhao, et al., Nat. Commun. 8 (2017) 14329.
Z. Su, Z. Yang, Y. Xu, et al., Mol. Cancer 14 (2015) 48.
A. Ambujakshan, B.D. Sahu, Biochem. Pharmacol. 231 (2025) 116642.
R. Yuan, W. Zhao, Q. -Q. Wang, et al., Pharmacol. Res. 170 (2021) 105748.
N. Wang, Y. Liu, D. Peng, et al., Adv. Healthc. Mater. 13 (2024) e2401646.
B. Perillo, M. Di Donato, A. Pezone, et al., Exp. Mol. Med. 52 (2020) 192–203. doi: 10.1038/s12276-020-0384-2
Y. Gao, P. Wang, Y. Chu, et al., Water Res. 248 (2024) 120826.
H.J. Kwon, L.H. Kim, C.H. Ahn, et al., J. Clin. Biochem. Nutr. 65 (2019) 193–202. doi: 10.3164/jcbn.19-28
J. Dan Dunn, L.A. Alvarez, X. Zhang, et al., Redox Biol. 6 (2015) 472–485.
W.D. Cook, D.M. Moujalled, T.J. Ralph, et al., Cell Death Differ. 21 (2014) 1600–1612. doi: 10.1038/cdd.2014.70
Z. Ding, R. Wang, Y. Li, X. Wang, Mol. Cell 85 (2025) 2610–2625. e5.
Y. Hong, W.Q. Xu, J. Feng, et al., Acta Pharmacol. Sin. 44 (2023) 561–572. doi: 10.1038/s41401-022-00968-6
D. Li, S. Liu, J. Zhu, et al., BMC Pharmacol. Toxicol. 22 (2021) 1.
J. Bi, Y. Zheng, L. Fang, et al., Organomet. Polym. Mater. 30 (2020) 3388–3395. doi: 10.1007/s10904-020-01548-z

Scheme 1 Schematic illustration of the fabrication of HD@NC@MOF and their synergistic therapeutic mechanisms for LUSC.

Figure 1 Preparation and characterization. (A) Schematic synthesis route of HD@NC@MOF. (B, C) FTIR spectra and UV–vis. spectra of HD@NC@MOF. (D–G) XPS spectra of HD@NC@MOF. (H, I) TEM image and particle size distribution of HD@NC@MOF. Scale bar: 100 nm. (J) TEM-mapping image of HD@NC@MOF. Scale bar: 200 nm.

Figure 2 Targeting studies of HD@NC@MOF. (A) Expression levels of CD44 in LUSC cells (SK-MES-1, H1703, LTEP-s, H2170) and normal cells (BEAS-2B, HBE). (B, C) Comparison of viability in HBE and SK-MES-1 cells after 48 h treatment with NC or HD@NC@MOF. (D–F) Comparison of uptake of HD@ICG@MOF in HBE and SK-MES-1 cells with or without HA pretreatment after 6 h exposure, along with corresponding quantitative results. Scale bar: 10 µm. (G, I) In vivo imaging of HD@ICG@MOF distribution in nude mice and related quantitative analyses. (H, J) Fluorescence images of major organs (tumor (T), heart (H), liver (Li), spleen (S), lung (Lu), and kidney (K) at 48 h after tail vein injection of HD@ICG@MOF and related quantitative analyses. (K, L) Fluorescence microscopy images of live/dead staining in SK-MES-1 cells and corresponding quantitative results. Scale bar: 100 µm. Data are presented as mean values ± SD (n = 3). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CD44, cluster of differentiation 44; ns, no significance. ###P < 0.001; P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group.

Figure 3 Study on the anti-LUSC mechanism of HD@NC@MOF. (A) Fluorescence microscopy images showing changes in intracellular ROS levels detected using the oxidant-sensitive fluorescent probe DCFH-DA. Scale bar: 100 µm. (B) Fluorescence co-localization images of HD@NC@MOF with mitochondria. Scale bar: 5 µm. (C) Morphological and organelle changes in SK-MES-1 cells after 48 h of HD@NC@MOF treatment with or without HA pretreatment. Green arrows indicate normal mitochondria, blue arrows indicate normal ER, red arrows indicate damaged mitochondria, pink arrows indicate intracellular vacuoles, orange arrows indicate blurred cell edges, and yellow arrows indicate slightly damaged mitochondria. (D–F) Changes in intracellular GSH, iron content, and MDA levels after different treatments. (G–N) WB analysis of changes in SLC7A11/GPX4 pathway and RIPK1/RIPK3/MLKL pathway-related proteins in SK-MES-1 cells after 12 h of HD@NC@MOF treatment, along with corresponding quantitative results. Data are presented as mean values ± SD (n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001; P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group.

Figure 4 Targeting via specific HA-CD44 binding is indispensable for the antitumor effect of HD@NC@MOF. (A, B) The effects of HD@NC@MOF on the viability of SK-MES-1 and HBE cells with or without HA pretreatment by CCK-8 assay. (C, D) Live/dead cell staining to observe the mortality of SK-MES-1 cells treated with HD@NC@MOF with or without HA pretreatment. (E, F) Fluorescence microscopy images (using oxidant-sensitive probe DCFH-DA) and quantitative analysis of intracellular ROS levels. (G-I) Effects of HD@NC@MOF on intracellular GSH, iron content, and MDA levels in SK-MES-1 cells with or without HA pretreatment. (J-Q) WB analysis and quantitative results of proteins related to the SLC7A11/GPX4 pathway and RIPK1/RIPK3/MLKL pathway in SK-MES-1 cells treated with HD@NC@MOF with or without HA pretreatment. Scale bar: 100 µm. Data are presented as mean values ± SD (n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001; ***P < 0.001 vs. the control group.

Figure 5 In vivo anticancer efficacy of HD@NC@MOF. (A) Schematic illustration of the treatment schedule in nude mice. (B) Tumor growth curves after different treatments. (C) Body weight changes of nude mice under different treatments. (D) Images of tumors isolated from tumor-bearing mice at the end of treatment. (E) Tumor weights at the end of treatment. (F) Tumor inhibition rates of different formulations (n = 5). (G) HE, Ki67, and TUNEL staining of tumor tissues after different treatments (n = 3). Scale bar: 50 µm. Data are presented as mean values ± SD. ##P < 0.01, ###P < 0.001; P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: