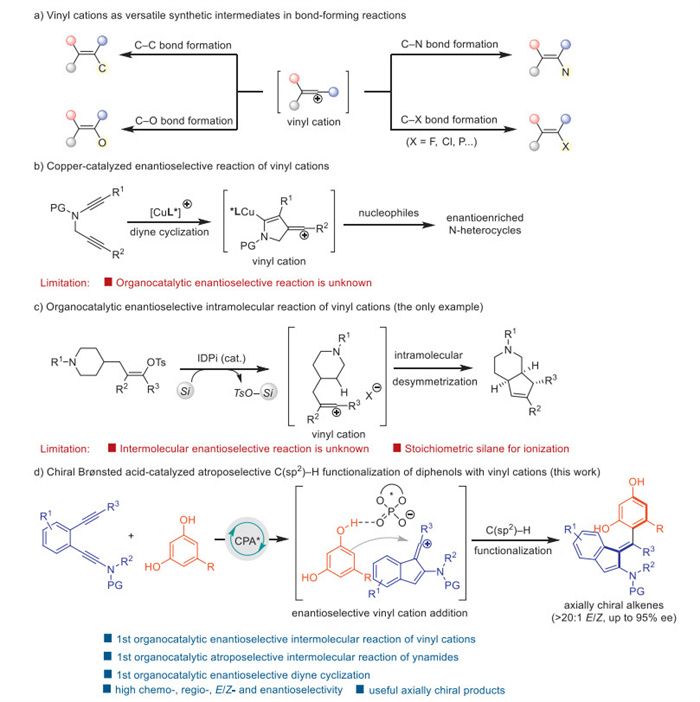
Scheme 1.
Catalytic enantioselective transformation of vinyl cations.
Atroposelective transformation of vinyl cations by chiral Brønsted acid catalysis
Da-Qiu Cui , Quan-Xin Li , Jia-Bo Huang , Ying-Qi Zhang , Xuan Wang , Shao-Fei Ni , Long-Wu Ye , Bo Zhou

Vinyl cations are powerful intermediates in organic synthesis, which have been employed in diverse bond-forming transformations due to their carbene-like properties. Over the past decades, a range of strategies have been developed to generate the highly reactive vinyl cation intermediates, including ionization of vinyl triflate analogues, addition of electrophiles to alkynes and allenes, cyclization of enynes and diynes, as well as decomposition of β–hydroxy-α-diazo ketones [1-4]. Based on these methodologies, C–H functionalization [5-9], rearrangement [10,11], addition [12,13] and other cascade reactions [14,15] could be realized using vinyl cation-mediated approach (Scheme 1a). However, the catalytic enantioselective transformation of vinyl cations has been rarely reported, especially for the related organocatalytic reaction. In 2019, our group developed a chiral copper-catalyzed asymmetric diyne cyclization reaction through vinyl cations via the remote control of enantioselectivity [16], which could be extended to divergent intramolecular [17-21] and intermolecular [22-26] reactions and led to the preparation of various chiral N-heterocycles (Scheme 1b). Of note, the organocatalytic enantioselective reaction is more challenging. In 2022, the only organocatalytic enantioselective reaction of vinyl cations was disclosed by Nelson and co-workers through imidodiphosphorimidate (IDPi)-catalyzed intramolecular desymmetric C–H insertion of piperidinyl fragments (Scheme 1c) [27]. Despite the efficacy of the established approaches, the organocatalytic enantioselective reaction of vinyl cations is still limited to intramolecular manner. Therefore, it is crucial to develop a new catalytic system to realize the metal-free enantioselective intermolecular transformation of vinyl cations, particularly with improved substrate flexibility, atom-economy, and potential utility.
During the past decades, chiral Brønsted acid (CBA) catalysis has been developed as an efficient method in asymmetric synthesis [28-32]. Motivated by the seminal contribution from Akiyama [33] and Terada [34] groups on CBA-catalyzed Mannich-type reactions, CBA catalysis was further incorporated into various transformations of imines and carbonyls by hydrogen bonding and ion-pairing interactions. Although these impressive advances achieved, the CBA-catalyzed reaction of C–C multiple bonds is less investigated [32]. Our group developed a CBA-catalyzed strategy for the enantioselective reaction of ynamides [35-41] by direct activation of C–C triple bonds [42-45], which enabled varieties of intramolecular transformations, including hydroalkoxylation/rearrangements [42,43], dearomative cyclization [44] and hydroamination [45]. The enantioselective intermolecular reaction of ynamides was reported by Tan group through an elegant multicomponent strategy using well-designed amines and aldehydes as reactants [46]. Nevertheless, the related atroposelective transformation is still unknown, probably due to the insufficient reactivity of sterically hindered substrates in atroposelective reactions. Thus, more reactive intermediates that controlled by effective catalytic system become imperative for the organocatalytic atroposelective transformation of ynamides.
Inspired by the recent development of vinyl cation-mediated asymmetric reactions [16-27] and CBA catalysis [42-46], we envisioned to explore the CBA-catalyzed enantioselective intermolecular transformation of vinyl cations. In this method, the CBA-catalyzed diyne cyclization could generate key vinyl cation intermediates, and the chiral environment created by CBA catalyst might control the enantioselectivity for further nucleophilic addition. Moreover, vinyl cations and sterically hindered nucleophiles could be the ideal synthons for the straightforward construction of axially chiral alkenes. The success of this organocatalytic enantioselective intermolecular reaction of vinyl cations relies on addressing the following challenges. (1) The feasibility for the generation of vinyl cations from CBA-catalyzed diyne cyclization is unknown, and the balance of stability and reactivity of vinyl cation intermediates in acidic conditions is challenging. (2) The combination of hydrogen bonding and ion-pairing interactions is essential for this CBA-catalyzed intermolecular reaction, which is unpresented in vinyl cation chemistry. (3) The requirement of sterically hindered substrates in atroposelective reactions could reduce their reactivity. Therefore, highly reactive catalytic systems are under great demand.
Axial chirality is a widespread chirality element in nature, and its construction [47-54] significantly promoted the applications in asymmetric catalysis, bioactive molecules and materials [55-59]. Among these, axially chiral alkenes are unique atropisomeric architectures bearing relatively distorted double bonds, and additional challenges exist for their preparation [60-63], particularly for acyclic tetrasubstituted alkenes [64-72]. The simultaneous control of E/Z selectivity and enantioselectivity requires reactive intermediates and efficient chiral induction model. Herein, we disclose the development of such a CBA-catalyzed atroposelective intermolecular C(sp2)–H functionalization of diphenols with vinyl cations by diyne cyclization pathway. This metal-free protocol leads to the practical and atom-economic preparation of axially chiral tetrasubstituted alkenes with high chemoselectivity, regioselectivity, E/Z selectivity and enantioselectivity, which could be rationalized by computational studies. The resulting atropisomeric framework can be derivatized to axially chiral ligand for asymmetric catalysis. Importantly, this transformation not only continues the first organocatalytic enantioselective intermolecular reaction of vinyl cations, but also features the first organocatalytic atroposelective intermolecular reaction of ynamides and the first organocatalytic enantioselective diyne cyclization (Scheme 1d).
At the outset, diyne 1a was employed as the vinyl cation precursor to explore this CBA-catalyzed atroposelective C(sp2)–H functionalization of diphenol 2a (Table 1, for more conditions, see Supporting information). The proof-of-concept experiment was performed with catalytic amount of BINOL-derived chiral phosphoric acid (CPA) A1 in DCM at 30 ℃, delivering the expected axially chiral alkene 3a in 28% yield and 21% ee albeit with poor chemoselectivity (entry 1). Other BINOL-derived CPAs A2–A6 containing polycyclic aryl groups and multisubstituted phenyl groups were then screened, and 2,4,6-trisubstituted phenyl groups were found to be beneficial for both chemoselectivity and enantioselectivity (entries 2–6). The replacement of BINOL with H8-BINOL skeleton resulted in similar enantioselectivities, and H8-BINOL-derived CPAs A7–A9 afforded the desired product in 11%–62% yields with 11%–73% ee (entries 7–9). To our delight, catalyst optimization using SPINOL-derived CPAs A10–A14 effectively improved the yield and enantioselectivity (entries 10–14). The bulky 2,4,6-triisopropylphenyl-substituted CPA catalyst A14 was determined as the best catalyst to produce atropisomeric alkene 3a in 80% yield and 93% ee, and the yield of O-attack byproduct 3a′ could be reduced to 5% (entry 14). The screening results of solvents revealed that DCM was the optimal solvent for this atroposelective reaction (entries 15 and 16). Further optimization of reaction conditions by lowering the catalyst loading as well as decreasing the concentration and temperature successfully improved the efficiency (entries 17–19), and the axially chiral tetrasubstituted alkene 3a was generated in 88% yield with 94% ee using A14 (15 mol%) as catalyst in DCM at 20 ℃ (entry 19). Of note, the chemoselectivity and E/Z selectivity could be well-controlled in this CPA-catalyzed transformation.
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Catalyst | Reaction conditions | Yield (%)b | ee of 3a (%)c | |
| 3a | 3a′ | ||||
| 1 | A1 | DCM, 30 ℃, 19 h | 28 | 20 | 21 |
| 2 | A2 | DCM, 30 ℃, 50 h | 47 | < 2 | < 2 |
| 3 | A3 | DCM, 30 ℃, 50 h | 58 | 15 | < 2 |
| 4 | A4 | DCM, 30 ℃, 19 h | 32 | 20 | 33 |
| 5 | A5 | DCM, 30 ℃, 5 h | 77 | 5 | 77 |
| 6 | A6 | DCM, 30 ℃, 3 h | 54 | 6 | 80 |
| 7 | A7 | DCM, 30 ℃, 57 h | 11 | 4 | 11 |
| 8 | A8 | DCM, 30 ℃, 1.5 h | 61 | 6 | 73 |
| 9 | A9 | DCM, 30 ℃, 1 h | 62 | 8 | 70 |
| 10 | A10 | DCM, 30 ℃, 72 h | 35 | 62 | 25 |
| 11 | A11 | DCM, 30 ℃, 46 h | 42 | 8 | 56 |
| 12 | A12 | DCM, 30 ℃, 72 h | 33 | 8 | 27 |
| 13 | A13 | DCM, 30 ℃, 50 h | 12 | < 2 | 55 |
| 14 | A14 | DCM, 30 ℃, 0.5 h | 80 | 5 | 93 |
| 15 | A14 | DCE, 30 ℃, 0.5 h | 76 | 7 | 90 |
| 16 | A14 | Toluene, 30 ℃, 12 h | 51 | 4 | 73 |
| 17d | A14 | DCM, 30 ℃, 1 h | 74 | 4 | 93 |
| 18d,e | A14 | DCM, 30 ℃, 1 h | 82 | 4 | 94 |
| 19d,e | A14 | DCM, 20 ℃, 1 h | 88 | 5 | 94 |
| a Reaction conditions: 1a (0.05 mmol), 2a (0.1 mmol), catalyst (0.01 mmol), solvent (2 mL), 20–30 ℃, 0.5–72 h, in vials. PMP = 4-methoxyphenyl, DCE = 1,2-dichloroethane. b Measured by 1H NMR using dimethyl terephthalate as the internal standard. c Determined by HPLC analysis. d A14 (0.0075 mmol). e DCM (4 mL). |
|||||
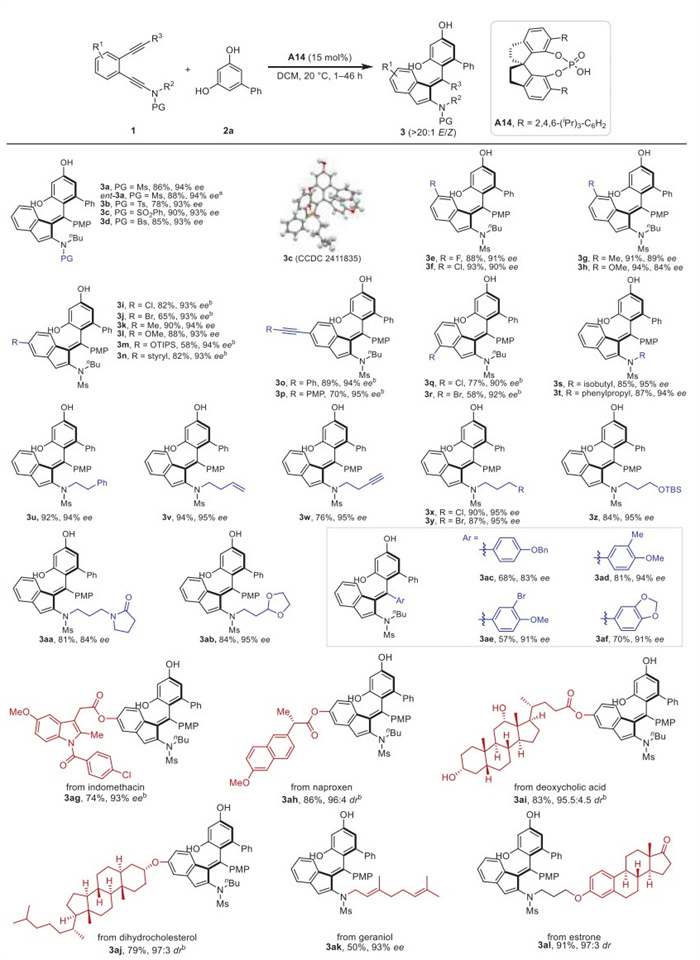
The scope of this atroposelective C(sp2)–H functionalization was then explored under the optimized reaction conditions (Table 1, entry 19). A broad range of diynes 1 and diphenols 2 equipped with various functional groups were compatible with this reaction to produce the corresponding axially chiral tetrasubstituted alkenes smoothly. As shown in Scheme 2, diynes bearing different sulfonyl protecting groups, including Ms (3a), Ts (3b), SO2Ph (3c) and Bs (3d), could be efficiently transformed into the desired atropisomeric alkenes in 78%–90% yields with 93%–94% ee. By replacing catalyst A14 with (R)-A14, the enantiomer of 3a was obtained with similar yield and enantioselectivity (ent–3a). Substituents with different electronic and steric properties (such as F, Cl, Br, Me, OMe, OTIPS, styryl and alkynyl groups) could be introduced into the different positions of the phenyl linker, allowing the assembly of products 3e–3r in 58%–94% yields with 84%–95% ee. Next, we turned our attention to the alkyl substituents on the ynamide moiety (R2), and the replacement of n–butyl group with isobutyl (3s), phenylpropyl (3t) and phenylethyl (3u) groups resulted in good to excellent yields and enantioselectivities. Benefit from the metal-free reaction conditions, a panel of alkyl ynamides containing different functional groups including alkene (3v), alkyne (3w), chloride (3x), bromide (3y), silyl ether (3z), amide (3aa) and acetal (3ab), were all compatible with this atroposelective reaction. Further exploration into the aryl moieties (R3 = Ar) connected with alkyne indicated that OBn (3ac), disubstituted electron-rich aryl groups (3ad and 3ae) and 1,3-benzodioxole (3af) could react well to produce the target products in 57%–81% yields with 83%–94% ee. Moreover, this catalytic protocol showed good compatibility with drug molecules and bioactive compounds. Diverse medicinally relevant molecules, such as indomethacin (3ag), naproxen (3ah), deoxycholic acid (3ai), dihydrocholesterol (3aj), geraniol (3ak) and estrone (3al), could be incorporated into the diyne substrates and converted into the expected axially chiral alkenes in 50%–91% yields with excellent enantiocontrol. Of note, the reaction proceeded with excellent E/Z ratios for all these examples. The absolute configuration of 3c was unambiguously determined by single crystal X-ray crystallographic analysis.
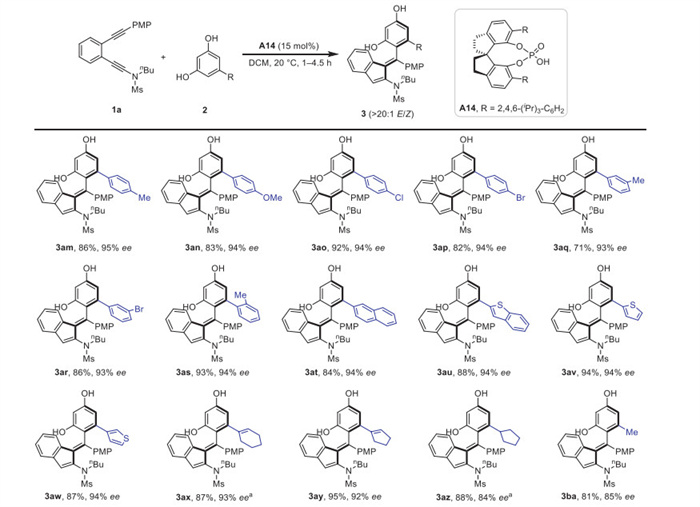
To further explore the capacity of this intermolecular atroposelective C(sp2)–H functionalization, the scope of diphenols 2 was then evaluated under the optimal reaction conditions. As depicted in Scheme 3, phenyl-substituted resorcinol derivatives bearing both electron-donating and -withdrawing groups at the para-, meta-, and ortho-positions of benzene ring were competent substrates for the intermolecular reaction, affording products 3am–3as in 71%–93% yields with 93%–95% ee. Diphenol nucleophiles with other aryl substituents, including 2-naphthyl, 2-benzothienyl, 2-thienyl and 3-thienyl groups, could undergo smooth C(sp2)–H functionalization to forge 3at–3aw in good to excellent yields with excellent ee values. In addition to (hetero)aryl diphenols, cyclohexenyl, cyclopentenyl, cyclopentyl, and methyl-substituted resorcinols were also well compatible, and the corresponding axially chiral alkenes 3ax–3ba were synthesized in 81%–95% yields with 84%–93% ee values. This reaction provided a convenient pathway for the assembly of axially chiral alkenes from various simple diphenols.
Based on the good configurational stability of constructed axially chiral tetrasubstituted alkenes 3 (for details, see Supporting information), the synthetic utility of this CPA-catalyzed atroposelective C(sp2)–H functionalization was then explored. As shown in Scheme 4a, the scale-up reaction of diyne 1c with diphenol 2a proceeded well to deliver the desired product 3c in 83% yield with 93% ee. Containing both alkene and phenol moieties, the axially chiral product could undergo various transformations, leading to diverse chiral architectures. For example, the enamide moiety on 3c could be hydrogenated to chiral amide 4 bearing a new carbon stereocenter, and the diastereoselectivity was effectively controlled (> 20:1 dr). The treatment of axially chiral alkene 3c with different electrophiles, including 4-phenyl-3H-1,2,4-triazole-3,5(4H)–dione (PTAD) [73], Tf2O and MeI, successfully functionalized the diphenol moiety at different positions to give products 5–7 regioselectively. The absolute configuration of 4 was determined by X-ray diffraction.
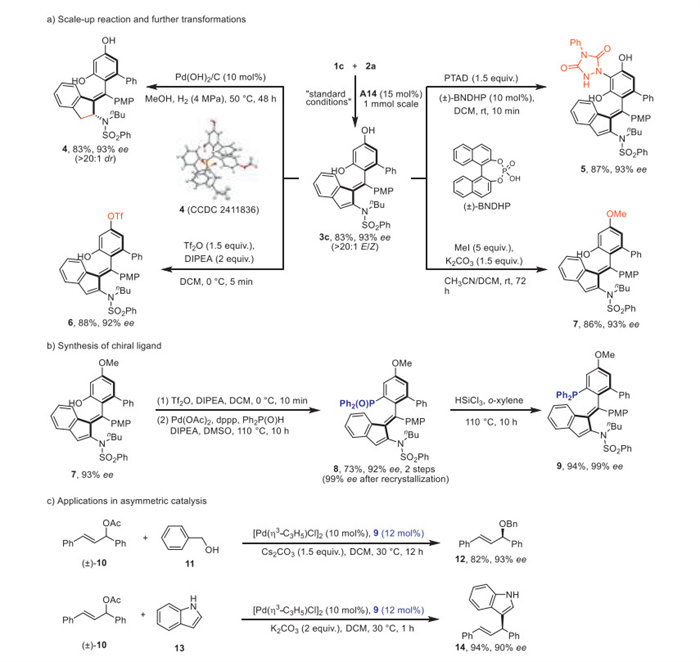
Further synthetic manipulations of the constructed scaffold provided an alternative way towards axially chiral ligand. Axially chiral alkene 7 could be diversified to atropisomeric phosphine oxide 8 through trifluoromethanesulfonylation and palladium-catalyzed C–P coupling, followed by a facile reduction to yield the target axially chiral phosphine 9 (Scheme 4b). Interestingly, phosphine 9 could be employed as an effective chiral ligand to control the enantioselectivity in the palladium-catalyzed asymmetric allylic functionalization. The reaction of rac-1,3-diphenylallyl acetate 10 with nucleophiles, such as benzyl alcohol 11 and indole 13, took place smoothly to forge products 12 and 14 in 82%–94% yields with 90%–93% ee (Scheme 4c). These results indicate the constructed axially chiral alkene skeletons to be potentially useful in asymmetric catalysis.
