Citation:
Chang Xu, Mengfan Luo, Jia Zhao, Jialong Yin, Can Feng, Heng Zhang, Peng Zhou, Zhaokun Xiong, Bo Lai. Ferrate(Ⅵ) combined with sulfur(Ⅳ) as an effective process for aromatic organoarsenic degradation: Essential role of Fe(Ⅲ) flocs[J]. Chinese Chemical Letters,
2026, 37(4): 111538.
doi:
10.1016/j.cclet.2025.111538
Ferrate(Ⅵ) combined with sulfur(Ⅳ) as an effective process for aromatic organoarsenic degradation: Essential role of Fe(Ⅲ) flocs
English
Ferrate(Ⅵ) combined with sulfur(Ⅳ) as an effective process for aromatic organoarsenic degradation: Essential role of Fe(Ⅲ) flocs
State Key Laboratory of Hydraulics and Mountain River Engineering, College of Architecture and Environment, Sichuan University, Chengdu 610065, China
b.
Sino-German Centre for Water and Health Research, Sichuan University, Chengdu 610065, China
c.
Key Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resource Reuse, Nanchang Hangkong University, Nanchang 330063, China
Received Date:
18 April 2025 Accepted Date:
02 July 2025 Revised Date:
06 May 2025 Available Online:
15 April 2026
Abstract:
This study employed the ferrate(Ⅵ)/sulfur(Ⅳ) (Fe(Ⅵ)/S(Ⅳ)) system to degrade aromatic organoarsenic compounds, with a focus on elucidating the role of in situ-formed Fe(Ⅲ) flocs. The introduction of S(Ⅳ) significantly enhanced oxidative degradation efficiency compared to Fe(Ⅵ) alone, achieving 94.8% p-arsanilic acid (p-ASA) degradation within 3 min. The evolution of active species under varying S(Ⅳ) dosages was systematically investigated via radical quenching experiments and probe compound analysis. SO4•−, •OH and Fe(Ⅳ)/Fe(Ⅴ) were identified as the dominant reactive species in the Fe(Ⅵ)/S(Ⅳ) system, with Fe(Ⅳ)/Fe(Ⅴ) serving as the primary driver of p-ASA degradation. Characterization revealed that Fe(Ⅲ) flocs contributed to arsenic (As) adsorption. While S(Ⅳ) addition altered the morphology and structure of Fe(Ⅲ) flocs, these changes exerted negligible effects on As adsorption capacity. A plausible degradation pathway for p-ASA was proposed, supported by density functional theory (DFT) calculations and degradation product analysis. The system demonstrated robust resistance to common interfering ions, while the low toxicity of degradation byproducts highlighted its potential as a sustainable technology for AOCs elimination. This work elucidated structural modifications in Fe(Ⅲ) flocs induced by S(Ⅳ) and underscored the pivotal role of Fe(Ⅳ)/Fe(Ⅴ), positioning the Fe(Ⅵ)/S(Ⅳ) system as a promising strategy for AOCs degradation.
Aromatic organoarsenic compounds (AOCs), including p-arsanilic acid (p-ASA) and 3-nitro-4-hydroxyphenylarsonic acid (ROX), are commonly employed in livestock feed additives to promote poultry growth and disease control [1]. Despite the 2013 ban on AOCs in animal feed by the United States, their use remains prevalent in developing countries [2]. Environmental surveys have detected substantial concentrations of p-ASA residues near agricultural regions in China [3]. Research indicates that organic arsenic in environmental matrices can undergo transformation into highly toxic inorganic species, such as arsenate (As(Ⅴ)) and arsenite (As(Ⅲ)) [4]. Addressing AOCs contamination is imperative, as these compounds pose significant ecological and public health hazards due to their persistence and toxic transformation products.
Adsorption techniques, recognized for their cost-effectiveness and operational simplicity, have been widely investigated for AOCs removal [5]. Materials such as metal-organic frameworks (MOFs) [6], aluminum oxides [7], iron oxides [8], manganese oxides [9], and nanomaterials [10] have demonstrated notable efficacy in this regard. However, adsorptive removal of AOCs is susceptible to fluctuating environmental conditions, while functional substituent groups on AOCs strongly modulate adsorption efficiency [11]. In contrast, chemical oxidation processes degrade AOCs into smaller organic fragments and inorganic arsenic, enabling true pollutant mineralization. Widely utilized oxidation methods, such as Fenton [12], Fenton-like [13], and ozone-based systems [14], efficiently mediate the conversion of p-ASA to inorganic arsenic species. Nonetheless, these methods achieve incomplete AOCs degradation, with suboptimal removal efficiencies for total and inorganic arsenic, underscoring the demand for more robust and efficient remediation strategies.
Ferrate(Ⅵ) (Fe(Ⅵ)) has gained prominence as a multifunctional oxidant-coagulant, synergizing potent oxidative capacity with coagulation properties to degrade diverse contaminants while mitigating secondary pollution via Fe(Ⅲ) flocs generation during its reduction [15,16]. However, its practical adoption is hindered by intrinsic limitations, such as sluggish reaction kinetics and rapid autodecomposition under acidic conditions, which restrict its scalability [17]. Recent studies reveal that integrating Fe(Ⅵ) with sulfite(Ⅳ) (S(Ⅳ)) markedly enhances system efficacy through synergistic redox interplay. This synergy facilitates the formation of reactive intermediates (Fe(Ⅳ)/Fe(Ⅴ)) and radical species (SO4•− and •OH), significantly augmenting oxidative potential compared to Fe(Ⅵ) alone system [18,19]. The Fe(Ⅵ)/S(Ⅳ) system enhances degradation kinetics for recalcitrant organics and broadens the operational pH range, effectively circumventing the pH-dependent activity constraints of conventional Fe(Ⅵ) treatment schemes. These advancements establish the Fe(Ⅵ)/S(Ⅳ) system as a robust candidate for integrated oxidation-coagulation processes in water remediation. Bradley et al. demonstrated that S(Ⅳ) activation modifies Fe(Ⅲ) flocs derived from the Fe(Ⅵ)/S(Ⅳ) system, inducing distinct structural properties (e.g., magnetic behavior, morphology, and particle size) compared to flocs generated via Fe(Ⅵ) autodecomposition [20]. These structural attributes directly impact the adsorptive performance of flocs. Consequently, structural variations in Fe(Ⅲ) flocs may critically influence arsenic removal efficiency, underscoring the need for comprehensive assessment of the Fe(Ⅵ)/S(Ⅳ) system's efficacy in AOCs removal.
This study investigates the efficacy of the Fe(Ⅵ)/S(Ⅳ) system for AOCs elimination, with a specific focus on elucidating the influence of floc morphology on arsenic (As) removal efficiency. The primary objectives of this study were to: (1) Assess the degradation efficiency of the Fe(Ⅵ)/S(Ⅳ) system toward AOCs, (2) identify reactive species through quenching experiments and probe compound analysis, (3) characterize Fe(Ⅲ) flocs structural modifications and evaluate their impact on As adsorption, and (4) elucidate the degradation mechanism of p-ASA. This work provides novel insights into AOCs removal via the Fe(Ⅵ)/S(Ⅳ) system, offering critical implications for optimizing arsenic-contaminated water treatment strategies.
Fe(Ⅵ) was synthesized via a solution-phase wet chemical synthesis method [21]. A complete list of chemicals used in this study is provided in Text S1 (Supporting information). Detailed descriptions of the analytical methods can be found in Text S2 (Supporting information). The residual Fe(Ⅵ) was analyzed using the ABTS method, as detailed in Text S3 (Supporting information). Organic constituents were quantified using high-performance liquid chromatography (HPLC). The specific conditions for HPLC analysis of each chemical are outlined in Table S1 (Supporting information). The degradation intermediates of p-ASA were studied using high-performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS/MS), as outlined in Text S4 (Supporting information). The flocs produced from the decomposition of Fe(Ⅵ) were gathered in both the presence and absence of S(Ⅳ) (Text S5 in Supporting information).
The degradation experiments were conducted as follows: A 200 µmol/L p-ASA stock solution was prepared by dissolving p-ASA in ultrapure water. A 500 µmol/L Na2SO3 solution was prepared in deionized water, and 20 mmol/L borate buffer was added to adjust the solution volume to 100 mL. Fe(Ⅵ) was prepared in a 2 mmol/L borate buffer solution, and its concentration was quantified via UV–vis spectrophotometry, as described previously [22]. Reactions were initiated by adding Fe(Ⅵ) under continuous stirring at 300 rpm. At predetermined time intervals, 2.0 mL aliquots were collected, filtered through a 0.22 µm membrane filter, quenched with 20 µL of 1 mol/L hydroxylamine hydrochloride (NH2OH·HCl), and subjected to analysis. All experiments were conducted in triplicate to ensure reproducibility.
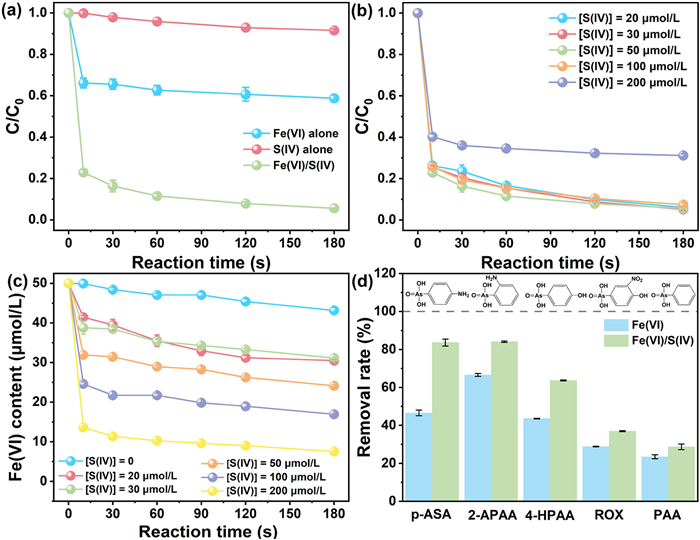
The removal efficiency of p-ASA was systematically compared across three systems: Fe(Ⅵ) alone, Fe(Ⅵ)/thiosulfate, and Fe(Ⅵ)/sulfite (Fig. S1 in Supporting information). Both sulfite (S(Ⅳ)) and thiosulfate significantly enhanced Fe(Ⅵ)-mediated p-ASA degradation at pH 8.0. The Fe(Ⅵ) alone system achieved 40.21% p-ASA removal, while the Fe(Ⅵ)/thiosulfate system increased this efficiency to 71.2%. In contrast, the Fe(Ⅵ)/sulfite system demonstrated near-complete p-ASA elimination. Sulfite exhibited superior efficacy in activating Fe(Ⅵ) for p-ASA oxidation. Consequently, sulfite was selected as the model S(Ⅳ) agent to assess the Fe(Ⅵ)/S(Ⅳ) system's efficacy in AOCs degradation. The Fe(Ⅵ)/S(Ⅳ) system was further benchmarked against established Fe(Ⅱ)-activated systems (Fe(Ⅱ)/S(Ⅳ), Fe(Ⅱ)/PMS, and Fe(Ⅱ)/PDS) under neutral conditions (Fig. S2 in Supporting information). Notably, the Fe(Ⅵ)/S(Ⅳ) system outperformed all Fe(Ⅱ)-based systems, achieving superior p-ASA removal efficiency under neutral pH. As shown in Fig. 1a, S(Ⅳ) alone exhibited negligible capacity for p-ASA elimination. The enhanced efficacy of S(Ⅳ) likely arises from synergistic generation of reactive intermediates and secondary contributions from Fe(Ⅲ) flocs.
Figure 1
Figure 1.
(a) Comparative performance of various systems in p-ASA removal. (b) Effect of S(Ⅳ) dosage on p-ASA degradation efficiency. (c) Fe(Ⅵ) decomposition under varying S(Ⅳ) dosages. (d) Comparative removal efficiencies of diverse AOCs in the Fe(Ⅵ)/S(Ⅳ) system. Experimental conditions: [AOC]0 = 5 µmol/L, [Fe(Ⅵ)]0 = 50 µmol/L, [S(Ⅳ)]0 = 50 µmol/L, pH 8.0.
The influence of S(Ⅳ) dosage on p-ASA degradation was systematically examined by maintaining Fe(Ⅵ) at 50 µmol/L while varying S(Ⅳ) concentrations from 20 µmol/L to 200 µmol/L. As shown in Fig. 1b, the addition of S(Ⅳ) (20–100 µmol/L) markedly improved p-ASA degradation efficiency, achieving removal rates of 52.7%, 53.6%, 53.2%, and 51.3% at respective S(Ⅳ) concentrations. This enhancement is attributed to the increased generation of reactive intermediates at higher S(Ⅳ) dosages, which facilitated p-ASA degradation. The consistent p-ASA degradation efficiency observed at 20–100 µmol/L S(Ⅳ) suggests an equilibrium between S(Ⅳ)-driven Fe(Ⅵ) activation and reactive oxygen species (ROS) generation. When the S(Ⅳ) concentration was further increased to 200 µmol/L, the elimination rate of p-ASA decreased. This reduced enhancement likely stems from competitive scavenging of reactive species by excess S(Ⅳ), which diverted oxidizing capacity from p-ASA degradation [23]. In parallel, temporal monitoring of Fe(Ⅵ) consumption revealed critical insights into system dynamics (Fig. 1c). Fe(Ⅵ) consumption increased from 6.84 µmol/L in the Fe(Ⅵ)-only system to 42.44 µmol/L in the presence of 200 µmol/L S(Ⅳ). Rapid Fe(Ⅵ) decomposition promotes the formation of high-valent iron intermediates (Fe(Ⅳ)/Fe(Ⅴ)), which are potent oxidants. Furthermore, S(Ⅳ) activates Fe(Ⅵ) to generate radical species (SO4•−, •OH) [24], which likely synergize with Fe(Ⅳ)/Fe(Ⅴ) to drive p-ASA degradation.
To comprehensively evaluate the synergistic enhancement of S(Ⅳ) on Fe(Ⅵ) oxidation, the Fe(Ⅵ)/S(Ⅳ) system's efficacy in degrading diverse AOCs (structures detailed in Table S2 in Supporting information) was investigated. As shown in Fig. 1d, S(Ⅳ) significantly accelerated Fe(Ⅵ)-mediated pollutant degradation, achieving near-complete AOCs elimination within 3 min. The addition of S(Ⅳ) improved the degradation efficiencies of p-ASA, 2-aminophenylarsonic acid (2-APAA), 4-hydroxyphenylarsonic acid (4-HPAA), ROX, and phenylarsonic acid (PAA) by 37.06%, 17.45%, 20.1%, 8.17%, and 5.23%, respectively, relative to Fe(Ⅵ) alone. The differential degradation efficiencies likely stem from Fe(Ⅵ)'s selective oxidation behavior and the distinct adsorption affinities of Fe(Ⅲ) flocs toward AOCs, modulated by electron-donating substituents (e.g., -NH2, -OH), steric hindrance, and molecular weight. These findings underscore the Fe(Ⅵ)/S(Ⅳ) system's broad-spectrum efficacy in AOCs degradation.
High-valent iron species are known to oxidize phenyl sulfoxide (PMSO) via oxygen atom transfer, yielding methyl phenyl sulfone (PMSO2). Conversely, reactive radicals induce PMSO transformation into polymeric or hydroxylated derivatives [25]. Therefore, PMSO was employed as a probe to differentiate the contributions of high-valent iron species and radical-mediated pathways. Fig. S3a (Supporting information) demonstrates that PMSO2 yields approached 100% in the Fe(Ⅵ) alone system, confirming the dominant role of high-valent iron species in PMSO oxidation. Increasing the S(Ⅳ) dosage from 20 µmol/L to 200 µmol/L progressively enhanced PMSO degradation rates, indicative of increased reactive species generation in the Fe(Ⅵ)/S(Ⅳ) system. Notably, PMSO2 yields in the Fe(Ⅵ)/S(Ⅳ) system progressively decreased from 79.5% to 61.2% with rising S(Ⅳ) dosages (Figs. S3b-f in Supporting information). The sub-100% PMSO2 yields signify the concurrent generation of radical species (e.g., SO4•−, •OH) upon S(Ⅳ) addition, which compete with high-valent iron species for PMSO oxidation [26].
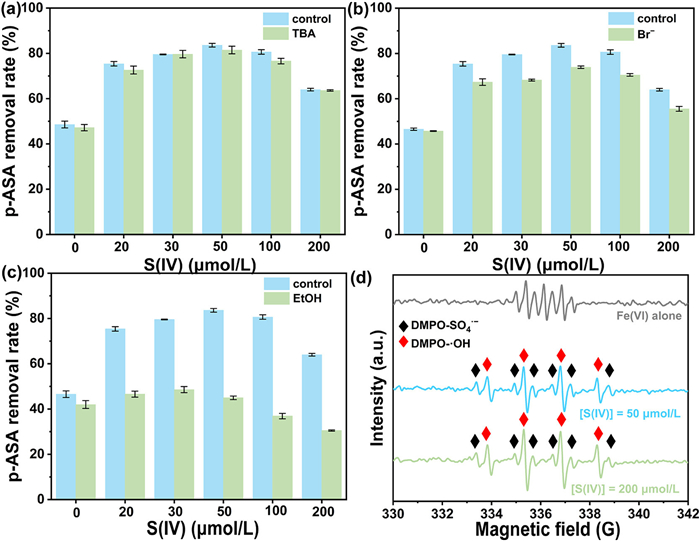
Chemical quenching experiments were conducted to elucidate the contributions of Fe(Ⅳ)/Fe(Ⅴ), SO4•−, and •OH to p-ASA degradation in the Fe(Ⅵ)/S(Ⅳ) system. tert-Butyl alcohol (TBA), bromide (Br−), and ethanol (EtOH) were employed as selective scavengers for •OH, SO4•−/•OH, and Fe(Ⅳ)/Fe(Ⅴ), respectively. The second-order rate constant for ·OH scavenging by TBA ranges from 3.8 × 108 L mol-1 s-1 to 7.6 × 108 L mol-1 s-1 [27]. Previous studies report second-order rate constants of 109–1010 L mol-1 s-1 for reactions of organic pollutants with SO4•− and •OH [28]. Under these conditions, 10 mmol/L TBA is expected to completely inhibit p-ASA oxidation via•OH scavenging. Fig. 2a demonstrates that TBA exerted only a modest inhibitory effect on p-ASA degradation in the Fe(Ⅵ)/S(Ⅳ) system, confirming that •OH is not the dominant reactive species. Using coumarin (COU) as a •OH probe [29], the concentration of 7-hydroxycoumarin (7-HOC) increased from 0.1 µmol/L to 0.5 µmol/L with rising S(Ⅳ) dosages, signaling progressive ·OH accumulation in the Fe(Ⅵ)/S(Ⅳ) system (Fig. S4 in Supporting information). Although •OH was generated during the reaction, it played a negligible role in p-ASA elimination, likely due to its low relative concentration or slower reaction kinetics with p-ASA compared to other species [27,28]. Br− exhibits rapid reactivity with SO4•− and •OH, contrasting with its inertness toward high-valent iron species [30]. The more significant inhibitory action of Br− compared to TBA indicates that the contribution of •OH is much less than that of SO4•− (Fig. 2b). Studies have established that Fe(Ⅳ)/Fe(Ⅴ) selectively oxidize electron-rich organic moieties but exhibit limited reactivity toward electron-deficient pollutants like benzoic acid (BA) [27]. BA degradation efficiency in the Fe(Ⅵ)/S(Ⅳ) system was evaluated across varying S(Ⅳ) concentrations (Fig. S5 in Supporting information). Elevated S(Ⅳ) dosages enhanced BA degradation, correlating with increased SO4•−/•OH generation. However, quenching and probe experiments revealed that SO4•−/•OH contributions to p-ASA removal remained largely unaffected by S(Ⅳ) concentration (Fig. 2b). The stronger inhibitory effect of EtOH compared to Br- is attributed to its competitive quenching of Fe(Ⅳ)/Fe(Ⅴ) (Fig. 2c) [31]. This aligns with PMSO experimental results, further corroborating the pivotal role of Fe(Ⅳ)/Fe(Ⅴ) in driving p-ASA degradation.
Figure 2
Figure 2.
Effects of TBA (a), Br− (b), and EtOH (c) on p-ASA removal. (d) EPR spectra of different systems. Experimental conditions: [Fe(Ⅵ)]0 = 50 µmol/L, [p-ASA]0 = 5 µmol/L, [TBA]0 = 10 mmol/L, [Br−]0 = 1 mmol/L, [EtOH]0 = 200 mmol/L, pH 8.0.
Electron paramagnetic resonance (EPR) experiments were performed to elucidate the generation mechanisms of reactive species in the Fe(Ⅵ)/S(Ⅳ) system. Previous studies have demonstrated that 5,5-dimethyl-1-pyrroline N-oxide (DMPO) is selectively oxidized to 5,5-dimethyl-2-pyrrolidinone-N-oxide (DMPOX) by Fe(Ⅳ)/Fe(Ⅴ) [32]. As shown in Fig. 2d, the Fe(Ⅵ) alone system exhibited characteristic DMPOX signals with a distinctive 1:2:1:2:1:2:1 intensity ratio. The detection of DMPOX conclusively confirms Fe(Ⅳ)/Fe(Ⅴ) formation in this system. In the Fe(Ⅵ)/S(Ⅳ) system with initial S(Ⅳ) concentrations of 50 µmol/L and 200 µmol/L, pronounced DMPO-•OH adduct signals were detected. Simultaneously, six satellite signals adjacent to the four primary peaks were identified and assigned to DMPO-SO4•− adducts. Similar spectral signatures have been reported in previous Fe(Ⅵ)-based oxidation studies [33]. Collectively, quenching experiments, PMSO conversion assays, and EPR analysis demonstrated that the Fe(Ⅵ)/S(Ⅳ) system generates Fe(Ⅳ)/Fe(Ⅴ), SO4•−, and •OH. Increasing S(Ⅳ) dosage enhanced •OH and SO4•− generation, though •OH played a negligible role in p-ASA elimination, with Fe(Ⅳ)/Fe(Ⅴ) and SO4•− emerging as the dominant oxidative species. SO4•− contributions remained consistent across S(Ⅳ) concentrations, likely due to inherently limited reaction kinetics between ROS and AOCs. Consequently, oxidation processes were predominantly mediated by Fe(Ⅳ)/Fe(Ⅴ), even at elevated S(Ⅳ) dosages.
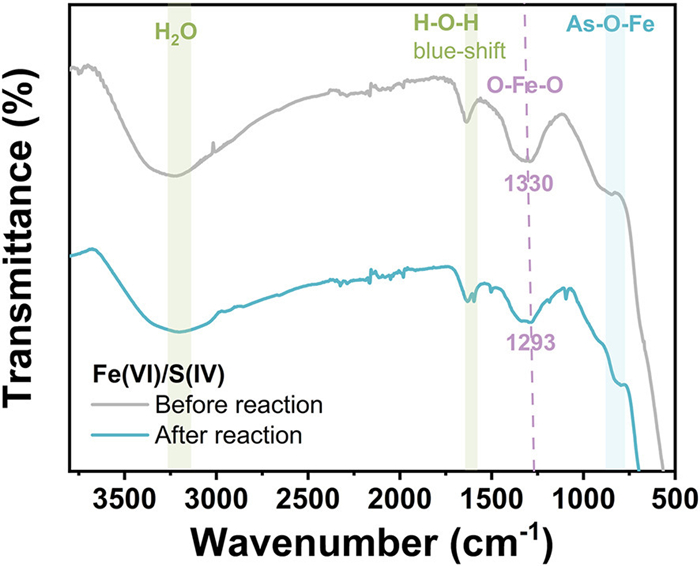
Fe(Ⅲ) flocs, formed during Fe(Ⅵ) decomposition, play a pivotal role in pollutant removal via dual mechanisms [34]. Primarily, Fe(Ⅲ) flocs mediate Fe(Ⅵ) reduction to generate reactive intermediates, enhancing pollutant degradation [35]. Secondarily, they adsorb organic pollutants and metal ions, synergistically augmenting water purification. Bzdyra et al. [20] reported that exogenous activators alter Fe(Ⅲ) flocs morphology and surface chemistry, thereby modulating their pollutant removal efficacy. Scanning electron microscopy (SEM) revealed Fe(Ⅲ) flocs with an irregular morphology in the absence of p-ASA (Fig. S6a in Supporting information). In the presence of p-ASA, Fe(Ⅲ) flocs developed pronounced surface protrusions (Fig. S6b in Supporting information), likely due to arsenic adsorption. Energy-dispersive X-ray spectroscopy (EDS) confirmed Fe(Ⅲ) flocs composition as 45.03% Fe and 41.31% O in the absence of p-ASA (Table S3 in Supporting information). Following p-ASA exposure, As was detected on floc surfaces. Notably, As and C accounted for 7.46% and 20.71% of floc composition, respectively, post-p-ASA adsorption (Figs. S6c and d in Supporting information). Fourier-transform infrared (FT-IR) spectra of Fe(Ⅲ) flocs provided further insights into arsenic interactions (Fig. 3). In the presence and absence of p-ASA, the obtained Fe(Ⅲ) flocs presented similar signals at 3250 cm−1 and 1620 cm−1, coming from the -OH in water molecules [36]. The O-Fe-O asymmetric stretching vibration shifted from 1330 cm−1 to 1293 cm−1 upon p-ASA adsorption, indicating structural Fe(Ⅲ) floc modification [37]. This shift likely reflects As(Ⅴ) adsorption-induced lattice distortion in Fe(Ⅲ) flocs. A distinct Fe-O-As vibrational peak at 820 cm−1 further confirmed arsenic binding to Fe(Ⅲ) flocs [38]. Collectively, these findings demonstrate that Fe(Ⅲ) flocs actively adsorb arsenic species, contributing to their immobilization.
Figure 3
Figure 3.
FT-IR spectra of Fe(Ⅲ) flocs in the presence and absence of p-ASA.
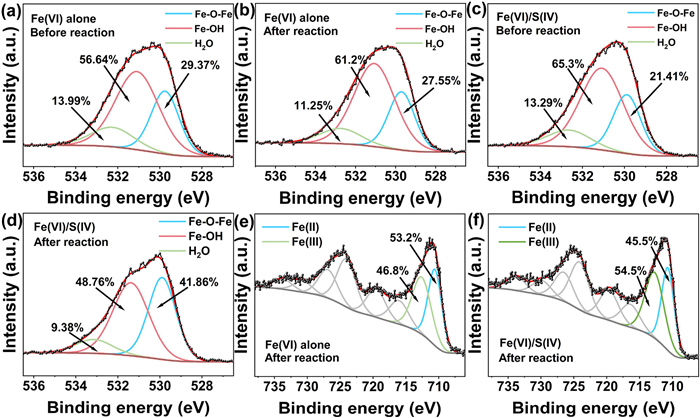
Subsequent analysis focused on S(Ⅳ)-induced modifications to Fe(Ⅲ) flocs structure and surface properties. Fig. S7 (Supporting information) demonstrates that S(Ⅳ) dosage modulates the zeta potential of Fe(Ⅲ) flocs, imparting a negative surface charge. Regardless of p-ASA presence, S(Ⅳ) addition induced a negative zeta potential in Fe(Ⅲ) flocs. This enhanced surface electronegativity likely facilitated electrostatic interactions with positively charged arsenic species. X-ray photoelectron spectroscopy (XPS) elucidated the surface valence states and chemical composition of Fe(Ⅲ) flocs. The O 1s spectrum exhibited three distinct peaks at 531.1 eV (Fe-OH), 532.5 eV (H2O), and 529.5 eV (Fe-O-Fe) [39]. In the Fe(Ⅵ) alone system, Fe(Ⅲ) flocs comprised 56.64% Fe-OH, 29.37% Fe-O-Fe, and 13.99% H2O (Fig. 4a). With p-ASA, Fe-OH increased to 61.2%, while H2O and Fe-O-Fe decreased to 11.25% and 27.55%, respectively (Fig. 4b). In the Fe(Ⅵ)/S(Ⅳ) system, Fe-OH decreased markedly, whereas Fe-O-Fe increased significantly after the reaction (Figs. 4c and d). This shift is attributed to S(Ⅳ)-mediated Fe(Ⅵ) activation, which promotes Fe-O-Fe bond formation. The distinctive peak at 711.3 eV in the Fe 2p3/2 spectrum corresponded to Fe(Ⅱ), whereas the peak at 713.1 eV indicated Fe(Ⅲ) [40]. Fe(Ⅱ) generation originated from two pathways: (1) S(Ⅳ)-driven Fe(Ⅵ) reduction to Fe(Ⅳ)/Fe(Ⅴ), followed by reductive/disproportionation reactions yielding Fe(Ⅱ); (2) S(Ⅳ)-ferric (oxyhydr)oxide interfacial electron transfer under near-neutral pH, fostering Fe(Ⅱ) regeneration. Fe(Ⅲ) content in the Fe(Ⅵ)/S(Ⅳ) system increased by 7.7% relative to Fe(Ⅵ) alone (Figs. 4e and f). This highlights S(Ⅳ)'s dual function: Enhancing Fe(Ⅵ) activation to Fe(Ⅳ)/Fe(Ⅴ) and sustaining Fe(Ⅲ)/Fe(Ⅱ) redox cycles, synergistically amplifying oxidative capacity. The As 3d XPS peak at 45.5 eV confirmed As(Ⅴ) formation [41]. As(Ⅴ) concentrations in the Fe(Ⅵ)/S(Ⅳ) system surpassed those in Fe(Ⅵ) alone (Figs. S8a and b in Supporting information), underscoring S(Ⅳ)'s role in enhancing oxidative capacity.
Figure 4
Figure 4.
(a-d) XPS spectra of Fe(Ⅲ) flocs O 1s before and after the different systems' reactions. (e, f) XPS spectra of Fe(Ⅲ) flocs Fe 2p after the different systems' reactions.
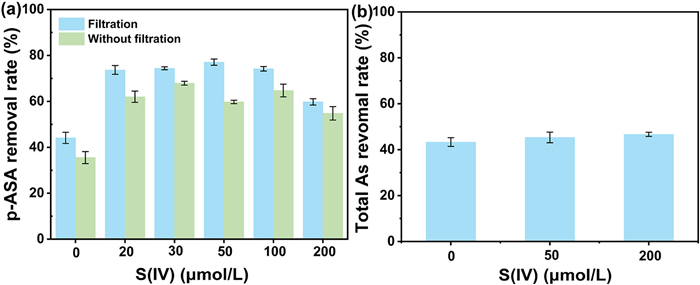
The data suggest that Fe(Ⅲ) flocs generated via the Fe(Ⅵ)/S(Ⅳ) system undergo significant structural modifications compared to those from Fe(Ⅵ) autodecomposition. However, the impact of these structural differences on arsenic removal efficiency remains ambiguous. To resolve this, we evaluated the effect of S(Ⅳ) on Fe(Ⅲ) flocs adsorption performance. Fig. S9 (Supporting information) reveals that varying borate concentrations minimally influence Fe(Ⅲ) flocs adsorption. Fig. 5a confirms Fe(Ⅲ) flocs-mediated p-ASA adsorption, yet total arsenic removal in both Fe(Ⅵ) alone and Fe(Ⅵ)/S(Ⅳ) systems exceeded p-ASA adsorption (Fig. 5b). This discrepancy arises because Fe(Ⅲ) flocs also adsorb inorganic arsenic and intermediates generated during p-ASA oxidation. XPS analysis of the As 3d spectra confirmed that adsorbed arsenic primarily existed as inorganic As(Ⅴ). Consequently, the higher total As removal indicates that Fe(Ⅲ) flocs preferentially immobilize As(Ⅴ) compared to undegraded p-ASA. The comparable total arsenic removal between systems indicates that arsenic adsorption is limited by the finite density of Fe-OH surface sites on Fe(Ⅲ) flocs, despite S(Ⅳ)-induced structural modifications. At higher S(Ⅳ) doses, accelerated Fe(Ⅵ) decomposition increased reactive species generation, enhancing As(Ⅴ) production. However, excess S(Ⅳ) introduced competitive ligands that reduced As(Ⅴ) adsorption via ligand exchange at Fe-OH sites. The counteracting effects of increased As(Ⅴ) production and competitive ligand adsorption negate each other, leading to consistent total arsenic removal rates irrespective of S(Ⅳ) concentration.
Figure 5
Figure 5.
(a) Effect of adsorption on p-ASA removal. (b) Effect of S(Ⅳ) concentration on total As removal. Experimental conditions: [Fe(Ⅵ)]0 = 50 µmol/L, [p-ASA]0 = 5 µmol/L, pH 8.0.
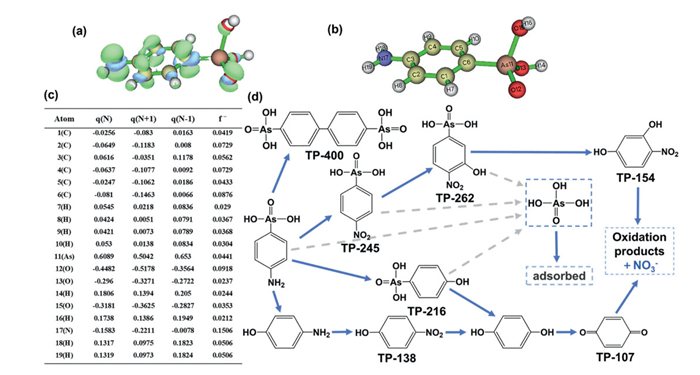
High-performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS/MS) was utilized to investigate the degradation and transformation mechanisms of p-ASA. Detailed characterization of the identified intermediates is provided in Table S4 (Supporting information) and Fig. S10 (Supporting information). Fig. 6a illustrates the electron cloud distribution in p-ASA, with pronounced density localized around the amino and arsenate functional groups. Fukui function analysis revealed elevated f-values at the C6, As11, O12, and N17 sites, indicating heightened susceptibility to electrophilic attack (Figs. 6b and c). Based on the Fukui indices and identified oxidation intermediates, the proposed decomposition pathways of p-ASA are schematically summarized in Fig. 6d. The degradation of p-ASA predominantly proceeds via two distinct mechanistic pathways. In the primary pathway, Fe(Ⅳ)/Fe(Ⅴ) reactive species mediate cleavage of the As-C bond in p-ASA, yielding p-aminophenol (p-AP) and As(Ⅲ) [42]. As(Ⅲ) is ultimately oxidized to As(Ⅴ), manifesting as inorganic arsenic species, while p-AP is rapidly oxidized by the active species to produce TP-138, followed by p-phenol and ultimately p-benzoquinone [43]. In the secondary pathway, oxidation of the amino group in p-ASA generates a nitro substituent, which undergoes further reactions with reactive oxygen species to produce TP-262. Concurrently, As-C bond cleavage releases As(Ⅲ) and generates TP-154. A minor pathway involves Fe(Ⅳ)/Fe(Ⅴ)-mediated electron transfer from p-ASA, generating radicals that dimerize to form TP-400 [44]. Arsenic-containing intermediates were either adsorbed or mineralized to CO2 and H2O via ROS, whereas inorganic arsenic species were sequestered by Fe(Ⅲ) flocs. To evaluate the environmental impact of by-products, acute toxicity, mutagenicity, and developmental toxicity of intermediates were assessed using the Toxicity Estimation Software Tool (T.E.S.T.) (Fig. S11 in Supporting information). By-products were categorized by toxicity levels, with p-ASA displaying pronounced acute toxicity. Although residual toxicity persists in by-products post-Fe(Ⅵ)/S(Ⅳ) treatment, a marked reduction in overall toxicity was observed. While mutagenicity and developmental toxicity were elevated in a minor subset of by-products relative to p-ASA, these constituted only a trace fraction of the total. Collectively, the Fe(Ⅵ)/S(Ⅳ) system significantly attenuates the toxicity of most p-ASA-derived by-products, underscoring its potential as an environmentally friendly strategy for organic arsenic removal. pH critically regulates the speciation and reactivity of the Fe(Ⅵ)/S(Ⅳ) system, underscoring the need to systematically evaluate its impact on pollutant removal efficiency. As illustrated in Fig. S12a (Supporting information), p-ASA removal efficiency by the Fe(Ⅵ)/S(Ⅳ) system increased progressively as pH decreased from 9.0 to 7.0, achieving optimal performance at neutral pH. However, further acidification to pH 6.5 resulted in a marked efficiency decline, owing to accelerated Fe(Ⅵ) self-decomposition under acidic conditions, which prematurely depleted oxidants and reduced their availability for pollutant degradation [45]. Conversely, under alkaline conditions, Fe(Ⅵ)/S(Ⅳ) reaction kinetics slowed significantly, inhibiting the generation of reactive species such as Fe(Ⅳ)/Fe(Ⅴ). This pH-dependent behavior highlights a trade-off between Fe(Ⅵ) stability and activation efficiency, with neutral pH optimizing oxidant longevity and reactive species generation, thereby maximizing p-ASA removal. As pH increased from 6.5 to 9.0, the removal efficiencies of both p-ASA and total arsenic declined (Fig. S12b in Supporting information), confirming that effective p-ASA oxidation is a prerequisite for total arsenic removal.
Figure 6
Figure 6.
Highest occupied molecular orbital (HOMO) of p-ASA (a). Fukui index of p-ASA (b, c). Degradation pathways of p-ASA (d).
To evaluate the practical applicability of the system, the effects of common anions and humic acid (HA) on p-ASA removal were systematically investigated. As shown in Fig. S12c (Supporting information), the introduction of anions suppressed p-ASA removal efficiency in the Fe(Ⅵ)/S(Ⅳ) system to varying degrees, with the strongest inhibition observed for CO32− and HA. The moderate inhibitory effects of Cl−, SO42−, and HCO3− likely arise from their competition with p-ASA for reactive species. These anions scavenge •OH and SO4•− radicals, forming less reactive species that diminish p-ASA degradation efficiency [46]. Notably, Fe(Ⅳ)/Fe(Ⅴ) species were established as the dominant reactive intermediates in the Fe(Ⅵ)/S(Ⅳ) system, with •OH and SO4•− playing negligible roles. Consequently, Cl−, SO42−, and HCO3− exerted minimal inhibitory effects on p-ASA removal. HA rapidly quenches Fe(Ⅵ), while Fe(Ⅵ)-derived adsorptive flocs remain effective in removing p-ASA [47]. However, HA introduction only partially inhibited p-ASA removal (Fig. S12c in Supporting information). Collectively, the Fe(Ⅵ)/S(Ⅳ) system demonstrates robustness against interference from most matrices.
In conclusion, the introduction of S(Ⅳ) enabled Fe(Ⅵ) to achieve 94.8% p-ASA removal within 3 min, demonstrating the Fe(Ⅵ)/S(Ⅳ) system's significant potential for rapid p-ASA degradation. Radical quenching and probe experiments elucidated the evolution of active species with S(Ⅳ) dosage, confirming SO4•−, •OH, and Fe(Ⅳ)/Fe(Ⅴ) as the dominant reactive intermediates. Increasing S(Ⅳ) dosage accelerated Fe(Ⅵ) conversion to Fe(Ⅳ)/Fe(Ⅴ) while concurrently promoting SO4•− and •OH generation. However, due to ROS concentration gradients and kinetic disparities, Fe(Ⅳ)/Fe(Ⅴ) played a pivotal role in p-ASA elimination. Zeta potential, FT-IR, and XPS analyses confirmed Fe(Ⅲ) flocs' role in arsenic sequestration, while S(Ⅳ) introduction altered floc morphology without compromising adsorption capacity. A proposed degradation mechanism involves Fe(Ⅳ)/Fe(Ⅴ)-mediated cleavage of the As-C bond in p-ASA, converting the predominant fraction of organic arsenic to inorganic species. The toxicity evaluation findings revealed that the decomposition products exhibited minimal toxicity. The Fe(Ⅵ)/S(Ⅳ) system also exhibited robust performance in neutral aqueous matrices and resilience to common coexisting ions, underscoring its viability for AOCs removal.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Chang Xu: Investigation, Data curation, Writing – original draft. Mengfan Luo: Methodology, Data curation. Jia Zhao: Investigation, Data curation. Jialong Yin: Methodology, Validation. Can Feng: Formal analysis, Data curation. Heng Zhang: Funding acquisition, Writing – review & editing, Supervision. Peng Zhou: Supervision, Software. Zhaokun Xiong: Methodology, Software. Bo Lai: Funding acquisition, Resources, Supervision.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111538.
[1]
Noor S. Abbood, N.S. Ali, E.H. Khader, et al., Res. Chem. Intermed. 49 (2023) 43–56. doi: 10.1007/s11164-022-04879-3
Figure 1
(a) Comparative performance of various systems in p-ASA removal. (b) Effect of S(Ⅳ) dosage on p-ASA degradation efficiency. (c) Fe(Ⅵ) decomposition under varying S(Ⅳ) dosages. (d) Comparative removal efficiencies of diverse AOCs in the Fe(Ⅵ)/S(Ⅳ) system. Experimental conditions: [AOC]0 = 5 µmol/L, [Fe(Ⅵ)]0 = 50 µmol/L, [S(Ⅳ)]0 = 50 µmol/L, pH 8.0.
Figure 4
(a-d) XPS spectra of Fe(Ⅲ) flocs O 1s before and after the different systems' reactions. (e, f) XPS spectra of Fe(Ⅲ) flocs Fe 2p after the different systems' reactions.
Figure 5
(a) Effect of adsorption on p-ASA removal. (b) Effect of S(Ⅳ) concentration on total As removal. Experimental conditions: [Fe(Ⅵ)]0 = 50 µmol/L, [p-ASA]0 = 5 µmol/L, pH 8.0.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
