School of Materials Science and Engineering, Shenyang University of Technology, Shenyang Key Laboratory of Advanced Energy Materials and Renewable Resources, Shenyang University of Technology, Shenyang 110870, China
b.
Key Laboratory of Superlight Materials and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150010, China
kzhu@hrbeu.edu.cn (K. Zhu). 1 These authors contributed equally to this work.
Received Date:
17 June 2025 Accepted Date:
15 August 2025 Revised Date:
05 August 2025 Available Online:
15 April 2026
Abstract:
NH4V4O10 has attracted significant attention as a cathode material for aqueous zinc-ion batteries (AZIBs) due to its adjustable interlayer spacing (~9.5 Å) and high theoretical specific capacity (~400 mAh/g). However, its development is hindered by sluggish Zn2+ kinetics and structural instability. In this work, a glycine (Gly) intercalation strategy is demonstrated, which establishes three stabilization mechanisms in the Gly-NVO cathode: (1) Gly pillars are shown to expand the interlayer spacing to 1.21 nm; (2) Hydrogen-bonding networks are formed between layers; (3) Reversible NH4+ (de)intercalation behavior is observed. The Gly-NVO cathode delivers a capacity of 520 mAh/g at 0.2 A/g (393 Wh/kg energy density), along with outstanding rate capability (400 and 150 mAh/g at 10 and 50 A/g, corresponding to power densities of 5688 and 24.3 kW/kg, respectively). A capacity retention of 88.2% is maintained after 10,000 cycles at 50 A/g. DFT calculations confirm that the introduction of Gly significantly enhances the electrical conductivity of NVO while effectively weakening electrostatic interactions, and energy barrier for Zn2+ intercalation and vanadium dissolution are reduced by Gly. Additionally, due to the reversible NH4+ (de)intercalation behavior in Gly-NVO, the assembled aqueous ammonium-ion batteries (AAIBs) exhibit stable cycling ability. This work highlights organic molecule pre-intercalation as a viable strategy for optimizing the durability of ammonium vanadate cathodes.
AZIBs have emerged as promising candidates for grid-scale energy storage due to their inherent safety, low cost, and environmental benignity [1-3]. However, the development of high-performance cathode materials remains a significant challenge. Layered ammonium vanadate (NH4V4O10) cathodes have attracted considerable attention owing to their high theoretical capacity (~400 mAh/g) and adjustable interlayer spacing (~9.5 Å) [4]. Nevertheless, their practical application is hindered by three intrinsic limitations. The strong electrostatic interactions between intercalated Zn2+ ions and the V4O10 framework are demonstrated to cause interlayer contraction, which subsequently induces V-O bond rupture and eventual structural collapse [5]. Vanadium dissolution is observed in weakly acidic electrolytes through the formation of soluble VO2+ and VO2+ species, leading to progressive capacity fading [6]. While NH4+ ions initially serve as structural templates for the layered architecture, their irreversible extraction during the first charge cycle is shown to trigger detrimental phase transformations [7]. Therefore, the development of advanced scientific strategies to fundamentally address these intrinsic limitations is critically required to advance the field of layered ammonium vanadate cathode materials.
In recent years, significant efforts have been made to address the inherent limitations of ammonium vanadate cathodes through various strategies including ion pre-intercalation [8-10], interface engineering [11,12], and defect engineering [13,14]. Bai et al. [8] demonstrated the "pillaring" effect of Rb+ intercalation in NH4V4O10, which effectively stabilizes the microstructure during cycling. Yuan et al. [11] developed VO2/NH4V4O10 heterostructures, where the multiphase interfaces and micron-flower architecture provide abundant Zn2+ storage sites. Bai et al. [14] involved V/O dual-defect engineering, which substantially reduces the Zn2+ diffusion barrier and ensures homogeneous ion distribution within the NH4V4O10 cathodes. Although numerous strategies have been proposed to address the limitations of NH4V4O10 cathodes, and the potential of organic molecular intercalation is still largely unexplored. Gly as the smallest amino acid (molecular length ~4.5 Å), exhibits low steric hindrance and can be readily intercalated into layered cathode materials (e.g., NH4V4O10 with an interlayer spacing of ~9.6 Å). Its zwitterionic form (NH3+-CH2—COO−) stabilizes the layered structure through electrostatic interactions, suppressing material dissolution. Additionally, its high thermal stability (~233 ℃) ensures molecular integrity during hydrothermal synthesis (180 ℃). The introduction of Gly molecules into NH4V4O10 cathodes is expected, it prompting a systematic investigation into organic molecular pre-intercalation engineering.
Herein, this work introduces Gly molecules into the interlayer structure of NH4V4O10 to form molecular pillars, expanding the interlayer spacing of Gly-NVO to 1.21 nm. A hydrogen bond network is established between the -NH2 groups of Gly molecules and the V4O10 framework. Ex-situ characterization reveals the reversible de(intercalation) behavior of NH4+ in Gly-NVO. Electrochemical test results demonstrate that the Gly-NVO cathode delivers an exceptionally high specific capacity of 520 mAh/g at 0.2 A/g and 400 mAh/g at 10 A/g. The rate performance shows that Gly-NVO can still provide a high discharge specific capacity of nearly 150 mAh/g even at 50 A/g, and maintains stable cycling for 10,000 cycles with 88.2% capacity retention. Moreover, its outstanding energy density (393 Wh/kg at 0.2 A/g) and power density (24.3 kW/kg at 50 A/g) exhibit competitive advantages among ammonium vanadate materials. Furthermore, the AAIBs (Gly-NVO||(NH4)2SO4||PTCDI) are assembled with Gly-NVO cathode delivers a specific capacity of 198 mAh/g at 0.2 A/g and maintains 82.1% capacity retention after 180 cycles. DFT calculations reveal that the introduction of Gly enhances the electronic/ionic conductivity of NH4V4O10, stabilizes the V-O bonds, and reduces the dissolution energy of VO2+/VO2+. In addition, NH4+ exhibits lower adsorption energy and intercalation formation energy in Gly-NVO. Therefore, Gly molecular pre-intercalation engineering is an effective strategy for improving NH4V4O10 cathodes. We anticipate that this study will further expand the scope of cathode modification for Ammonium vanadate cathodes.
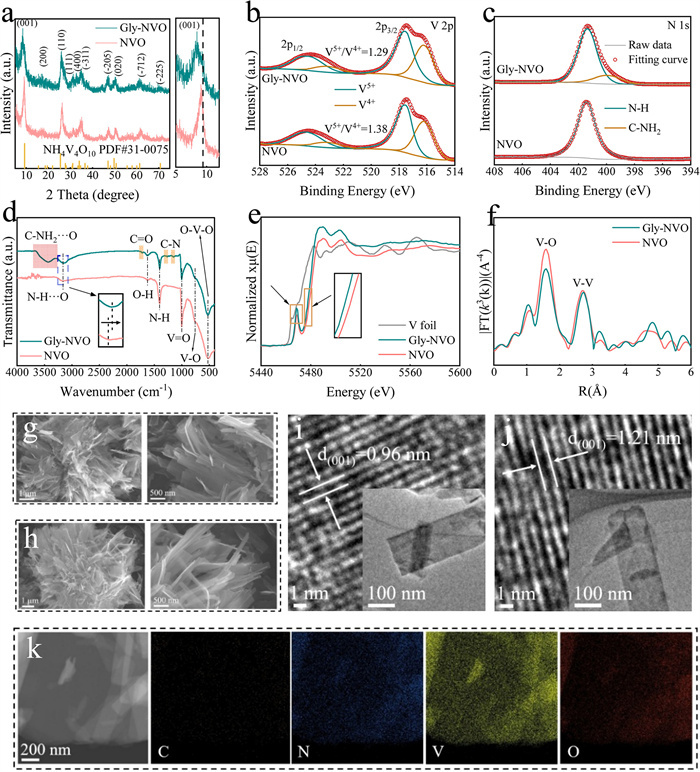
NH4V4O10 (NVO) and Gly-modified NH4V4O10 (Gly-NVO) are successfully synthesized through a one-step hydrothermal method (Fig. S1), with detailed preparation procedures provided in the Supporting Information. In Fig. 1a, the diffraction peaks of both NVO and Gly-NVO are perfectly indexed to the monoclinic NH4V4O10 phase (PDF #31–0075, space group: P2/m) without any impurity peaks, confirming that the crystal structure is well preserved after Gly modification. Notably, a slight lattice shift is observed in the (001) plane of Gly-NVO, which may be attributed to the intercalation of Gly molecules into the interlayer structure, resulting in an expanded interlayer spacing. This phenomenon can be explained by Bragg's law (2dsinθ = nλ), where the increased d-spacing leads to corresponding shifts in diffraction angles. The systematic variation in peak positions observed in the X-ray diffraction patterns further supports the structural modifications induced by intercalation, confirming the expansion of interlayer spacing due to the incorporation of organic molecules between inorganic layers. This observation is well consistent with previous reports on similar intercalation compounds [8,15,16]. The valence states of the constituent elements in the materials are analyzed by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1b, the V 2p spectra of NVO and Gly-NVO exhibit mixed-valence characteristics, with the peaks located at 517.5 and 524.9 eV corresponding to V5+, while the minor peaks at 516.2 and 523.2 eV are corresponded to V4+ [17,18]. The coexistence of these oxidation states is crucial for enhancing electrochemical performance, as it facilitates multiple electron transfers during charge/discharge processes, thereby contributing to improved specific capacity. This mixed-valence behavior is consistent with previous reports on vanadium-based electrode materials, where such electronic configurations have been demonstrated to promote reversible redox reactions and enhance electrochemical kinetics [19]. Through quantitative fitting of the V 2p XPS spectra, the V5+/V4+ ratio in Gly-NVO is significantly reduced compared to pristine NVO, demonstrating that Gly molecular pre-intercalation promotes the reduction of V5+ to V4+. Gly as a bifunctional molecule containing both amino (-NH2) and carboxyl (-COOH) groups, creates a reducing environment during synthesis where the amino group releases electrons under high-temperature/hydrothermal conditions, reducing partial V5+ to V4+. Simultaneously, the carboxyl group forms stable coordination with V4+, further promoting the reduction of V4+. Additionally, Gly-induced interlayer expansion in the (100) plane alleviates distortion in VO6 octahedra, reducing the need for some V5+ to maintain high valence states for lattice energy balance. The O 1s spectrum (Fig. S2 in Supporting information) is deconvoluted into three characteristic peaks at 530.1, 531.4 and 532.5 eV, corresponding to V-O bonds, oxygen vacancies (Od) and interlayer crystalline water, respectively [12]. Further investigation by electron paramagnetic resonance (EPR) reveals a distinct difference in oxygen vacancy concentration between the samples (Fig. S3 in Supporting information) [20]. While negligible oxygen vacancies are detected in pristine NVO, suggesting that the oxygen vacancies observed by XPS may be primarily surface-localized, measurable oxygen vacancies are confirmed in Gly-NVO. Previous studies [6,12,13] have demonstrated that an appropriate concentration of oxygen vacancies can effectively facilitate charge carrier transport and enhance ion migration rates in electrode materials. The N 1s spectra of Gly-NVO further confirms the successful incorporation of Gly molecules with the material (Fig. 1c). The spectrum exhibits two distinct peaks, a characteristic binding energy at 400.2 eV corresponding to the C—NH2 bond of Gly molecules [21] and while the 401.5 eV feature belongs to N—H, likely associated with NH4+ [22]. These spectral features provide direct evidence for the presence of intact Gly molecules in the composite structure. The appearance of the C—NH2 characteristic specifically confirms that Gly maintains molecular integrity during intercalation rather than undergoing decomposition. The chemical bonding characteristics of the materials are further investigated by Fourier transform infrared spectroscopy (FTIR) (Fig. 1d). The characteristic absorption bands observed at 522.8, 761.4, and 1007.6 cm-1 are assigned to the bending vibration of O-V-O bonds, asymmetric stretching vibration of V-O bonds, and stretching vibration of V = O bonds, respectively, confirming the presence of characteristic vanadium-oxygen frameworks [8,23]. Additional vibrational features appearing at 1408.6 and 1626.9 cm-1 correspond to the bending and stretching modes of N—H (NH4+) and O—H (Lattice H2O) bonds, respectively, indicating the presence of NH4+ species in the materials [21]. A broad absorption band in the range of 3100–3200 cm-1 is observed, which can be attributed to hydrogen bonding interactions between NH4+ and the V4O10 framework [22]. And a distinct redshift in the N—H···O vibrational mode of Gly-NVO compared to pristine NVO. This redshift (typically indicative of altered chemical environments or strengthened intermolecular interactions) suggests the formation of dynamic hydrogen bonding between intercalated Gly molecules and NH4+. Notably, a particularly significant broad absorption band in the 3300–3600 cm-1 region, which is characteristic of hydrogen bonding interactions between the C—NH2 groups of Gly molecules and the O2- in the V4O10 framework [2,24]. This observation provides compelling evidence for the formation of additional hydrogen-bonding networks, which serve as structural bridges between organic and inorganic components. The existence of these intermolecular interactions not only confirms the successful incorporation of Gly into the layered structure, but also suggests a stabilization mechanism where Gly molecules act as molecular pillars through hydrogen bonding with inorganic layers. Such hydrogen-bond formation has been reported to enhance the structural stability of similar hybrid materials during electrochemical processes by reducing interlayer repulsion and maintaining the integrity of the layered framework [25,26]. To further elucidate the impact of Gly pre-intercalation on NH4V4O10, we conducted synchrotron-based X-ray absorption spectroscopy (XAS) investigations. The V K-edge X-ray absorption near-edge structure (XANES) spectra are shown in Fig. 1e. The absorption-edge energy positions of Gly-NVO (5465–5470 eV) exhibit significantly higher intensity compared to NVO, indicating a reduction in the coordination number of V (octahedral → square pyramidal) after the intercalation of Gly molecules, which is attributed to the interaction between the 1s and bound hybridized d-orbitals. Comparing the absorption-edge positions of Gly-NVO and NVO, the V K-edge XANES of Gly-NVO shifts toward lower energy, suggesting a decrease in the average oxidation state of V atoms. This implies partial electron transfer from Gly to the layered structure of the material [15]. The k3-weighted Fourier-transformed V K-edge extended X-ray absorption fine structure (EXAFS) spectra of Gly-NVO and NVO are shown in Fig. 2f. Comparative analysis reveals that the first-shell V-O intensity of Gly-NVO is significantly lower than that of NVO, indicating a reduction in the V-O coordination number. This suggests a possible transformation in the coordination geometry from octahedral to square pyramidal, accompanied by increased local disorder, which may enhance the stability of the layered structure. Moreover, the second-shell V-V distance in Gly-NVO (2.75 Å) is elongated compared to that in NVO (2.68 Å), providing direct evidence for the successful intercalation of Gly molecules into the layered framework [27].
Figure 1
Figure 1.
Structural and elemental properties of NVO and Gly-NVO. (a) XRD patterns of NVO and Gly-NVO. (b) V 2p spectra of NVO and Gly-NVO. (c) N 1s spectra of NVO and Gly-NVO. (d) FTIR spectra of NVO and Gly-NVO. (e) XANES spectra of V K-edge. (f) V K-edge of Fourier-transform EXAFS spectra. (g, h) SEM images of NVO and Gly-NVO. (i, j) HRTEM images of NVO and Gly-NVO. (k) TEM-mapping of Gly-NVO.
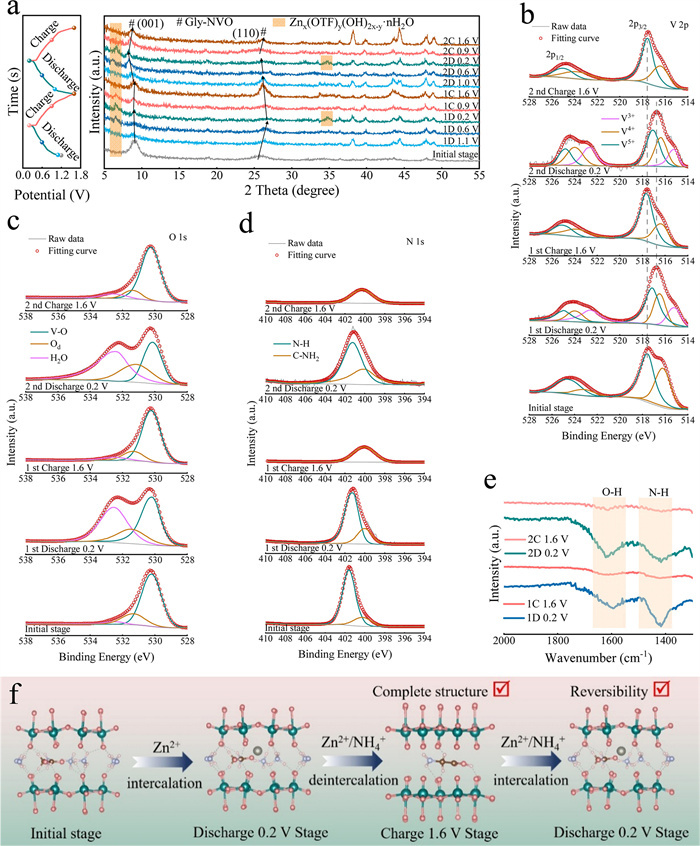
Figure 2.Ex-situ characterization of Gly-NVO in the first two cycles. (a) XRD patterns; (b-d) V 2p, O 1s and N 1s spectra. (e) FTIR spectra, f schematic image of Zn2+/NH4+ (de)intercalation in Gly-NVO.
The surface morphologies of NVO and Gly-NVO are examined by scanning electron microscopy (SEM), revealing similar nanobelt structures for both materials (Figs. 1g and h). This morphological similarity indicates that the introduction of Gly does not significantly alter the overall nanostructure of the material, suggesting that the organic molecules are primarily intercalated into the interlayer spaces rather than adsorbed on the surfaces. This observation demonstrates that any enhancement in the electrochemical performance of Gly-NVO cannot be attributed to morphological changes. High-resolution transmission electron microscopy (HRTEM) provides definitive evidence for the successful intercalation of Gly molecules, as demonstrated by the expansion of the (001) interplanar spacing from 0.96 nm in the pristine material to 1.21 nm in Gly-NVO (Figs. 1i and j). The increased interlayer distance is particularly advantageous for electrochemical applications as it facilitates faster ion diffusion kinetics while maintaining structural stability during charge/discharge cycles [16,19]. In Fig. 1k, elemental mapping analysis conducted by transmission electron microscopy (TEM) clearly demonstrates the homogeneous distribution of Gly throughout the Gly-NVO composite, rather than forming isolated aggregates, as evidenced by the uniform spatial distribution of carbon signals.
The Gly-NVO electrode is systematically analyzed by ex situ XRD and XPS at different charge/discharge states during the first two cycles. The XRD patterns show completely reversible structural evolution, where all characteristic peaks are maintained in both position and intensity after cycling, demonstrating excellent crystallinity retention (Fig. 2a). Notably, the (001) plane of Gly-NVO is observed to exhibit distinct structural evolution during electrochemical cycling. The first cycle is found to display smaller reversible peak shifts compared to subsequent cycles. A subtle low-angle shift is detected for the (001) plane during initial discharge to 0.2 V, which is subsequently restored to its original position upon charging to 1.6 V. Remarkably, more pronounced but completely reversible peak shifts are exhibited in the second cycle. This behavior is contrasted with reported ammonium vanadate materials, where strong Zn2+-host electrostatic interactions are typically found to induce opposite peak-shift trends (high-angle shifts following Zn2+ intercalation) [8,12,20,22]. The anomalous shift pattern observed in Gly-NVO indicates that strong electrostatic interactions may be effectively mitigated through Gly-mediated charge screening. Interestingly, the (110) plane is observed to exhibit an opposite shift trend during the first cycle, which is potentially caused by its smaller interplanar spacing compared to the (001) plane, while consistent behavior is maintained in subsequent cycles. These findings collectively demonstrate that the electrostatic interactions between Zn2+ and host material are alleviated by Gly intercalation. Moreover, the comparative results clearly demonstrate that pristine NVO exhibits an opposite peak shift behavior compared to Gly-NVO during the first cycle, as evidenced by the ex-situ XRD patterns in Fig. S4 (Supporting information). Specifically, during the discharge phase at 0.2 V, the (001) and (110) diffraction planes shift toward higher angles, and then return to their original positions during the charging phase at 1.6 V. This opposite peak shift behavior is attributed to the strong interaction between Zn2+ and NVO, which precisely highlights the role of Gly in modulating the Zn2+ intercalation behavior in conventional NH4V4O10 materials. Generally, Zn2+ exhibits strong electrostatic interactions with NH4V4O10, which typically leads to contraction of interplanar spacing in certain crystal planes upon Zn2+ intercalation due to electrostatic attraction. In this work, we have demonstrated through comprehensive characterization that Gly molecules are intercalated into the layered structure of NH4V4O10, serving to expand the (001) interplanar spacing and weaken the electrostatic interaction with Zn2+. This explains the observed behavior in ex-situ XRD pattern where the (001) lattice spacing first increases and then decreases during Zn2+ intercalation/deintercalation, indicating effective suppression of Zn2+ electrostatic interactions. However, the (110) plane, with its smaller interplanar spacing and absence of Gly molecules to mitigate Zn2+ electrostatic effects, exhibits opposite displacement behavior compared to the (001) plane. The reversible peaks observed near 7° and 34° are attributed to the formation/dissolution of basic zinc salts (such as Znx(OTF)y(OH)2x-y·nH2O or similar compounds), which is well-documented in AZIBs [15]. These characteristic peaks are observed to exhibit excellent reversibility throughout the cycling process, indicating that the zinc salt byproducts are involved in a dynamic equilibrium process during charge/discharge cycles. Importantly, the complete reversibility of these peaks is confirmed to demonstrate the absence of detrimental side reactions that could cause permanent electrode passivation. Fig. S6 (Supporting information) is shown to provide direct evidence for the reversible Zn2+ (de)intercalation behavior in Gly-NVO, although residual Zn signals are persistently observed even at the fully charged state (1.6 V). This phenomenon is attributed to the presence of structurally stabilized Zn2+. Fig. 2b clearly demonstrates the multivalent redox behavior of vanadium ions during the electrochemical cycling process. In the fully discharged state (0.2 V), a significant reduction in V5+ content is observed, accompanied by the emergence of V3+ species, which confirms the involvement of multiple electron transfer processes (V5+ ↔ V4+ ↔ V3+) [20]. It is noteworthy that when charged back to 1.6 V, the valence state of vanadium is fully restored to its initial distribution, demonstrating excellent redox reversibility. However, the observed shift in the V 2p spin-orbit doublet can be attributed to the reduction of vanadium oxidation states during the electrochemical process (gradually shifting toward lower binding energy during discharge) [28]. The evolution of O 1s spectra (Fig. 2c) reveals reversible changes in H2O content that can be attributed to the formation/dissolution of basic zinc salts (Znx(OTF)y(OH)2x-y·nH2O) during cycling [26]. This dynamic process maintains excellent reversibility, with complete restoration of the original spectral characteristics observed after full charging, in full agreement with ex-situ XRD observations. Moreover, the XRD characterization on the Gly-NVO electrode after 10 cycles is shown in Fig. S5 (Supporting information). The results confirm that the basic zinc salt byproduct Znx(OTF)y(OH)2x-y·nH2O maintains highly reversible behavior even after prolonged cycling, with no evidence of detrimental side reactions. Interestingly, the N 1s spectrum of Gly-NVO reveals a previously unreported reversible NH4+ (de)intercalation behavior (Fig. 2d), which contrasts sharply with reported ammonium vanadate cathodes where NH4+ de intercalation is typically irreversible [8,12,21,29]. The complete disappearance of the NH4+ peak at 401.5 eV indicates effective deintercalation of ammonium ions from the host structure. The subsequent reappearance of this characteristic during discharge demonstrates remarkable reversibility. This reversible NH4+ (de)intercalation process is observed to provide exceptional protection for the structural integrity of the host framework throughout the electrochemical cycling. The nearly unchanged intensity of the C—NH2 characteristic peak at 400.2 eV, observed during the entire electrochemical cycling process, further confirms the outstanding stability of Gly pillars in the interlayer structure. The stabilization mechanism is further investigated through ex situ FTIR characterization (Fig. 2e). The reversible appearance and disappearance of NH4+ bending vibration at 1410 cm-1 confirms the highly reversible (de)intercalation behavior of ammonium ions, which shows excellent agreement with XPS analysis results. Additionally, the H2O-related absorption peak at 1630 cm-1 exhibits reversible intensity variations, corresponding to the formation and dissolution of Znx(OTF)y(OH)2x-y·nH2O complexes during cycling. Fig. 2f illustrates the deintercalation behavior of Zn2+ and NH4+ in Gly-NVO. Specifically, during the initial charging stage to 1.6 V, the intercalated Zn2+ and NH4+ in Gly-NVO are simultaneously deintercalated. However, due to the pillar effect of Gly molecules, the structural stability of Gly-NVO is well maintained. Upon subsequent discharging to 0.2 V, Zn2+ and NH4+ are reintercalated into the material simultaneously, demonstrating excellent reversibility.
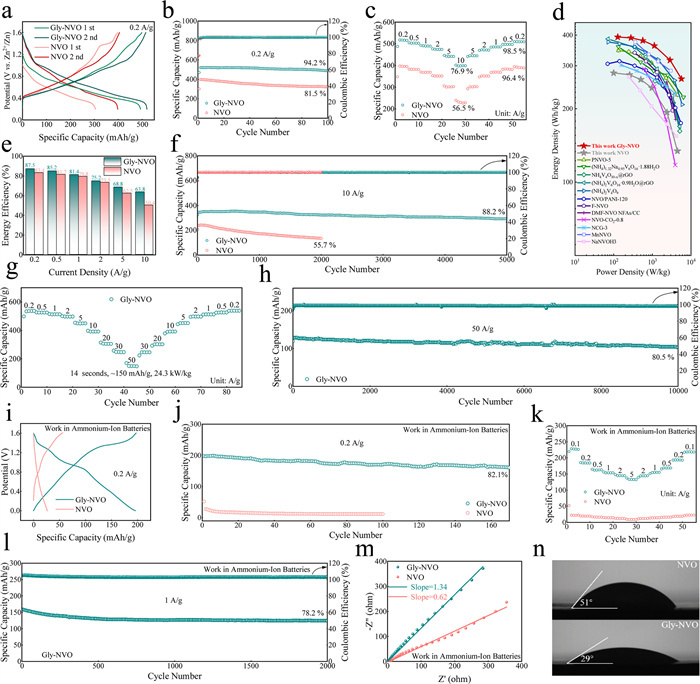
To comprehensively evaluate the cathode performance of the Gly-modified material, CR2032 coin cells are assembled using both NVO||ZnOTf||Zn and Gly-NVO||ZnOTf||Zn configurations. The cyclic voltammetry (CV) profiles of NVO and Gly-NVO recorded at a scan rate of 0.1 mV/s for the initial five cycles is presented in Fig. S7 (Supporting information), respectively. A remarkable difference in electrochemical reversibility is observed between the two materials. The Gly-NVO electrodes demonstrate excellent reproducibility of CV curves after the first cycle, with nearly overlapping redox peaks in subsequent scans, indicating highly reversible electrochemical reactions. In contrast, the NVO electrodes show progressively shifting peak positions and varying peak intensities, suggesting relatively poor reaction reversibility. The GCD curves of both materials at 0.2 A/g is presented in Fig. 3a, revealing significant performance differences. Gly-NVO delivers an impressive initial discharge capacity of 492 mAh/g, which further increases to 520 mAh/g in the second cycle, demonstrating exceptional capacity activation behavior. In contrast, the pristine NVO exhibits substantially lower capacities of 301 and 398 mAh/g for the first and second discharges, respectively. It is particularly noteworthy that while a distinct charging plateau at 1.4 V is observed in the first cycle of NVO, corresponding to the irreversible phase transition induced by NH4+ deintercalation (as previously reported in literature), this characteristic plateau is completely absent in Gly-NVO. This striking difference clearly demonstrates the enhanced structural stability achieved through Gly modification. The remarkably high initial Coulombic efficiency of 98.8% observed for Gly-NVO in Fig. S8 (Supporting information), significantly surpassing that of pristine NVO (77.3%). This exceptional efficiency is attributed to the stabilized coordination environment created by Gly molecules, which enables nearly complete NH4+ reintercalated during discharge while preventing irreversible structural rearrangements. The minimal efficiency loss (1.2%) contrasts sharply with conventional NH4V4O10 systems where NH4+ extraction typically induces permanent capacity loss (> 10%) due to phase transitions, as reported in previous studies [9,13,14,21,28]. The self-discharge characteristics of both materials are systematically evaluated in Fig. S9 (Supporting information). When fully charged to 1.6 V and subsequently stand for 24 h, Gly-NVO demonstrates superior performance with a coulombic efficiency of 96%, significantly exceeding the 91% efficiency observed for pristine NVO. Furthermore, Gly-NVO maintains a remarkably high open-circuit voltage of 1.439 V after the standing, compared to NVO lower retained voltage. These results clearly demonstrate the enhanced stability through Gly modification, which effectively suppresses spontaneous charge dissipation [29]. The rate capability of both materials is systematically evaluated in Fig. 3c. Remarkably, even at a high current density of 10 A/g, Gly-NVO maintains a superior discharge capacity of approximately 400 mAh/g, representing 76.9% retention relative to its capacity at 0.2 A/g. This outstanding rate performance is attributed to significantly enhanced Zn2+ diffusion kinetics facilitated by the expanded interlayer spacing and optimized charge transport pathways resulting from Gly intercalation. Furthermore, when the current density is returned to 0.2 A/g after the rate test, Gly-NVO exhibits excellent capacity recovery with 98.5% retention of its initial capacity, demonstrating remarkable structural stability and reversible Zn2+ storage capability. Notably, Gly-NVO cathode achieves an exceptionally high energy density of 393 Wh/kg at 0.2 A/g and maintains 1128 Wh/kg at the high current density of 10 A/g with an impressive power density of 5688 W/kg maintains 265 Wh/kg (Fig. 3d). These remarkable metrics reflect the rapid Zn2+ storage kinetics and excellent charge/discharge capability enabled by the Gly-modified structure. Gly-NVO cathodes have demonstrated excellent competitiveness among ammonium vanadate cathode materials reported in recently (Fig. 3d) [12,24,29-36]. Furthermore, the energy efficiency highlights the characteristics of Gly-NVO as a high-rate cathode (Fig. 3e). Within the current density range of 0.2–10 A/g, Gly-NVO demonstrates significantly superior energy efficiency compared to NVO. This reflects the higher electrochemical reaction efficiency and reduced polarization of Gly-NVO [15]. The cycle performance of Gly-NVO at 0.2 and 10 A/g is evaluated in Figs. 3b and f. Gly-NVO demonstrates exceptional cycling stability, maintaining a high discharge capacity of 490 mAh/g after 100 cycles at 0.2 A/g, corresponding to an impressive capacity retention of 94.2%. By comparing the first and 50th charge-discharge curves of NVO and Gly-NVO (Fig. S10 in Supporting information), the polarization voltage is obviously different. The NVO cathode has a serious polarization, the polarization voltage grows from 0.21 V to 0.26 V, indicating that the internal resistance of the material polarization increases, and the electron and ion migration is restricted. On the contrary, Gly-NVO does not exhibit significant polarization, which leads to a reduction of energy loss during charging and discharging, and is conducive to the stable cycling performance of the materials. More remarkably, the material exhibits outstanding long-term cyclability at a high current density of 10 A/g, it delivering 88.2% capacity retention after 5000 cycles. This exceptional cycling stability can be attributed to the dual stabilizing effects of Gly molecules serving as structural pillars and the formation of an extensive hydrogen-bonding network following their intercalation. The Gly pillars effectively mitigate layer stacking and prevent structural collapse during repeated cycling, while the three-dimensional hydrogen-bonding framework between the organic molecules and inorganic host matrix enhances structural integrity [37]. In addition, the effect of Gly intercalation on the cycling performance of the materials is further evaluated, and charge-discharge tests are performed on two materials with different doping amounts of 2Gly-NVO and 6Gly-NVO, and the test results are shown in Figs. S11 and S12 (Supporting information). The specific capacity of the two materials is improved and they also have good cycling stability at 0.2 and 10 A/g. These findings collectively demonstrate that moderate Gly intercalation effectively enhances the cycling stability of electrode materials while improving both reversible Zn2+ storage capability and specific capacity. However, when the Gly addition amount is relatively low, the interlayer spacing expansion of the material becomes insignificant. Consequently, 2Gly-NVO demonstrates lower specific capacity compared to Gly-NVO. Conversely, when Gly is excessively added, 6Gly-NVO exhibits Coulombic efficiency fluctuations during later cycles at 0.2 A/g, which may be attributed to side product formation on the material surface caused by Gly overloading, thereby compromising cycling stability. Given that Gly-NVO previously exhibits a high specific capacity of 400 mAh/g at 10 A/g, its rate performance is further tested at 50 A/g (Fig. 3g). At 20, 30 and 50 A/g, Gly-NVO can deliver the specific capacities of 310, ~255 and ~150 mAh/g, respectively. Notably, at a superhigh current density of 50 A/g, the discharge process is completed in only 14 s, with a remarkable power density of 24.3 kW/kg achieved. And the Gly-NVO maintains a capacity retention of 80.5% at 50 A/g, it demonstrates certain advantages compared to other related battery systems [17-19]. Further tests on the rate capability and cycling performance of pristine NVO at 50 A/g are conducted (Fig. S13 in Supporting information). The results demonstrate that, due to the poor stability and slow Zn2+ kinetics of pristine NVO, it delivers almost no measurable capacity at 30 or 50 A/g. Even at 20 A/g, its discharge capacity (~55 mAh/g) is significantly lower than that of Gly-NVO. These findings further confirm that Gly modification effectively enhances the electrochemical kinetics and structural stability of the material. The superior rate capability of Gly-NVO remains unmatched by other reported cathodes for AZIBs, in other words, the Gly-NVO cathode is expected to be regarded as a prime candidate for next-generation AZIBs energy storage systems.
Figure 3
Figure 3.
Electrochemical performance of NVO and Gly-NVO. (a) GCD curves of NVO and Gly-NVO in the first two cycles at 0.2 A/g. (b) Cycling performance of NVO and Gly-NVO at 0.2 A/g. (c) Rate performance of NVO and Gly-NVO. (d) ragone image of NVO, Gly-NVO and reported ammonium vanadate cathodes. (e) Energy effciency of Gly-NVO. (f) Cycling performance of NVO and Gly-NVO at 10 A/g. (g) Rate performance of Gly-NVO at 50 A/g. (h) Cycling performance of Gly-NVO at 50 A/g (aqueous ammonium-ion batteries system). (i) GCD curves of NVO and Gly-NVO at 0.2 A/g. (j) cycling performance of NVO and Gly-NVO at 0.2 A/g. (k) Rate performance of NVO and Gly-NVO. (l) NH4+ intercalated energy of NVO and Gly-NVO. (m) EIS of NVO and Gly-NVO. (n) Contact angles of NVO and Gly-NVO after 4 h of immersion in 2 mol/L (NH4)2SO4 aqueous solution.
In Gly-NVO, NH4+ (de)intercalation behavior is observed, prompting its exploration as a cathode material for AAIBs. Furthermore, a full-cell configuration (Gly-NVO||(NH4)2SO4||PTCDI) is assembled using PTCDI as the anode and a 2 mol/L (NH4)2SO4 aqueous solution as the electrolyte. As shown in Figs. 3i and j, the cell delivers a specific capacity of 198 mAh/g at 0.2 A/g, with 82.1% capacity retention after 180 cycles. In contrast, due to irreversible NH4+ deintercalation in NVO, the first-cycle charge capacity is only 52 mAh/g, and negligible capacity is observed in subsequent cycles. In Fig. S14 (Supporting information), the charge curves of Gly-NVO during the first five cycles at 0.2 A/g demonstrate efficient NH4+ deintercalation. Rate performance tests (Fig. 3k) reveal that Gly-NVO maintains a capacity of nearly 140 mAh/g even at 5 A/g. Additionally, after 2000 cycles at 1 A/g (Fig. 3l), Gly-NVO retains 78.2% of its initial capacity, highlighting its cycling stability and feasibility as an AAIB cathode material. Electrochemical impedance spectroscopy (EIS, Fig. 3m) indicates that the interfacial diffusion resistance dominates the low-frequency region in Gly-NVO, with a high linear slope (1.34), suggesting favorable NH4+ diffusion kinetics. Contact angle measurements (Fig. 3n) further confirm enhanced electrolyte wettability on Gly-NVO (contact angle: 29° after 4 h of immersion in 2 mol/L (NH4)2SO4), indicating a more intimate electrode/electrolyte interface and reduced impedance, consistent with EIS analysis. In summary, Gly-NVO exhibits excellent electrochemical performance in AAIBs, demonstrating its potential as a high-performance cathode for aqueous ammonium-ion batteries.
The electrochemical kinetics of Gly-NVO are systematically investigated by differential scanning voltammetry (DSCV) at various scan rates, with the detailed calculation procedure provided in Supporting information. Within the scan rate range of 0.1–1 mV/s, the CV curves exhibit highly reversible characteristics, featuring three distinct reduction peaks (R3, R2, and R1, corresponding to V5+ → V4+, V4+ → V3+, and H+ insertion, respectively) and two oxidation peaks (O1 and O2, representing V3+ → V4+ and V4+ → V5+, respectively). A slight polarization effect is observed as the scan rate increases (Fig. S15a in Supporting information). The kinetic behavior is further quantified through the linear fitting of log(i) versus log(v) plots to determine the b-values for each redox couple. The charge storage mechanism is identified as being capacitive (b-value approaching 1.0) or diffusion-controlled (b-value of 0.5) [18]. The determined b-values confirm that the electrochemical process in Gly-NVO is predominantly capacitive-controlled, which facilitates rapid Zn2+ diffusion kinetics (Fig. S15b in Supporting information). Furthermore, the capacitive contribution is quantitatively analyzed as shown in Fig. S15c (Supporting information), revealing a significant increase from 74.6% to 90.4% with increasing scan rates from 0.1 mV/s to 1.0 mV/s. The contribution rate of capacitance at different scan rate is shown in Fig. S16 (Supporting information). The high capacitive contribution and its progressive enhancement with scan rate demonstrate the exceptional rate capability of Gly-NVO [24], in excellent agreement with the rate performance results presented in Fig. 3c. The CV curves of NVO at different scan rates are shown in Fig. S17 (Supporting information). The kinetic behavior of NVO is dominated by a diffusion-controlled process. Moreover, it is found through pseudocapacitance fitting that the pseudocapacitive contribution of VO2 is significantly lower than that of Gly-NVO as the scan rate increases, which also reflects the superior rate capability of Gly-NVO. The Zn2+ diffusion kinetics in both materials are quantitatively evaluated through galvanostatic intermittent titration technique (GITT, Fig. S18 in Supporting information), with detailed calculation procedures provided in Supporting Information (Fig. S19 in Supporting information). Gly-NVO demonstrates significantly enhanced Zn2+ diffusion coefficients (DZn2+= 10-5.8–10-6.7 cm2/s), nearly two orders of magnitude higher than those of pristine NVO (10-7.7–10-8.6 cm2/s), indicating substantially improved ion transport kinetics through Gly modification. This observation is further corroborated by electrochemical impedance spectroscopy (EIS) analysis (Fig. S20 in Supporting information), where Gly-NVO exhibits a considerably lower charge transfer resistance (Rct = 34.3 Ω) in the high-frequency semicircle region [38], representing more than 50% reduction compared to NVO. Further fitting analysis of the low-frequency region slope in the EIS spectra reveals that Gly-NVO exhibits a significantly higher slope (0.995) compared to pristine NVO (0.539). Since the low-frequency linear region typically represents Warburg diffusion resistance, this result clearly demonstrates enhanced Zn2+ diffusion kinetics at the solid-liquid interface of Gly-NVO (Fig. S21 in Supporting information) [39].
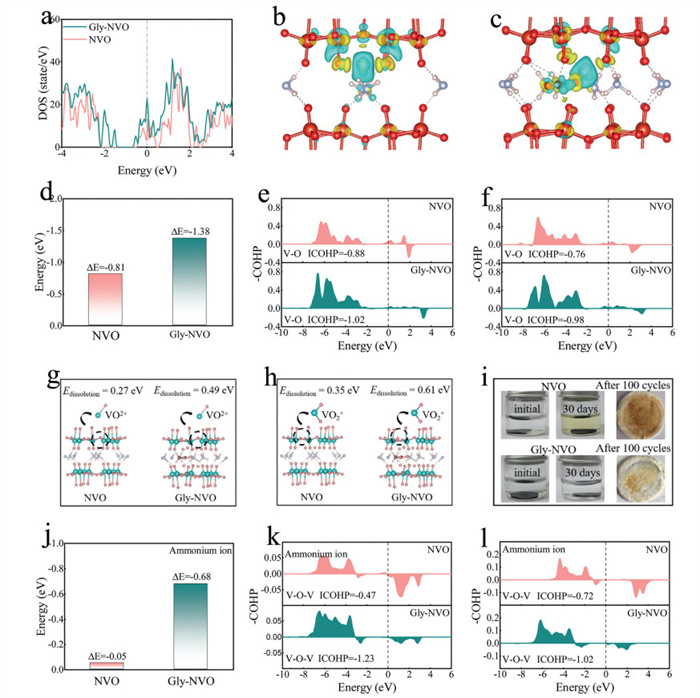
The structural and electronic modifications induced by Gly intercalation are further elucidated through density functional theory (DFT) calculations. As shown in Fig. 4a, the density of states (DOS) analysis reveals metallic characteristics near the Fermi level for both materials, indicating their intrinsic conductivity. Notably, compared with the pristine material, Gly-NVO exhibits a significantly enhanced DOS contribution near the Fermi level, suggesting improved electronic conductivity upon Gly incorporation [15]. The increased electronic states near the Fermi level arise from the formation of additional charge transport pathways through Gly-mediated electronic coupling between vanadium oxide layers [40]. The charge distribution characteristics during Zn2+ intercalation is systematically investigated through electron density difference analysis (Figs. 4b and c). Distinct charge redistribution patterns were observed between the two materials, where blue and yellow electron clouds represent charge accumulation and depletion, respectively. In pristine NVO, significant charge accumulation is detected between Zn2+ ions and the V4O10 framework, suggesting strong Coulombic interactions that may impede ion migration. In contrast, Gly-NVO exhibits markedly different electronic behavior: the variation in electron density within the V4O10 framework is reduced, accompanied by a pronounced weakening of charge interactions with Zn2+. Instead, prominent charge transfer activity is observed near Gly molecules, indicating their active participation in the charge compensation process. This altered charge distribution pattern demonstrates that Gly intercalation effectively screens the strong electrostatic interactions between Zn2+ and the host framework, thereby promoting faster Zn2+ migration by reducing the diffusion energy barrier [16,36]. The thermodynamic stability of Zn2+ intercalation is quantitatively evaluated through enthalpy calculations, where the intercalating energy (ΔH) is defined as ΔH = Et - E1 - E2, with Et representing the total energy of the Zn2+-materials system, while E1 and E2 correspond to the energies of the host material and isolated Zn2+, respectively. As shown in Fig. 4d, Gly-NVO exhibits a significantly more negative intercalation energy (−1.38 eV) compared to pristine NVO (−0.81 eV), demonstrating enhanced thermodynamic stability for Zn2+ storage in the Gly-modified structure [41,42]. Although the exothermic ΔH indicates thermodynamically favorable Zn2+ binding, the kinetic barrier for deintercalation is equally critical. In Gly-NVO, the Gly molecules facilitate Zn2+ insertion/extraction by reducing charge transfer resistance and weakening the electrostatic interactions of Zn2+, even under strong overall binding [43]. This is consistent with the GITT results, which demonstrate a high Zn2+ diffusion coefficient (DZn2+) and excellent rate performance. Furthermore, the long cycling stability of Gly-NVO further confirms its robust Zn2+ intercalation/deintercalation capability. The structure of the Zn2+ intercalated materials is shown in Fig. S22 (Supporting information). This energy difference reflects the favorable structural modifications induced by Gly intercalation, which create more stable coordination environments for Zn2+ ions while reducing the energy penalty associated with ion insertion. The more exothermic ΔH value of Gly-NVO suggests stronger binding interactions between the host material and Zn2+, contributing to improved cycling stability without compromising the kinetic properties, also evidenced by the previously discussed charge distribution analysis. The bonding characteristics and their influence on structural stability are investigated through crystal orbital Hamilton population (COHP) analysis. Following established methodology [20,22], the -COHP values below the Fermi level (positive values) are identified as corresponding to bonding states, while those above the Fermi level (negative values) represent antibonding interactions. The |ICOHP| value serves as a direct measure of interatomic bond strength, a larger |ICOHP| value indicates a stronger interatomic force (the sites of V and O in Fig. S23 in Supporting information). The modified material demonstrates significantly reduced antibonding states and enhanced bonding states compared to its pristine counterpart (Figs. 4d and f). Quantitative analysis shows that the V-O bond strength in Gly-NVO, represented by |ICOHP| values of 1.02 eV before intercalation and 0.98 eV after intercalation, substantially exceeds that in NVO (0.88 and 0.76 eV, respectively). These results demonstrate that Gly intercalation not only strengthens the intrinsic V-O bonding within the host framework but also maintains better bond integrity during Zn2+ (de)intercalating processes. The vanadium dissolution behavior, a critical degradation mechanism in vanadium oxide-based cathodes caused by weak acid environments or disproportionation reactions (etc.) leading to VO2+ or VO2+ dissolution [44,45], is systematically investigated through dissolution energy calculations (Figs. 4g and h). The dissolution energy calculations reveal significantly enhanced stability against vanadium dissolution in Gly-NVO, with VO2+ or VO2+ exhibiting substantially higher dissolution energies of 0.49 and 0.61 eV, respectively, compared to the pristine material. Combine the above, these elevated energy barriers demonstrate that Gly intercalation effectively suppresses the vanadium leaching process through multiple stabilization mechanisms (in theory): (1) The strong coordination between Gly molecule and vanadium ions reinforces the V-O framework; (2) The hydrogen-bonding network creates a protective microenvironment around active vanadium sites; (3) The modified electronic structure reduces the propensity for disproportionation reactions. To experimentally verify the suppression of vanadium dissolution, times and cycles analyses are performed by examining both electrolyte solutions and cycled separators (Fig. 4i). The Gly-NVO electrodes demonstrate markedly reduced vanadium leaching compared to pristine NVO, as evidenced by significantly less vanadium deposition on separators and lower vanadium concentration in post-cycling electrolytes. This visual confirmation complements the theoretical dissolution energy calculations, showing that the Gly-modified material maintains excellent structural integrity during prolonged electrochemical cycling.
Figure 4
Figure 4.
DFT calculations of NVO and Gly-NVO. (a) DOS of NVO and Gly-NVO. (b, c) Differential charge density of NVO and Gly-NVO. (d) Zn2+ intercalated energy of NVO and Gly-NVO. (e, f) COHP and ICOHP of the V-O bond in NVO and Gly-NVO before and after Zn2+ intercalation. (g, h) Dissolution energy of VO2+ and VO2+ in NVO and Gly-NVO. (i) optical photo of electrolyte immersed electrodes and separator after cycling. (j) NH4+ intercalated energy of NVO and Gly-NVO. (k, l) COHP and ICOHP of the V-O-V bond in NVO and Gly-NVO after NH4+ intercalation and deintercalation.
The NH4+ (de)intercalation behavior is further investigated based on DFT calculations. In Fig. 4j, the formation energy of NH4+ intercalation is displayed, where Gly-NVO exhibits a lower formation energy (E = −0.68 eV), while NH4+ intercalation in NVO is more difficult (E = −0.05 eV). The structure of the NH4+ intercalated material is shown in Fig. S24 (Supporting information). The COHP analysis of NH4+ intercalation is compared in Fig. 4k, where Gly-NVO demonstrates more bonding states, while NVO shows more antibonding states. The |ICOHP| value of Gly-NVO is 1.23 eV, indicating greater structural stability after NH4+ intercalation (the sites of V and O are shown in Fig. S25 in Supporting information). Due to the irreversible deimmunization in conventional ammonium vanadate, the structural energy of NVO after NH4+ deintercalation is −474.62 eV, while that of Gly-NVO is −531.56 eV, suggesting that the structure of Gly-NVO is more stable owing to the pillar effect of Gly molecules (Fig. S26 in Supporting information). Additionally, Fig. 4l presents the COHP analysis of both materials after NH4+ deintercalation. Similarly, Gly-NVO exhibits more bonding states, and its |ICOHP| value (1.02 eV) is higher than that of NVO (0.72 eV). The sites of V and O are shown in Fig. S27 (Supporting information). These results further confirm the feasibility of Gly-NVO as a cathode material for AAIBs.
In summary, a triply stabilized Gly-NVO cathode has been successfully constructed through a Gly pre-intercalation strategy, featuring: (1) Gly molecular pillars, (2) hydrogen-bond networks between -NH2 groups and V4O10 frameworks, and (3) reversible NH4+ (de)intercalation behavior. As a cathode for AZIBs, Gly-NVO demonstrates a high specific capacity of 520 mAh/g along with exceptional rate performance (400 and 150 mAh/g at 10 and 50 A/g, respectively), outstanding energy density (393 Wh/kg at 0.2 A/g and power density (24.3 kW/kg at 50 A/g), and electrochemical kinetics have been significantly enhanced. The reversible (de)intercalation behavior of NH4+ in Gly-NVO enables its application as an AAIBs cathode, exhibiting excellent stability (82.1% capacity retention after 180 cycles at 0.2 A/g). Theoretical calculations validate the Gly-induced enhancement in the kinetics and stability of Gly-NVO. Consequently, the proposed Gly-NVO cathode can serve as a promising candidate for next-generation AZIBs cathodes and may potentially function as a universal cathode for rechargeable aqueous battery systems.
Declaration of competing interest
The authors declare no competing financial interest.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Zihan Wang: Writing – original draft. Meizhen Dai: Data curation. Pengcheng Song: Conceptualization. Wenxuan Liu: Data curation. Junhua You: Funding acquisition. Fang Hu: Writing – review & editing. Yusheng Wu: Funding acquisition. Kai Zhu: Writing – review & editing.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 51974188), the Natural Science Foundation of Liaoning Province (No. 2025-MS-116), the Liaoning Applied Basic Research Program (No. 2023JH2/101300011) and the Shenyang Science and Technology Project (No. 23–407–3–13).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111729.
[1]
T. Wang, Y. Wang, X. Wang, et al., Adv. Funct. Mater. 34 (2024) 2314157. doi: 10.1002/adfm.202314157
[2]
T. Wang, Y. Wang, P. Cui, et al., Energy Environ. Sci. 18 (2025) 2546–2558. doi: 10.1039/d4ee05894a
H. Xu, T. Xue, K. Ouyang, et al., J. Energy Storage 119 (2025) 116436. doi: 10.1016/j.est.2025.116436
[45]
D. Zhang, J. Cao, C. Yang, et al., Adv. Energy Mater. 15 (2024) 2404026.
Figure 1
Structural and elemental properties of NVO and Gly-NVO. (a) XRD patterns of NVO and Gly-NVO. (b) V 2p spectra of NVO and Gly-NVO. (c) N 1s spectra of NVO and Gly-NVO. (d) FTIR spectra of NVO and Gly-NVO. (e) XANES spectra of V K-edge. (f) V K-edge of Fourier-transform EXAFS spectra. (g, h) SEM images of NVO and Gly-NVO. (i, j) HRTEM images of NVO and Gly-NVO. (k) TEM-mapping of Gly-NVO.
Figure 2Ex-situ characterization of Gly-NVO in the first two cycles. (a) XRD patterns; (b-d) V 2p, O 1s and N 1s spectra. (e) FTIR spectra, f schematic image of Zn2+/NH4+ (de)intercalation in Gly-NVO.
Figure 3
Electrochemical performance of NVO and Gly-NVO. (a) GCD curves of NVO and Gly-NVO in the first two cycles at 0.2 A/g. (b) Cycling performance of NVO and Gly-NVO at 0.2 A/g. (c) Rate performance of NVO and Gly-NVO. (d) ragone image of NVO, Gly-NVO and reported ammonium vanadate cathodes. (e) Energy effciency of Gly-NVO. (f) Cycling performance of NVO and Gly-NVO at 10 A/g. (g) Rate performance of Gly-NVO at 50 A/g. (h) Cycling performance of Gly-NVO at 50 A/g (aqueous ammonium-ion batteries system). (i) GCD curves of NVO and Gly-NVO at 0.2 A/g. (j) cycling performance of NVO and Gly-NVO at 0.2 A/g. (k) Rate performance of NVO and Gly-NVO. (l) NH4+ intercalated energy of NVO and Gly-NVO. (m) EIS of NVO and Gly-NVO. (n) Contact angles of NVO and Gly-NVO after 4 h of immersion in 2 mol/L (NH4)2SO4 aqueous solution.
Figure 4
DFT calculations of NVO and Gly-NVO. (a) DOS of NVO and Gly-NVO. (b, c) Differential charge density of NVO and Gly-NVO. (d) Zn2+ intercalated energy of NVO and Gly-NVO. (e, f) COHP and ICOHP of the V-O bond in NVO and Gly-NVO before and after Zn2+ intercalation. (g, h) Dissolution energy of VO2+ and VO2+ in NVO and Gly-NVO. (i) optical photo of electrolyte immersed electrodes and separator after cycling. (j) NH4+ intercalated energy of NVO and Gly-NVO. (k, l) COHP and ICOHP of the V-O-V bond in NVO and Gly-NVO after NH4+ intercalation and deintercalation.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