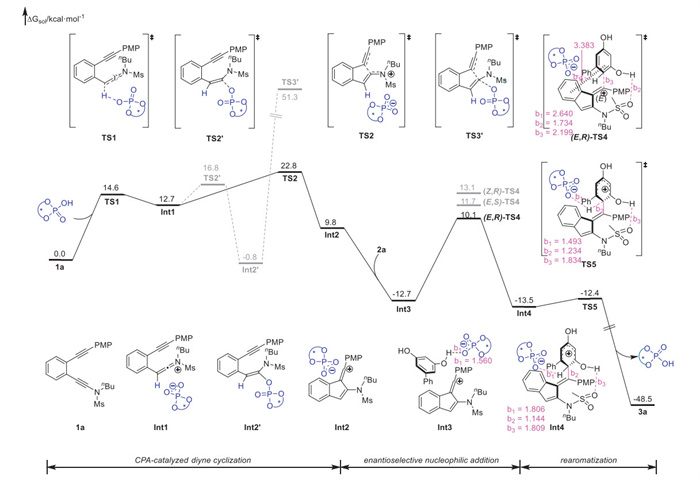
To illustrate the reaction mechanism and the origin of selectivities, density functional theory (DFT) calculations were carried out based on the non-linear effect investigations (for details, see Supporting information) using diyne 1a and diphenol 2a as substrates. As depicted in Scheme 5, the reaction starts with the activation of C≡C bond of ynamide moiety with CPA catalyst through transition state TS1, followed by the generation of keteniminium intermediate Int1 through proton transfer. Then, both phosphoryloxylation and ion-pairing pathways were investigated for the following cyclization step. For the phosphoryloxylation pathway, although the enamide intermediate Int2′ is thermodynamically stable, the following cyclization step is kinetically unfavorable (52.1 kcal/mol), which could be excluded. For the ion-pairing pathway, direct cyclization takes place to give vinyl cation intermediate Int2 via ion pair transition state TS2 with a free energy barrier of 10.1 kcal/mol, which further captures diphenol 2a to afford a more stable intermediate Int3. Subsequently, the nucleophilic attack of diphenol onto vinyl cation moiety generates the dearomatized chiral intermediate Int4 through (E,R)-TS4 with a free energy barrier of 22.8 kcal/mol, which is the rate-, E/Z- and enantioselectivity-determining step. An analysis of other transition states indicates that the free energy barrier of (E,R)-TS4 is 3.0 kcal/mol lower than that of (Z,R)-TS4 (leading to Z isomer), and 1.6 kcal/mol lower than that of (E,S)-TS4 (leading to the minor enantiomer). Thus, the dearomatized chiral intermediate Int4 is kinetically more favorable than other intermediates, and the calculated E/Z selectivity and enantioselectivity are consistent with the experimental results. Finally, the catalytic cycle culminates in an exothermic rearomatization, delivering the axially chiral alkene 3a and releasing the CPA catalyst.
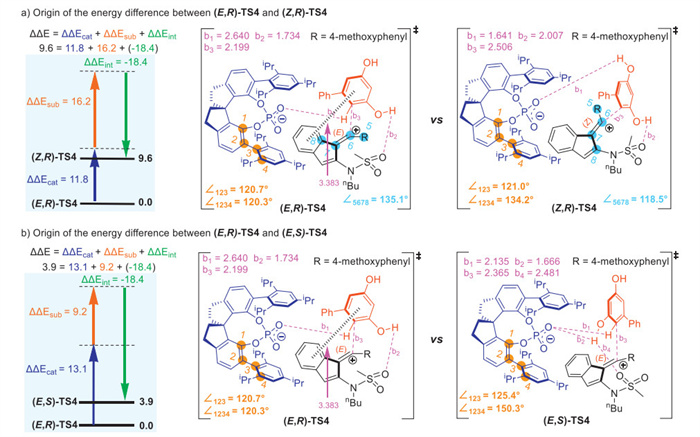
Furthermore, energy decomposition analysis was performed to elucidate the source of E/Z selectivity and enantioselectivity. As illustrated in Scheme 6a, the inspection of transition states (E,R)-TS4 and (Z,R)-TS4 shows that the C=C bond of the planar alkene in the substrate is distorted to 118.5° in (Z,R)-TS4, which is larger than that of (E,R)-TS4 (135.1°). In addition, the angle and dihedral angle of the arms in the free catalyst (∠123 = 120.4°, ∠1234 = 112.9°) are distorted to 121.0° and 134.2° in (Z,R)-TS4, which are more significant than that of (E,R)-TS4 (120.7°, 120.3°). Thus, the obtained E/Z selectivity arises from the distortion of catalyst and substrate. Scheme 6b demonstrates that the angle and dihedral angle change from ∠123 = 120.4° and ∠1234 = 112.9° in the free catalyst to ∠123 = 125.4° and ∠1234 = 150.3° in (E,S)-TS4, and (E,S)-TS4 exhibits shorter distances between catalyst and substrates (b1 = 2.135 Å and b2 = 1.666 Å) than (E,R)-TS4 (b1 = 2.640 Å and b2 = 1.734 Å), which could result in greater steric repulsion. Therefore, the distortion of catalyst as well as the steric effects provide a higher free energy of (E,S)-TS4, and lead to the observed enantioselectivity. Further analysis of (E,R)-TS4 disclosed additional π-π stacking interactions between resorcinol ring and indene moiety, which was evidenced by NCI analysis (see Supporting information for details).
In conclusion, a CBA-catalyzed enantioselective intermolecular transformation of vinyl cations has been realized by C(sp2)–H functionalization of diphenols, providing a modular platform for the atom-economic synthesis of axially chiral tetrasubstituted alkenes. The effective control mode of vinyl cations enables high chemoselectivity, regioselectivity, E/Z selectivity and enantioselectivity. Importantly, this protocol represents the first organocatalytic enantioselective intermolecular reaction of vinyl cations, as well as the first organocatalytic atroposelective intermolecular transformation of ynamides. By taking advantage of the facile scalability and product derivatizations, the constructed atropisomeric skeleton could be transformed into chiral phosphine ligand, and applied to asymmetric catalysis. Moreover, experimental and computational mechanistic studies provide insights into the reaction mechanism, origin of E/Z selectivity and enantioselectivity. We believe this CBA-catalyzed enantioselective intermolecular reaction of vinyl cations not only enriches the chemical space of axial chirality but also inspires the new aspects of vinyl cation chemistry and alkyne chemistry.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Da-Qiu Cui: Data curation. Quan-Xin Li: Data curation. Jia-Bo Huang: Data curation. Ying-Qi Zhang: Data curation. Xuan Wang: Data curation. Shao-Fei Ni: Data curation, Conceptualization. Long-Wu Ye: Writing – review & editing, Supervision. Bo Zhou: Writing – review & editing, Writing – original draft, Supervision, Project administration, Investigation, Conceptualization.
We are grateful for financial support from the National Natural Science Foundation of China (Nos. 22301250, 22125108, and 22331004), the Natural Science Foundation of Fujian Province of China (No. 2023J05005), Guangdong Basic and Applied Basic Research Foundation (Nos. 2024A1515010323, 2025A1515011907), the Natural Science Foundation of Xiamen, China (No. 3502Z202371002), the Fundamental Research Funds for the Central Universities (No. 20720230003), the open research fund of Songshan Lake Materials Laboratory (No. 2023SLABFN16).
Supplementary material associated with this article can be found, in the online version, at doi:
M. Hanack, Angew. Chem. Int. Ed. 17 (1978) 333–341. doi: 10.1002/anie.197803331
T. Okuyama, Acc. Chem. Res. 35 (2002) 12–18. doi: 10.1021/ar0100374
M. Niggemann, S. Gao, Angew. Chem. Int. Ed. 57 (2018) 16942–16944. doi: 10.1002/anie.201810701
X.J. Liu, Y. Xu, C. Tang, P.C. Qian, L.W. Ye, Sci. China Chem. 65 (2022) 20–30. doi: 10.1007/s11426-021-1117-2
T. Jin, M. Himuro, Y. Yamamoto, J. Am. Chem. Soc. 132 (2010) 5590–5591. doi: 10.1021/ja101633x
A.J. Walkinshaw, W. Xu, M.G. Suero, M.J. Gaunt, J. Am. Chem. Soc. 135 (2013) 12532–12535. doi: 10.1021/ja405972h
T. Wurm, J. Bucher, S.B. Duckworth, et al., Angew. Chem. Int. Ed. 56 (2017) 3364–3368. doi: 10.1002/anie.201700057
S. Popov, B. Shao, A.L. Bagdasarian, et al., Science 361 (2018) 381–387. doi: 10.1126/science.aat5440
S.E. Cleary, X. Li, L.C. Yang, et al., J. Am. Chem. Soc. 141 (2019) 3558–3565. doi: 10.1021/jacs.8b12420
T. Okuyama, T. Takino, T. Sueda, M. Ochiai, J. Am. Chem. Soc. 117 (1995) 3360–3367. doi: 10.1021/ja00117a006
M. Kreuzahler, A. Daniels, C. Wölper, G. Haberhauer, J. Am. Chem. Soc. 141 (2019) 1337–1348. doi: 10.1021/jacs.8b11501
S. Schroeder, C. Strauch, N. Gaelings, M. Niggemann, Angew. Chem. Int. Ed. 58 (2019) 5119–5123. doi: 10.1002/anie.201810916
B. Wigman, W. Lee, W. Wei, K.N. Houk, H.M. Nelson, Angew. Chem. Int. Ed. 61 (2022) e202113972. doi: 10.1002/anie.202113972
T. Ikeuchi, S. Inuki, S. Oishi, H. Ohno, Angew. Chem. Int. Ed. 58 (2019) 7792–7796. doi: 10.1002/anie.201903384
H. Komatsu, T. Ikeuchi, H. Tsuno, et al., Angew. Chem. Int. Ed. 60 (2021) 27019–27025. doi: 10.1002/anie.202111267
F.L. Hong, Z.S. Wang, D.D. Wei, et al., J. Am. Chem. Soc. 141 (2019) 16961–16970. doi: 10.1021/jacs.9b09303
F.L. Hong, C.Y. Shi, P. Hong, et al., Angew. Chem. Int. Ed. 61 (2022) e202115554. doi: 10.1002/anie.202115554
H.H. Chen, Y.B. Chen, J.Z. Gao, L.W. Ye, B. Zhou, Angew. Chem. Int. Ed. 63 (2024) e202411709. doi: 10.1002/anie.202411709
Y.X. Zheng, L.G. Liu, T.Q. Hu, et al., Nat. Commun. 15 (2024) 9227. doi: 10.1038/s41467-024-53605-7
F.S. Li, X.Y. Zou, T.Q. Hu, et al., Sci. Adv. 10 (2024) eadq7767. doi: 10.1126/sciadv.adq7767
Y.B. Chen, L.G. Liu, Z.Q. Wang, et al., Nat. Commun. 15 (2024) 2232. doi: 10.1038/s41467-024-46288-7
F.L. Hong, Y.B. Chen, S.H. Ye, et al., J. Am. Chem. Soc. 142 (2020) 7618–7626. doi: 10.1021/jacs.0c01918
L.J. Qi, C.T. Li, Z.Q. Huang, et al., Angew. Chem. Int. Ed. 61 (2022) e202210637. doi: 10.1002/anie.202210637
C.T. Li, L.J. Qi, L.G. Liu, et al., Nat. Commun. 14 (2023) 7058. doi: 10.1038/s41467-023-42805-2
Y.B. Chen, L.G. Liu, C.M. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202303670. doi: 10.1002/anie.202303670
C.Y. Weng, L.G. Liu, M. Sun, et al., Angew. Chem. Int. Ed. 64 (2025) e202418254. doi: 10.1002/anie.202418254
S.K. Nistanaki, C.G. Williams, B. Wigman, et al., Science 378 (2022) 1085–1091. doi: 10.1126/science.ade5320
T. Akiyama, Chem. Rev. 107 (2007) 5744–5758. doi: 10.1021/cr068374j
D. Parmar, E. Sugiono, S. Raja, M. Rueping, Chem. Rev. 114 (2014) 9047–9153. doi: 10.1021/cr5001496
T. James, M. van Gemmeren, B. List, Chem. Rev. 115 (2015) 9388–9409. doi: 10.1021/acs.chemrev.5b00128
R. Maji, S.C. Mallojjala, S.E. Wheeler, Chem. Soc. Rev. 47 (2018) 1142–1158. doi: 10.1039/c6cs00475j
J.L. Kennemur, R. Maji, M.J. Scharf, B. List, Chem. Rev. 121 (2021) 14649–14681. doi: 10.1021/acs.chemrev.1c00620
T. Akiyama, J. Itoh, K. Yokota, K. Fuchibe, Angew. Chem. Int. Ed. 43 (2004) 1566–1568. doi: 10.1002/anie.200353240
D. Uraguchi, M. Terada, J. Am. Chem. Soc. 126 (2004) 5356–5357. doi: 10.1021/ja0491533
X.N. Wang, H.S. Yeom, L.C. Fang, et al., Acc. Chem. Res. 47 (2014) 560–578. doi: 10.1021/ar400193g
G. Evano, C. Theunissen, M. Lecomte, Aldrichimica. Acta 48 (2015) 59–70.
Y.B. Chen, P.C. Qian, L.W. Ye, Chem. Soc. Rev. 49 (2020) 8897–8909. doi: 10.1039/d0cs00474j
F.L. Hong, L.W. Ye, Acc. Chem. Res. 53 (2020) 2003–2019. doi: 10.1021/acs.accounts.0c00417
C.C. Lynch, A. Sripada, C. Wolf, Chem. Soc. Rev. 49 (2020) 8543–8583. doi: 10.1039/d0cs00769b
Y.C. Hu, Y. Zhao, B. Wan, Q.A. Chen, Chem. Soc. Rev. 50 (2021) 2582–2625. doi: 10.1039/d0cs00283f
L. Hu, J. Zhao, Acc. Chem. Res. 57 (2024) 855–869. doi: 10.1021/acs.accounts.3c00743
B. Zhou, Y.Q. Zhang, K. Zhang, et al., Nat. Commun. 10 (2019) 3234. doi: 10.1038/s41467-019-11245-2
P.F. Chen, B. Zhou, P. Wu, B. Wang, L.W. Ye, Angew. Chem. Int. Ed. 60 (2021) 27164–27170. doi: 10.1002/anie.202113464
Y.Q. Zhang, Y.B. Chen, J.R. Liu, et al., Nat. Chem. 13 (2021) 1093–1100. doi: 10.1038/s41557-021-00778-z
Z.S. Wang, L.J. Zhu, C.T. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202201436. doi: 10.1002/anie.202201436
J. Wei, J. Zhang, J.K. Cheng, S.H. Xiang, B. Tan, Nat. Chem. 15 (2023) 647–657. doi: 10.1038/s41557-023-01179-0
E. Kumarasamy, R. Raghunathan, M.P. Sibi, J. Sivaguru, Chem. Rev. 115 (2015) 11239–11300. doi: 10.1021/acs.chemrev.5b00136
J.K. Cheng, S.H. Xiang, S. Li, L. Ye, B. Tan, Chem. Rev. 121 (2021) 4805–4902. doi: 10.1021/acs.chemrev.0c01306
J.A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos, J.M. Lassaletta, Chem. Soc. Rev. 50 (2021) 2968–2983. doi: 10.1039/d0cs00870b
G. Centonze, C. Portolani, P. Righi, G. Bencivenni, Angew. Chem. Int. Ed. 62 (2023) e202303966. doi: 10.1002/anie.202303966
S.H. Xiang, W.Y. Ding, Y.B. Wang, B. Tan, Nat. Catal. 7 (2024) 483–498. doi: 10.1038/s41929-024-01138-z
T.A. Schmidt, V. Hutskalova, C. Sparr, Nat. Rev. Chem. 8 (2024) 497–517. doi: 10.1038/s41570-024-00618-x
A.G. Woldegiorgis, X. Lin, Beilstein J. Org. Chem. 17 (2021) 2729–2764. doi: 10.3762/bjoc.17.185
J.K. Cheng, B. Tan, Chem. Rec. 23 (2023) e202300147. doi: 10.1002/tcr.202300147
Q.L. Zhou, Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim Germany, 2011.
G. Bringmann, T. Gulder, T.A.M. Gulder, M. Breuning, Chem. Rev. 111 (2011) 563–639. doi: 10.1021/cr100155e
X. Zhang, J. Yin, J. Yoon, Chem. Rev. 114 (2014) 4918–4959. doi: 10.1021/cr400568b
J.E. Smyth, N.M. Butler, P.A. Keller, Nat. Prod. Rep. 32 (2015) 1562–1583. doi: 10.1039/C4NP00121D
M. Basilaia, M.H. Chen, J. Secka, J.L. Gustafson, Acc. Chem. Res. 55 (2022) 2904–2919. doi: 10.1021/acs.accounts.2c00500
J. Feng, Z. Gu, SynOpen (2021) 68–85 05.
S. Wu, S.H. Xiang, J.K. Cheng, B. Tan, Tetrahedron Chem. 1 (2022) 100009. doi: 10.1016/j.tchem.2022.100009
P.F. Qian, T. Zhou, B.F. Shi, Chem. Commun. 59 (2023) 12669–12684. doi: 10.1039/d3cc03592a
J.Y. Zou, W.Y. Xu, J. Wang, Q. Liu, Y. He, Synthesis 56 (2024) 1862–1872. doi: 10.1055/a-2230-0759
Y. Tan, S. Jia, F. Hu, et al., J. Am. Chem. Soc. 140 (2018) 16893–16898. doi: 10.1021/jacs.8b09893
C. Ma, F.T. Sheng, H.Q. Wang, et al., J. Am. Chem. Soc. 142 (2020) 15686–15696. doi: 10.1021/jacs.0c00208
L. Jin, Q.J. Yao, P.P. Xie, et al., Chem 6 (2020) 497–511.
D. Yokose, Y. Nagashima, S. Kinoshita, J. Nogami, K. Tanaka, Angew. Chem. Int. Ed. 61 (2022) e202202542. doi: 10.1002/anie.202202542
W. Li, S. Chen, J. Xie, et al., Nat. Synth. 2 (2023) 140–151.
J. Wang, J. Gu, J.Y. Zou, et al., Nat. Commun. 15 (2024) 3254. doi: 10.3390/polym16233254
P. Hu, L. Hu, X.X. Li, et al., Angew. Chem. Int. Ed. 63 (2024) e202312923. doi: 10.1002/anie.202312923
Q.-H. Wu, M. Duan, Y. Chen, et al., Nat. Catal. 7 (2024) 185–194. doi: 10.1038/s41929-023-01097-x
F. Wu, Y. Zhang, R. Zhu, Y. Huang, Nat. Chem. 16 (2024) 132–139. doi: 10.1038/s41557-023-01358-z
Y.H. Miao, Z.X. Zhang, X.Y. Huang, et al., Chin. Chem. Lett. 35 (2024) 108830.

Scheme 2 Substrate scope for the atroposelective synthesis of axially chiral tetrasubstituted alkenes from different diynes 1. Reaction conditions: 1 (0.1 mmol), 2a (0.2 mmol), A14 (0.015 mmol), DCM (8 mL), 20 ℃, 1–46 h, in vials; yields were those for the isolated products; ee values were determined by HPLC analysis. a (R)-A14 (0.015 mmol) was used as catalyst. b A14 (0.02 mmol). PG = protecting group, PMP = 4-methoxyphenyl, Bs = 4-bromobenzenesulfonyl.

Scheme 3 Substrate scope for the atroposeclective synthesis of axially chiral tetrasubstituted alkenes from different diphenols 2. Reaction conditions: 1a (0.1 mmol), 2 (0.2 mmol), A14 (0.015 mmol), DCM (8 mL), 20 ℃, 1–4.5 h, in vials; yields were those for the isolated products; ee values were determined by HPLC analysis. a A14 (0.02 mmol).

Scheme 5 DFT calculated Gibbs free energy profiles for the reaction mechanism. Relative free energies (ΔG, in kcal/mol) of all the transition states and intermediates were computed at the B3LYP-D3/6–311++G(d,p)-SMD(DCM)//B3LYP-D3/6–31G(d) level of theory.
Table 1. Optimization of reaction conditions for the CBA-catalyzed atroposelective C(sp2)–H functionalization of diphenol 2a.a
|
|||||
| Entry | Catalyst | Reaction conditions | Yield (%)b | ee of 3a (%)c | |
| 3a | 3a′ | ||||
| 1 | A1 | DCM, 30 ℃, 19 h | 28 | 20 | 21 |
| 2 | A2 | DCM, 30 ℃, 50 h | 47 | < 2 | < 2 |
| 3 | A3 | DCM, 30 ℃, 50 h | 58 | 15 | < 2 |
| 4 | A4 | DCM, 30 ℃, 19 h | 32 | 20 | 33 |
| 5 | A5 | DCM, 30 ℃, 5 h | 77 | 5 | 77 |
| 6 | A6 | DCM, 30 ℃, 3 h | 54 | 6 | 80 |
| 7 | A7 | DCM, 30 ℃, 57 h | 11 | 4 | 11 |
| 8 | A8 | DCM, 30 ℃, 1.5 h | 61 | 6 | 73 |
| 9 | A9 | DCM, 30 ℃, 1 h | 62 | 8 | 70 |
| 10 | A10 | DCM, 30 ℃, 72 h | 35 | 62 | 25 |
| 11 | A11 | DCM, 30 ℃, 46 h | 42 | 8 | 56 |
| 12 | A12 | DCM, 30 ℃, 72 h | 33 | 8 | 27 |
| 13 | A13 | DCM, 30 ℃, 50 h | 12 | < 2 | 55 |
| 14 | A14 | DCM, 30 ℃, 0.5 h | 80 | 5 | 93 |
| 15 | A14 | DCE, 30 ℃, 0.5 h | 76 | 7 | 90 |
| 16 | A14 | Toluene, 30 ℃, 12 h | 51 | 4 | 73 |
| 17d | A14 | DCM, 30 ℃, 1 h | 74 | 4 | 93 |
| 18d,e | A14 | DCM, 30 ℃, 1 h | 82 | 4 | 94 |
| 19d,e | A14 | DCM, 20 ℃, 1 h | 88 | 5 | 94 |
| a Reaction conditions: 1a (0.05 mmol), 2a (0.1 mmol), catalyst (0.01 mmol), solvent (2 mL), 20–30 ℃, 0.5–72 h, in vials. PMP = 4-methoxyphenyl, DCE = 1,2-dichloroethane. b Measured by 1H NMR using dimethyl terephthalate as the internal standard. c Determined by HPLC analysis. d A14 (0.0075 mmol). e DCM (4 mL). |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: