Citation:
Linan Wu, Shenghua Gao, Peng Zhan. Breakthrough progress in the structural determination of the monkeypox virus I7L protease and the design of targeted inhibitors[J]. Chinese Chemical Letters,
2026, 37(4): 111845.
doi:
10.1016/j.cclet.2025.111845
Breakthrough progress in the structural determination of the monkeypox virus I7L protease and the design of targeted inhibitors
English
Breakthrough progress in the structural determination of the monkeypox virus I7L protease and the design of targeted inhibitors
Department of Medicinal Chemistry, Shandong Key Laboratory of Druggability Optimization and Evaluation for Lead Compounds, School of Pharmaceutical Sciences, Shandong University, Ji'nan 250012, China
*Corresponding authors. E-mail addresses: shh_gao@163.com (S. Gao)
Monkeypox virus (MPXV), a member of the orthopoxvirus genus, has been endemic in parts of Africa for decades. Since 2022, MPXV has spread globally, with over 142,000 confirmed cases reported across 133 countries by 2025 [1]. The World Health Organization (WHO) has declared the outbreak a Public Health Emergency of International Concern (PHEIC) twice [1].
Proteases play a crucial role in the viral life cycle, especially in facilitating maturation and generating infectious particles, making them ideal drug targets [2]. Successful development has been achieved for antiviral agents targeting proteases of human immunodeficiency virus (HIV) and hepatitis C virus (HCV), effectively curbing viral infections [3]. Notably, nirmatrelvir, which acts on the main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is currently extensively utilized in the treatment of corona virus disease 2019 (COVID-19) [4]. Similarly, proteases are also essential for orthopoxvirus. As an orthopoxvirus, MPXV relies on the core protease I7L (CorePro, the following text will be uniformly referred to as I7L protease) to mediate viral protein maturation during replication. The I7L protease is highly conserved among orthopoxviruses. MPXV I7L protease shares 98.82% and 98.58% sequence homology with variola virus (VARV) and vaccinia virus (VACV), respectively. This high degree of conservation makes I7L protease a promising target for broad-spectrum anti-poxvirus therapies [5–8].
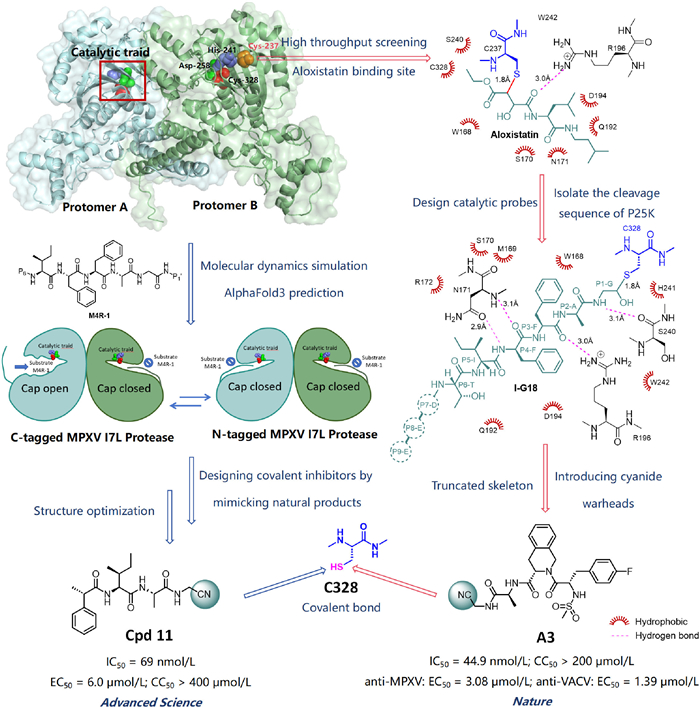
However, the three-dimensional structure and catalytic mechanism of the I7L protease have long remained unknown, significantly hindering drug development. Recently, two studies, published in Advanced Science (Xu et al.) and Nature (Yang et al.), using X-ray crystallography and cryo-electron microscopy (cryo-EM) to reveal the structural dynamic of the MPXV I7L protease and developed nanomolar covalent inhibitors based on these insights, making a major breakthrough in anti-MPXV therapy (Fig. 1) [9,10].
Figure 1
Figure 1.
Structure-guided discovery of MPXV I7L protease inhibitors (PDB code: 9LIK). Red sphere: Cys328; green sphere: Asp258; purple sphere: His241; orange sphere: Cys237. The protein in the upper left corner is visualized using PyMOL, and the compound structure and 2D binding mode are created with ChemDraw.
Both studies provide valuable insights into the structure of the MPXV I7L protease. Xu and Zhang's team reported the first crystal structure of the protease at 2.6 Å resolution (PDB code: 9LIK) in a study published in Advanced Science. They found that the cap regions (residues 125–168) of the two subunits in the dimer adopt two different conformations: “cap-open” and “cap-closed”. In the cap-closed state, residues 125–168 block the substrate from reaching the catalytic triad, keeping the protease inactive. In contrast, in the cap-open state, the cap is stabilized by inter-dimer hydrogen bonds, such as those between residues 280–287 of protomers A/B and the cap region. Molecular dynamics (MD) simulations confirmed that the cap can switch between these two states, showing that its flexibility is a key mechanism for regulating substrate binding.
The structure of the MPXV I7L protease and its aloxistatin-bound complex was determined by Yang and colleagues, as reported in Nature. A unique “dance-partner” homodimeric state of the protease was revealed by cyo-EM, in which each subunit was found to be organized into three discrete domains, the catalytic domain (CD), the N-terminal domain (NTD), and the C-terminal domain (CTD). The catalytic domain exhibits the characteristic fold of CE family/Ulp1-like proteases, while the NTD and CTD have unique folds that may confer specific functions to I7L protease. The active site of I7L protease contains the catalytic triad of His241-Asp258-Cys328, with Cys328 located in the upper helix of the catalytic domain and His241 and Asp258 in the loop connecting the central β-sheet. Structural analysis further indicated that the active site remains in a closed conformation in the absence of substrate but undergoes a conformational transition to an open state upon substrate binding, thereby facilitating substrate entry and subsequent catalytic activity.
Both studies provided insights into substrate recognition mechanisms. Xu et al. used AlphaFold3 to predict the enzyme-substrate complex structure and analyzed the substrate recognition and catalytic mechanism of the I7L protease. They analyzed the key S1–S5 subsites of the protease using M4R-1 as the substrate. S1–S4 were located inside the enzyme, while S3 and S5 were solvent-exposed. S1 had a small cavity; substrate binding caused side-chain conformation shift of Trp168, making space for the substrate. S2 was formed by Ser240, His241, Cys237, and Trp242, creating a hydrophobic pocket. S4 had a larger cavity and prefers aromatic amino acids at the substrate’s P4 position (with P1–P5 positions corresponding to S1–S5 subsites, respectively). S3 and S5 were flexible and could accommodate residues with different polarities. These findings supported the design of protease inhibitors based on subsite features and substrate preference. In addition, during catalysis, Cys328 acts as a nucleophile that attacks the substrate's peptide bond, His241 serves as an acid-base catalyst to transfer protons, and Asp258 stabilizes His241 through hydrogen bonds. Quantum mechanics/molecular mechanics (QM/MM) simulations showed that the catalytic process includes three steps: formation of a non-covalent complex, nucleophilic attack by the catalytic cysteine on the P1 glycine to form a covalent acyl-enzyme intermediate (acylation), and hydrolysis of the intermediate to release the product and regenerate the active site (deacylation). These findings offer atomic-level insights into the I7L protease mechanism.
In another study, Yang et al. discovered aloxistatin, which provided a “proof of concept”, but its binding site was deviated from the catalytic center and did not trigger conformational changes, thus failing to reveal the true substrate recognition mechanism of the I7L protease. To address this issue, the team isolated the cleavage sequence of the natural substrate P25K of the MPXV I7L protease, truncating it into a nine-peptide EEDTIFFAG, and replaced the original amide bond at the cleavage site G18 with an aldehyde group (-CHO), resulting in the catalytic probe I-G18. Yang et al. analyzed the complex structure of I7L protease with the nonapeptide I-G18 to reveal its substrate-binding mode. Studies have demonstrated that I-G18 binding to I7L protease stabilizes the protease in an open conformation, exposes the S1–S4 pocket, promotes covalent bond formation at Cys328, and positions the P1-P6 groups within the substrate binding site. The P1–P6 residues of I-G18 fit tightly into the substrate-binding pocket. P1 and P2 are located in a deep cavity where the S1 subsite, formed by Trp168, Met169, and His241, selectively binds small amino acids such as glycine. The S2 subsite, made up of residues like Cys237 and Trp168, also prefers small residues. The P3 residue is stabilized through hydrophobic interactions with Trp168 and Met169. P4 extends into the hydrophobic S4 subsite formed by Gln192 and Phe193, which favors large hydrophobic residues. Additionally, P4 forms a hydrogen bond with Asn171, further stabilizing the enzyme-substrate interaction (Fig. 1). All these structural details provide crucial information for the design of inhibitors.
Both research teams designed inhibitors based on the structural features and substrate recognition mechanism of the I7L protease. Based on the exposed active site in the “cap-open” state and the distinct roles of each subsite in the I7L protease during M4R-1 binding, Xu et al. designed covalent inhibitors that mimic natural substrates, include covalent warheads, reduce peptide bonds, and match the S1–S4 subsites to inhibit protease activity effectively. They used a fluorescence resonance energy transfer (FRET) assay to identify several I7L inhibitors with nanomolar potency. Among them, compound 11 showed strong inhibition of the MPXV I7L protease (concentration of inhibitory 50% (IC50) = 69 nmol/L), significantly suppressed VACV replication in cells (concentration of effective 50% (EC50) = 6.0 µmol/L), and exhibited no significant cytotoxicity (concentration of cytotoxicity 50% (CC50) > 400 µmol/L). Compound 11 binds similarly to M4R-1 and fits well into the I7L protease subsites. It forms a covalent bond between its nitrile warhead and Cys328. Its P2-alanine occupies the hydrophobic S2 subsite, and P3-isoleucine resides in the solvent-exposed S3 subsite. Compared to M4R-1, the P4 group binds deeper into S4. This structure-based, substrate-mimicking strategy provides a strong foundation for developing more drug-like inhibitors.
Based on the binding mode of I-G18, Yang et al. developed peptide-mimetic inhibitors containing nitrile warheads. The nitrile group at the P1 position covalently binds to Cys328, while the tetrahydroisoquinoline at P3 and the 4-fluorobenzyl group at P4 are designed to target the S3 and S4 subsites, respectively. These inhibitors show potent activity against the I7L protease (IC50 = 44.9–100.3 nmol/L) and exhibit strong antiviral effects against both MPXV and VACV. Among them, compound A3 is the most effective, demonstrating broad-spectrum antiviral activity against MPXV and VACV at the cellular level (anti-MPXV: EC50 = 3.08 µmol/L; anti-VACV: EC50 = 1.39 µmol/L), along with minimal cytotoxicity (CC50 > 200 µmol/L). Cryo-EM results confirm that interactions in the A3-I7L complex mirror those in the I7L-I-G18 complex, validating the inhibitor’s design concept.
Both studies confirm that the “open” conformation is the warhead-accessible active state and provide quantifiable, evidence-based structure-function strategies. Xu et al. identified the “cap-open” as pharmacologically active and optimized the S1–S5 subsites. Using the substrate as a template, they refined side chains and introduced a nitrile warhead, completing a closed design loop from “conformational switch” to nanomolar inhibitors. Yang et al. used the “closed-to-open” transition as a core design principle, optimizing P1–P4 side chains and warheads to stabilize the protease's open state, yielding nanomolar potency, broad-spectrum, low-toxicity leads. Together, these studies show that substrates or inhibitors can drive the enzyme from a “closed and inactive” to an “open and catalytic” state, explaining specificity and efficiency while offering a precise, transferable template for antipoxvirus drug development.
In summary, both studies have systematically clarified the structural dynamics and catalytic mechanism of the MPXV I7L protease, filling a critical gap in the structural biology of orthopoxvirus proteases. However, the two studies differ in their methodological strengths. Xu et al. used X-ray crystallography to resolve the MPXV I7L protease structure, offering high-resolution insights into static structural details, particularly the influence of local conformations such as the “cap” region on substrate binding. Yang et al. combined cryo-EM with X-ray crystallography, enabling the visualization of complex dimeric structures and dynamic conformational changes, making their approach well-suited for revealing how global structural features, such as NTD/CTD folding, regulate catalytic function. Furthermore, the novel covalent inhibitors developed by both research teams provide promising lead structures for the identification and optimization of preclinical drug candidates. Additionally, the integrated “structure-function” research approach combining cryo-EM and artificial intelligence (AI)-based prediction offers valuable methodological insights for the development of protease-targeted therapeutics against other highly pathogenic viruses, including Ebola virus and influenza virus.
However, despite exhibiting antiviral activity at the cellular level, significant challenges remain in translating these findings into clinically viable therapeutic strategies. Both studies provide limited data on the in vivo pharmacokinetic properties, such as absorption, distribution, metabolism, and excretion (ADME), as well as the potential toxicological profiles of the inhibitors. Furthermore, the viral models employed in cell-based assays may not fully recapitulate the complexity of human infection. Critical parameters, including effective dosing, optimal administration routes, and the therapeutic window in humans, require thorough investigation through rigorous clinical research.
Future research should focus on studying the pharmacokinetic properties, bioavailability, and stability of these inhibitors. Additionally, their metabolic pathways and in vivo toxicological profiles need thorough investigation to support clinical development [11]. Additionally, research can be expanded to new target or broad-spectrum antiviral targets, developing combination drug strategies to enhance efficacy and reduce drug resistance, while establishing appropriate animal models to accelerate the clinical translation of antiviral drugs against MPXV [12–16]. Notably, drawing inspiration from the development of anti-COVID-19 drugs, the emergency research and development of therapeutic agents during the COVID-19 pandemic prioritized molecules with previously reported anti-coronavirus activity and drugs with established broad-spectrum antiviral properties, with remdesivir and azvudine serving as successful examples [17]. Therefore, repurposing existing drugs based on similarities (e.g., viral family relatedness or target homology) represents a viable shortcut for identifying anti-MPXV agents. For instance, focused exploration of molecules previously reported to exhibit anti-poxvirus activity may yield novel discoveries [18,19]. As understanding of MPXV's biological characteristics grows and drug development technologiesadvance, more effective and safer antiviral agents are expercted to emerge, providing powerful tools to combat this global public health threat [20].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors are supported by the Key Research and Development Program, Ministry of Science and Technology of the People’s Republic of China (No. 2023YFC2606500), and Supported By Young Talent of Lifting engineering for Science and Technology in Shandong, China (No. SDAST2025 QTB057).
[1]
World Health Organization, Global mpox trends. https://worldhealthorg.shinyapps.io/mpx_global/, 2025.
Figure 1
Structure-guided discovery of MPXV I7L protease inhibitors (PDB code: 9LIK). Red sphere: Cys328; green sphere: Asp258; purple sphere: His241; orange sphere: Cys237. The protein in the upper left corner is visualized using PyMOL, and the compound structure and 2D binding mode are created with ChemDraw.

 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载:
