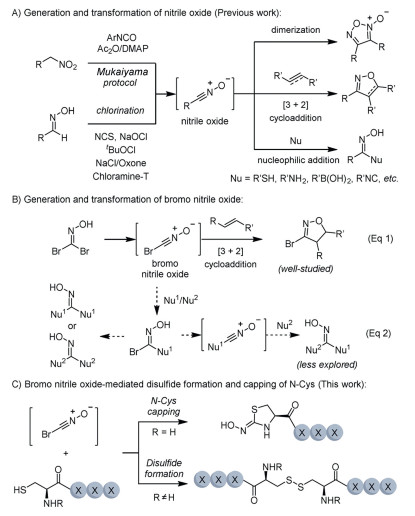
Figure 1.
Nitrile oxide-mediated transformations.
Bromo nitrile oxide-mediated disulfide formation and capping of N-terminal cysteine in peptides
Jingrong Huang , Xin Ding , Zihan Lin , Yu Lin , Yongqi Liao , Wang Gu , Fa-Jie Chen , Fen-Er Chen

Nitrile oxide [1-3] is a highly reactive organic species, and it has broad applications in organic synthetic methodologies and total synthesis of natural products [4-6]. Conventionally, nitrile oxide is involved in nucleophilic additions or cycloaddition reactions, such as 1,3-dipolar [2 + 3] cycloaddition with alkenes [7] or alkynes [8-10], and dimerization to yield furoxans [11-13] (Fig. 1A). Due to their high reactivity, nitrile oxides are commonly generated and used in situ from precursors such as chlorooxime, oxime, and nitro compounds [14-22]. Recent studies revealed that nitrile oxide can be generated in aqueous media and serves as a valuable handle for bioconjugation, enabling chemo-selective modifications of native peptides, proteins, and bacteriophage [23-28]. For example, Gao [26] and Wang [29] independently reported the fast chlorooxime-cysteine bioconjugation using nitrile oxide and achieved efficient Cys-Cys stapling of native peptides using bifunctional bischlorooxime reagents. Non-symmetric stapling [30] and bicyclization of native peptides were also achieved using activated esters bearing bischlorooxime motifs [31].
In addition to C-substituted nitrile oxides, bromo nitrile oxide, generated from 1,1-dibromoformaldoxime, has been studied in [3 + 2] cycloaddition reactions to afford 3-bromo isoxazolines, which serve as valuable intermediates for the construction of hydroxy esters, nitriles, and their corresponding α,β-unsaturated derivatives (Fig. 1B, Eq. 1). We envisioned that oximes bearing two halogen substituents, such as 1,1-dibromoformaldoxime, could undergo sequential elimination to generate 2 equiv. of nitrile oxide, enabling the crosslinking of two nucleophiles (Fig. 1B, Eq. 2). While this strategy is conceptually appealing, the application of bromo nitrile oxide for the incorporation of two distinct nucleophiles remains less explored, likely due to the inherent challenges of achieving crosslinking over competitive dimerization of the same nucleophiles (Fig. 1B, Eq. 2). For example, bromo nitrile oxide generated from 1,1-dibromoformaldoxime could couple with the first nucleophile (Nu1) to afford an adduct. Subsequently, the adduct could be transferred into substituted nitrile oxide through the HBr elimination, followed by the addition of the second nucleophile, either Nu1 or Nu2, to afford the dimerization or crosslinking products.
Herein, we developed a bromo nitrile oxide-mediated method for capping N-terminal cysteine in peptides via crosslinking of sulfhydryl and N-terminal amino groups using 1,1-dibromoformaldoxime (Fig. 1C). This two-fold nucleophilic addition to nitrile oxide resulted in the formation of a five-membered thiazolidin-2-one oxime. Interestingly, when internal cysteines or thiols were treated with bromo nitrile oxide, disulfides were obtained, offering a complementary approach to conventional oxidative disulfide bond formation methods, such as those employing N-chlorosuccinimide (NCS) [32], dimethyl sulfoxide (DMSO) [33], hydrogen peroxide (H2O2) [34], molecular oxygen (O2), or SO2F2 [35]. The reactions proceed with fast kinetics under mild conditions, demonstrating broad applicability for cysteine-containing peptides, unprotected native peptides, and both aromatic and aliphatic thiols. Moreover, oxidant-sensitive residues were well tolerated, and selective modification of N-terminal cysteines [36-41] was successfully achieved in native peptides, highlighting the potential of this method for late-stage modification of native peptides and the derivatization of biologically active molecules.
To test the hypothesis of two-fold nucleophilic addition to bromo nitrile oxide, we conducted the model reaction of cysteine methyl ester (1a) using 1,1-dibromoformaldoxime (2) as the nitrile oxide precursor (Table 1). When the reaction was performed in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 35 ℃ for 1 h, cycloaddition product 3a featuring a thiazolidin-2-one oxime motif was obtained in 80% yield, along with 20% of disulfide 4a (Table 1, entry 1). This five-membered heterocyclic compound can be considered to be generated from the replacements of bromo substituents of 1,1-dibromoformaldoxime by the unprotected sulfhydryl and amino groups at N-terminal cysteines, alternatively, through two-fold nucleophilic addition to bromo nitrile oxide, as we proposed. Further optimization of the reaction conditions revealed that lowering the reaction temperature to 25 ℃ or 15 ℃ decreased yields (Table 1, entries 2 and 3). Cyclized product 3a was not observed when the reaction was conducted in THF or using THF/H2O as the mixed solvent (Table 1, entries 4 and 5). Interestingly, this transformation can be carried out under physiological conditions (phosphate buffered saline, pH 7.4), providing product 3a with a moderate yield (56%) (Table 1, entry 6). The tolerance of aqueous media implied the potential application of this method for the modification of N-terminal cysteine in biomolecules such as native peptides.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Temp (℃) | Solvent | Yield of 3a (%) | Yield of 4a (%) |
| 1 | 35 | HFIP | 80 | 20 |
| 2 | 25 | HFIP | 65 | 32 |
| 3 | 15 | HFIP | 50 | 50 |
| 4 | 35 | THF | 0 | Trace |
| 5 | 35 | THF/H2O (1:1) | 0 | Trace |
| 6 | 35 | PBS, pH 7.4 | 56 | 34 |
| a Reaction conditions: cysteine 1a (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (1.0 equiv.) in HFIP (2 mL) at 35 ℃ for 1 h, unless otherwise noted. Isolated yield. | ||||
With the success of thiol/amine-involved cycloaddition, we investigated the addition using thiol as the single nucleophile with Boc-protected cysteine methyl ester (1b) as the model substrate (Table 2). When HFIP was used as the solvent, the addition of 1b with 2 afforded the oxime product 5 in 43% yield (Table 2, entry 1), which was presumably formed through a similar pathway as thiazolidin-2-one oxime 3a. Unfortunately, further optimization of the reaction conditions did not provide a higher yield of adduct 5. On the contrary, disulfide 4b was observed as the major product. The optimized reaction conditions for disulfide formation involved 1.0 equiv. of 1b and 0.5 equiv. of 2 in the presence of 5 equiv. of triethylamine in THF/H2O (1:1) at room temperature for 15 min, which afforded product 4b in 74% yield (Table 2, entry 2). In the absence of 2, only a trace amount of disulfide was observed (Table 2, entry 4). Other reaction conditions tested were less effective (Table 2, entries 3-11, and Table S7 in Supporting information). Notably, the reaction can proceed in PBS buffer (pH 7.4) without the need for an external base, albeit with a moderate yield (49%) (Table 2, entry 11). This protocol featuring mild conditions could serve as a complementary method for disulfide formation with oxidants like NCS and H2O2, minimizing side reactions like over-oxidation.
DownLoad:
CSV
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Entry | Equiv. of 2 | Base (equiv.) | Solvent | Yield of 4b (%) | Yield of 5 (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1.5 | - | HFIP | 15 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 0.5 | Et3N (5.0) | THF/H2O (1:1) | 74 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1.0 | Et3N (5.0) | THF/H2O (1:1) | 74 | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | - | Et3N (5.0) | THF/H2O (1:1) | Trace | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 0.5 | - | THF/H2O (1:1) | Trace | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 0.5 | DIPEA (5.0) | THF/H2O (1:1) | 49 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 0.5 | DMAP (5.0) | THF/H2O (1:1) | 65 | 11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 0.5 | NaHCO3 (5.0) | THF/H2O (1:1) | 62 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 0.5 | Et3N (5.0) | THF/H2O (4:1) | 72 | 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 0.5 | Et3N (5.0) | THF/H2O (1:4) | 60 | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 0.5 | - | PBS, pH 7.4 | 49 | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: cysteine 1b (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (0.5 equiv.), base (5.0 equiv.) in solvent (2 mL) at r.t. for 15 min, unless otherwise noted. Isolated yield. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
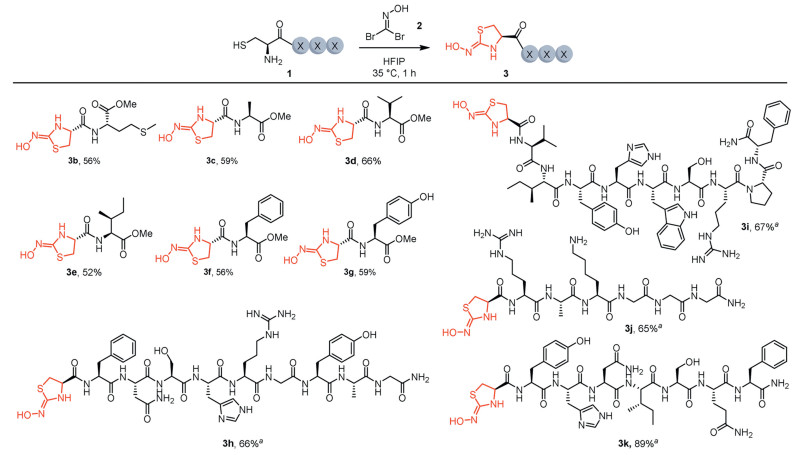
With the optimized reaction conditions in hand, we explored the substrate scope for N-terminal cysteine capping mediated by bromo nitrile oxide. As shown in Fig. 2, dipeptides reacted smoothly with 1,1-dibromoformaldoxime in HFIP, affording the cyclized products (3b-3g) in moderate yields. Notably, oxidant-sensitive methionine was well tolerated (3b). A peptide containing an unprotected tyrosine residue reacted with 2, yielding product 3g in 59% yield, with no observed oxidation of the phenol side chain. Compared to short peptides, longer peptides exhibited improved reactivity, achieving moderate to good capping efficiency (3h–3k). Additionally, nucleophilic residues such as tryptophan, lysine, serine, arginine, and histidine did not interfere with the reaction. No labeling or oxidation at these reactive residues was observed, highlighting the excellent chemo- and site-selectivity of this method for N-terminal cysteine in native peptides. Given its broad substrate scope and functional group compatibility, this capping strategy holds potential for the modification and derivatization of complex native peptides and biomolecules, including proteins and antibodies.
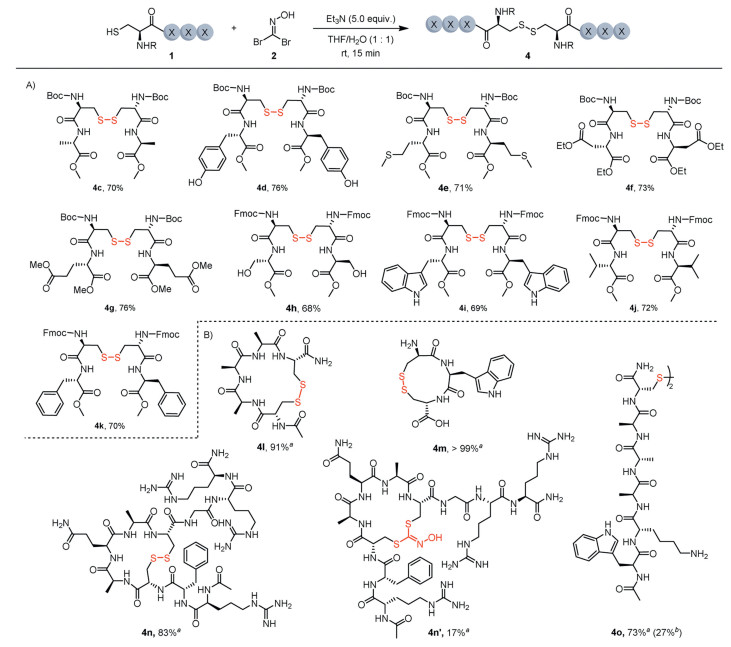
Next, we explored the substrate scope for disulfide formation mediated by bromo nitrile oxide (Fig. 3). A wide range of dipeptides bearing cysteine residues were efficiently converted into disulfides with good yields under the standard reaction conditions. Notably, residues with oxidant-sensitive side chains, such as tyrosine (4d), tryptophan (4i), and methionine (4e), were well tolerated, with no observed oxidation of these functional groups. Additionally, serine-containing peptide 4h, featuring an unprotected hydroxyl group, was obtained smoothly. Both the acid-sensitive Boc group and the base-sensitive Fmoc group were well tolerated under the reaction conditions. Furthermore, intramolecular disulfide bond formation enabled the synthesis of disulfide-bridged cyclic peptides. Peptides with 11-membered (4m), 17-membered (4l), and 17-membered rings (4n) were obtained in excellent conversions in the presence of 1 equiv. of 1,1-dibromoformaldoxime 2 in HFIP, as determined by high-performance liquid chromatography (HPLC). Interestingly, a small amount (17% conversion) of Cys-Cys stapling product 4n' was observed, which could be the result of two-fold cysteine-nitrile oxide conjugation. Similarly, disulfide 4o was obtained in 73% conversion with the formation of stapled peptides as side products (Fig. S8 in Supporting information for details).
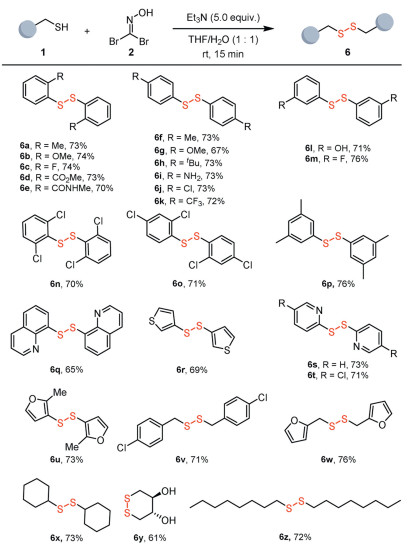
Encouraged by the broad substrate scope of peptides for disulfide formation, we investigated the applicability of this method to small molecules. As shown in Fig. 4, 1,1-dibromoformaldoxime efficiently mediated disulfide bond formation for both aromatic and aliphatic thiols. Thiophenols bearing either electron-donating or electron-withdrawing groups afforded good yields. Notably, the reaction proceeded smoothly in the presence of unprotected amino (6i) and hydroxyl (6l) groups, without significantly affecting the yields. Disulfides containing heteroaromatic rings were obtained in comparable yields, including those derived from quinoline (6q), thiophene (6r), pyridine (6s and 6t), and furan (6u). Aliphatic disulfides were also synthesized in moderate to good yields (6v–6z). Additionally, intramolecular disulfide bond formation provided the six-membered ring-containing product 6y in 61% yield. These results highlight the broad substrate scope and excellent functional group tolerance of this novel disulfide bond formation strategy.
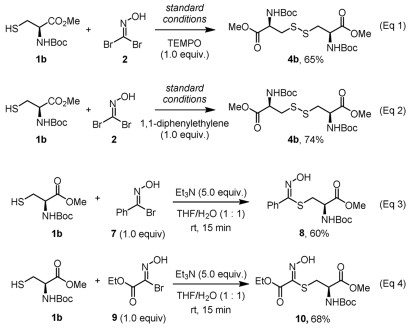
To gain deeper insight into the reaction mechanism, we conducted control experiments (Fig. 5). Under standard reaction conditions, the addition of radical scavengers (TEMPO or 1,1-diphenylethylene) provided comparable yields of disulfide 4b (Fig. 5, Eqs. 1 and 2), suggesting that disulfide formation is unlikely to proceed via a radical pathway. To elucidate the role of the bromo substituent in bromo nitrile oxide for the disulfide formation, we performed comparative studies with C-linked nitrile oxides. As shown in Fig. 5, Eqs. 3 and 4, replacing one of the bromo atoms of 1,1-dibromoformaldoxime with a phenyl group (7) or ester (9) led to the generation of thiohydroximates 8 and 10 in 60% and 68% yields, respectively. Only trace amounts of disulfide were detected in both cases. The generation of thiohydroximates 8 and 10 from bromooximes is consistent with our previous observation for the cysteine conjugations mediated by chlorooxime [26,28,31]. These results implied that the bromo substituent is likely to act as a leaving group rather than an electron-withdrawing group to enable the disulfide bond formation. The observation of oxime product 5 further supports the crucial role of both bromo atoms of 1,1-dibromoformaldoxime for the efficient disulfide bond formation.
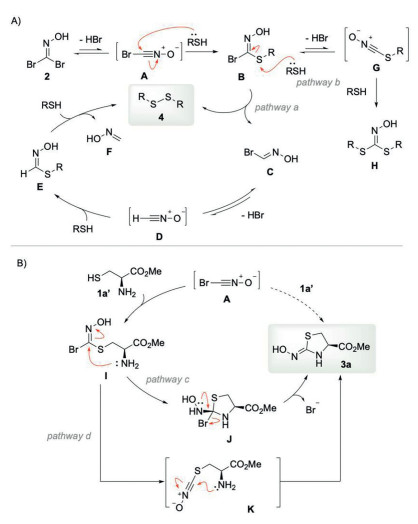
Although the reaction mechanism is not clear at this stage, we propose a plausible mechanism for disulfide bond formation and N-terminal cysteine capping based on the experimental data and previous studies (Fig. 6) [26]. Disulfide formation is initiated by the in situ generation of bromo nitrile oxide A via the elimination of HBr from 1,1-dibromoformaldoxime 2. Conjugation of nitrile oxide A with a thiol affords thiohydroximate B, which subsequently reacts with another thiol to afford disulfide 4 and bromooxime C (Fig. 6A, pathway a). Bromooxime C may serve as a precursor to nitrile oxide D, which undergoes disulfide formation through a similar mechanism. Alternatively, intermediate B undergoes elimination to form nitrile oxide G and subsequently generates adduct H (Fig. 6A, pathway b). Due to the slow formation of nitrile oxide from trans-bromooxime [42], the generation of thiohydroximate H is disfavored. In the case of N-terminal cysteine modification, bromo nitrile oxide A reacts with cysteine 1a' to generate adduct I, which undergoes intramolecular addition of the proximal amino group (Fig. 6B, pathway c) to form intermediate J. Subsequent elimination of HBr from J furnishes the cyclized product 3a. Alternatively, the cyclization could proceed via nitrile oxide intermediate K (Fig. 6B, pathway d) through intramolecular nucleophilic addition of the amino group to the nitrile oxide intermediates.
In conclusion, we have developed a novel method for disulfide bond formation and N-terminal cysteine capping in peptides mediated by bromo nitrile oxide. These transformations proceed under mild conditions with fast kinetics, efficiently converting a wide range of thiophenols, thiols, and cysteine-containing native peptides into disulfides, cyclic disulfides, or thiazolidin-2-one oximes. Notably, this protocol is compatible with oxidant-sensitive residues such as methionine, tyrosine, and tryptophan, serving as a valuable alternative for methods with strong oxidants. Furthermore, the compatibility of this method with aqueous media highlights its potential for the late-stage modification of biologically active molecules, including native peptides and proteins. Limitations of this strategy include the relatively lower yields compared to the conventional disulfide formation protocols, the use of organic co-solvent, and the low conversion to stapling product, which needs further optimization to achieve higher efficiency, diversity, and biocompatibility.
The authors declare no competing interest.
Jingrong Huang: Writing – review & editing, Writing – original draft, Methodology, Investigation. Xin Ding: Writing – review & editing, Investigation. Zihan Lin: Investigation. Yu Lin: Investigation. Yongqi Liao: Investigation. Wang Gu: Writing – review & editing. Fa-Jie Chen: Writing – review & editing, Writing – original draft, Validation, Supervision, Project administration, Funding acquisition, Conceptualization. Fen-Er Chen: Supervision.
The authors gratefully acknowledge the grant support from the Natural Science Foundation of Fujian Province, China (No. 2024J08025), Fuzhou University (No. 511264), and Fuzhou University Testing Fund of Precious Apparatus (No. 2024T014).
Supplementary material associated with this article can be
found, in the online version, at doi:
S. Roscales, J. Plumet, Org. Biomol. Chem. 16 (2018) 8446-8461. doi: 10.1039/c8ob02072h
J. Plumet, ChemPlusChem 85 (2020) 2252-2271. doi: 10.1002/cplu.202000448
P.Y. Ushakov, A.Y. Sukhorukov, Nat. Prod. Rep. 42 (2025) 876-910. doi: 10.1039/d4np00069b
C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat, P. Kittakoop, Angew. Chem. Int. Ed. 55 (2016) 3997-4001. doi: 10.1002/anie.201511730
J.K. Eaton, R.A. Ruberto, A. Kramm, V.S. Viswanathan, S.L. Schreiber, J. Am. Chem. Soc. 141 (2019) 20407-20415. doi: 10.1021/jacs.9b10769
J.K. Eaton, L. Furst, R.A. Ruberto, et al., Nat. Chem. Biol. 16 (2020) 497-506. doi: 10.1038/s41589-020-0501-5
D. Muri, J. Bode, E. Carreira, Org. Lett. 2 (2000) 539-541. doi: 10.1021/ol991396q
F. Heaney, Eur. J. Org. Chem. 2012 (2012) 3043-3058. doi: 10.1002/ejoc.201101823
M. Gao, Y. Li, Y. Gan, B. Xu, Angew. Chem. Int. Ed. 54 (2015) 8795-8799. doi: 10.1002/anie.201503393
S.L. Bartlett, Y. Sohtome, D. Hashizume, et al., J. Am. Chem. Soc. 139 (2017) 8661-8666. doi: 10.1021/jacs.7b03782
Z.X. Yu, P. Caramella, K.N. Houk, J. Am. Chem. Soc. 125 (2003) 15420-15425. doi: 10.1021/ja037325a
T. Pasinszki, B. Hajgato, B. Havasi, N.P. Westwood, Phys. Chem. Chem. Phys. 11 (2009) 5263-5272. doi: 10.1039/b823406j
N.N. Makhova, L.L. Fershtat, Tetrahedron Lett. 59 (2018) 2317-2326. doi: 10.1016/j.tetlet.2018.04.070
L.D. Angelis, A.M. Crawford, Y.L. Su, et al., Org. Lett. 23 (2021) 925-929. doi: 10.1021/acs.orglett.0c04130
S. Bhosale, S. Kurhade, S. Vyas, V.P. Palle, D. Bhuniya, Tetrahedron 66 (2010) 9582-9588. doi: 10.1016/j.tet.2010.10.029
M.F. Hartmer, S.R. Waldvogel, Chem. Commun. 51 (2015) 16346-16348. doi: 10.1039/C5CC06437F
S. Maiti, P. Samanta, G. Biswas, D. Dhara, ACS Omega 3 (2018) 562-575. doi: 10.1021/acsomega.7b01632
A.Y. Sukhorukov, A.A. Sukhanova, S.G. Zlotin, Tetrahedron 72 (2016) 6191-6281.
G. Zhao, L. Liang, C.H.E. Wen, R. Tong, Org. Lett. 21 (2019) 315-319. doi: 10.1021/acs.orglett.8b03829
F.T. Gao, Z. Fang, R.R. Su, et al., Org. Biomol. Chem. 16 (2018) 9211-9217. doi: 10.1039/c8ob02721h
S. Minakata, S. Okumura, T. Nagamachi, Y. Takeda, Org. Lett. 13 (2011) 2966-2969. doi: 10.1021/ol2010616
E. Inokuchi, A. Yamada, K. Hozumi, et al., Org. Biomol. Chem. 9 (2011) 3421-3427. doi: 10.1039/c0ob01193b
R.J.B. Schäfer, M.R. Monaco, M. Li, et al., J. Am. Chem. Soc. 141 (2019) 18644-18648. doi: 10.1021/jacs.9b07632
K. Yang, F. Zhang, T. Fang, G. Zhang, Q. Song, Angew. Chem. Int. Ed. 58 (2019) 13421-13426. doi: 10.1002/anie.201906057
S. Zhu, G. Samala, E.T. Sletten, J.L. Stockdill, H.M. Nguyen, Chem. Sci. 10 (2019) 10475-10480. doi: 10.1039/c9sc04079j
F.J. Chen, M. Zheng, V. Nobile, J. Gao, Chem. Eur. J. 28 (2022) e202200058. doi: 10.1002/chem.202200058
F.J. Chen, J. Gao, Chem. Eur. J. 28 (2022) e202201843. doi: 10.1002/chem.202201843
W. Gu, J. Huang, Y. Lu, et al., J. Org. Chem. 89 (2024) 6364-6370. doi: 10.1021/acs.joc.4c00356
Q. Chen, T. Long, J. Zheng, et al., CCS Chem. 4 (2022) 3355-3363. doi: 10.31635/ccschem.021.202101386
F.J. Chen, W. Lin, F.E. Chen, Nat. Rev. Chem. 8 (2024) 304-318. doi: 10.1038/s41570-024-00591-5
F.J. Chen, N. Pinnette, F. Yang, J. Gao, Angew. Chem. Int. Ed. 62 (2023) e202306813. doi: 10.1002/anie.202306813
Y. Xing, Y. Wang, D. Ma, et al., Tetrahedron Lett. 120 (2023) 154459. doi: 10.1016/j.tetlet.2023.154459
J.P. Tam, C.R. Wu, W. Liu, J.W. Zhang, J. Am. Chem. Soc. 113 (1991) 6657-6662. doi: 10.1021/ja00017a044
E.V. Kudryavtseva, M.V. Sidorova, M.V. Ovchinnikov, Z.D. Bespalova, J. Pept. Sci. 6 (2000) 208-216. doi: 10.1002/(SICI)1099-1387(200005)6:5<208::AID-PSC241>3.0.CO;2-V
H. Li, M. Peng, J. Li, et al., Nat. Commun. 15 (2024) 8325.
H. Ren, F. Xiao, K. Zhan, et al., Angew. Chem. Int. Ed. 48 (2009) 9658-9662. doi: 10.1002/anie.200903627
X. Zheng, Z. Li, W. Gao, et al., J. Am. Chem. Soc. 142 (2020) 5097-5103. doi: 10.1021/jacs.9b11875
K. Li, W. Wang, J. Gao, Angew. Chem. Int. Ed. 59 (2020) 14246-14250. doi: 10.1002/anie.202000837
F. Ghorbani, S. You, G.A. Grabovyi, et al., J. Am. Chem. Soc. 146 (2024) 32333-32342. doi: 10.1021/jacs.4c05711
P. Hartmann, K. Bohdan, M. Hommrich, et al., Nat. Chem. 16 (2024) 380-388. doi: 10.1038/s41557-023-01388-7
M. Djalo, M. Silva, H. Faustino, et al., Chem. Commun. 58 (2022) 7928-7931. doi: 10.1039/d2cc02204d
A.F. Hegarty, M. Mullane, J. Chem. Soc., Chem. Commun. (1984) 229-230.

Figure 2 Substrate scope of peptides for capping of N-terminal cysteine. Reaction conditions: peptide 1 (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (1.0 equiv.) in HFIP (2.0 mL) at 35 ℃ for 1 h, unless otherwise noted. Isolated yield. aReaction conditions: peptide 1 (1.0 mmol/L), 1,1-dibromoformaldoxime 2 (1.0 mmol/L) in HFIP, 35 ℃ for 1 h. Conversion determined by HPLC.

Figure 3 Substrate scope for disulfide formation. Reaction conditions: Peptide 1 (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (0.1 mmol, 0.5 equiv.), triethylamine (1.0 mmol, 5.0 equiv.) in THF/H2O (1:1) (2 mL) at room temperature for 15 min, unless otherwise noted. Isolated yield. a Reaction conditions: peptide 1 (1 mmol/L), 1,1-dibromoformaldoxime 2 (1 mmol/L) in HFIP at room temperature for 30 min. Conversion determined by HPLC. b Conversion of cysteine stapling product.

Figure 4 Substrate scope of small molecules for disulfide formation. Reaction conditions: Thiol 1 (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (0.1 mmol, 0.5 equiv.), triethylamine (1.0 mmol, 5.0 equiv.) in THF/H2O (1:1) (2 mL) at room temperature for 15 min. Isolated yield.
Table 1. Optimization of reaction conditions for N-Cys capping.a
|
||||
| Entry | Temp (℃) | Solvent | Yield of 3a (%) | Yield of 4a (%) |
| 1 | 35 | HFIP | 80 | 20 |
| 2 | 25 | HFIP | 65 | 32 |
| 3 | 15 | HFIP | 50 | 50 |
| 4 | 35 | THF | 0 | Trace |
| 5 | 35 | THF/H2O (1:1) | 0 | Trace |
| 6 | 35 | PBS, pH 7.4 | 56 | 34 |
| a Reaction conditions: cysteine 1a (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (1.0 equiv.) in HFIP (2 mL) at 35 ℃ for 1 h, unless otherwise noted. Isolated yield. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimization of reaction conditions for disulfide formation.a
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Entry | Equiv. of 2 | Base (equiv.) | Solvent | Yield of 4b (%) | Yield of 5 (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1.5 | - | HFIP | 15 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 0.5 | Et3N (5.0) | THF/H2O (1:1) | 74 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1.0 | Et3N (5.0) | THF/H2O (1:1) | 74 | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | - | Et3N (5.0) | THF/H2O (1:1) | Trace | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 0.5 | - | THF/H2O (1:1) | Trace | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 0.5 | DIPEA (5.0) | THF/H2O (1:1) | 49 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 0.5 | DMAP (5.0) | THF/H2O (1:1) | 65 | 11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 0.5 | NaHCO3 (5.0) | THF/H2O (1:1) | 62 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 0.5 | Et3N (5.0) | THF/H2O (4:1) | 72 | 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 0.5 | Et3N (5.0) | THF/H2O (1:4) | 60 | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 0.5 | - | PBS, pH 7.4 | 49 | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: cysteine 1b (0.2 mmol, 1.0 equiv.), 1,1-dibromoformaldoxime 2 (0.5 equiv.), base (5.0 equiv.) in solvent (2 mL) at r.t. for 15 min, unless otherwise noted. Isolated yield. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: