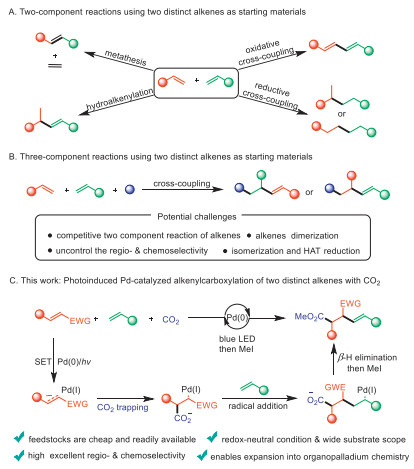
Scheme 1.
Cross-coupling of alkenes.
Three-component alkenylcarboxylation of two distinct alkenes with CO2 via photoinduced palladium catalysis
Jie Zhou , Luping Hu , Rui Wang , Ruijie Wang , Jun Xu , Huajian Xu

C–C bonds formation is a key challenge in modern organic chemistry [1,2], and the development of novel strategies for constructing C–C bonds remains a focus of intensive research. Alkenes, as the most abundant and readily accessible feedstocks in the chemical industry, serve as key building blocks for synthesizing complex functional molecules [3-5]. In the past decades, the formation of C–C bonds by two-component reaction of two distinct alkenes has been extensively studied (Scheme 1A) [6-18], for example, alkenes metathesis reactions, hydroalkenylation, oxidative or reductive cross-coupling. Based on the nature of atom- and step-economy, multicomponent reactions (MCRs) will provide a valuable strategy for simultaneous construction of multiple C–C and C-X (X = heteroatom) bonds in a one-pot operation. This approach offers an operationally simple and highly modular method for the synthesis of structurally diverse molecules from abundant feedstocks [19,20]. However, three-component reactions of two distinct alkenes face several challenges: (1) Control of the regio- & chemoselectivity; (2) Various competitive two-component reaction; (3) Alkenes dimerization (Scheme 1B).
To date, only a few examples of three-component reactions involving two different alkenes have been reported [21-25]. For example, in 2023, the Glorius group employed metal-free photosensitized radical relay 1,4-carboimination across two distinct olefins, with alkyl carboxylic acid-derived bifunctional oxime esters [21]. The same year, the Yin group reported a three-component cross coupling of two distinct olefins and arylboronic acids, using strategy of an organometallic-radical relay, to achieved alkenes 1,1-difunctionalizations [23]. Recently, the Huang group developed an approach for palladium-catalyzed hydrocarbonylative cross-coupling between electron-rich and electron-deficient olefins in the presence of CO to produce structurally diverse β-ketoaldehyde surrogates [24]. Despite the progress achieved in the field, it is still necessary to develop diversified three-component coupling catalytic systems to enrich the MCR types of two distinct alkenes.
CO2, widely recognized as a greenhouse gas, is inexpensive, non-toxic, and has been considered as an ideal one-carbon feedstock for chemical transformations aimed at synthesizing high-value-added chemicals [26-28]. The development of photocatalytic transformations of readily available alkenes and CO2 has recently attracted substantial attention [29-35]. In this field, many groups have reported photoredox-catalyzed regioselective silacarboxyaltion [36], thiocarboxylation [37], phosphonocarboxylation [38], arylcarboxylation [39], dicarboxylation [40], aminocarboxylation [41], cyancarboxylation [42] and alkynylcarboxylation [43] of alkenes and CO2 under mild conditions. These methods serve as powerful tools for the synthesis of structurally diverse carboxylic acid derivatives. So far, the alkenylcarboxylation of two distinct alkenes with CO2 has not been reported, primarily due to the competitive two component reaction of alkenes and uncontrollable of regio- & chemoselectivity.
In last few years, visible-light-induced palladium catalysis has emerged as a highly vibrant and promising field of research [44]. Since the first report of photoexcited Pd and aryl iodides generation of carbon radicals by the Gevorgyan group in 2016 [45], a wide variety of transformations have been established based on excited-state Pd complexes, which efficiently reduce various electrophiles, such as C-X (X = F, Cl, Br, I, OTf) [46-67], oxime esters [68-71], diazo compounds [72], N-hydroxyphthalimide esters [73-75] and ketones [76], as radical precursors via single-electron transfer (SET). Inspired by these works, we envisioned that photoinduced excited-state Pd(0) species could selectively reduce electron-deficient alkenes through SET to generate alkene radical anions. It would be trapped by CO2, leading to new carbon radicals, followed by Heck-type coupling with electron-rich or electron-neutral alkenes (Scheme 1C). The successful realization of this method, which overcomes the undesired two-component reactions of alkenes, will open up new chemical spaces in cross-coupling reactions and enable the alkenylcarboxylation of densely functionalized and complex alkenes.
To validate our hypotheses, we commenced our study with examining the three-component reaction of readily available ethyl acrylate (S1), styrene (S2) and CO2 as the model substrates (Table 1). The reaction was carried out using palladium as a photocatalyst, Cs2CO3 as the base, and HMPA as the solvent under 1 atm of CO2, with irradiation by a 455 nm blue LEDs at 40–50 ℃. After extensive screening of reaction parameters, we were pleased to obtain a 70% isolated yield of 1 (Table 1, entry 1). When the reaction conditions were modified, a significant increase in the amounts of several side products was observed, including hydroalkenylation (1b), hydrocarboxylation (1c), isomerization (1d), and oligomerization (1e) [10]. Reducing the Pd catalyst loading to 5 mol% and the ligand loading to 15 mol% led to a slightly lower yield (Table 1, entry 2). Pd(OAc)2 could also act as a catalyst, but a lower yield (32%) was obtained (Table 1, entry 3). For Pd(Ⅱ) precatalysts, the anions are expected to influence the reduction rate of Pd(Ⅱ) to Pd(0) species [53], thus affecting the reaction outcome. Replacing PPh₃ with other ligands led to either complete cessation of the reaction or a substantial reduction in yield (Table 1, entries 4–6). Alternative solvents, such as DMSO and DMAc, gave lower yields (Table 1, entries 7 and 8), which confirmed HMPA as the optimal solvent. Using K2CO₃ as the base, the yield of 1 decreased (Table 1, entry 9). Additionally, the absence of ligands, additives, or bases in the reaction system significantly decreased the yield of 1 (Table 1, entries 10–12). The use of equimolar amounts of S1 and S2 leads to an increase in oligomerization products 1e (Table 1, entry 13). Control experiments confirmed the importance of both the palladium catalyst and blue light in this photoinduced palladium-catalyzed systems (Table 1, entries 14 and 15).
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Variations from standard conditions | Yield (%) |
| 1 | None | 78 (70)b |
| 2 | Pd(PPh3)4 (5 mol%), PPh3 (15 mol%) | 71 |
| 3 | Pd(OAc)2 as Pd catalyst | 32 |
| 4 | Xantphos as a ligand | N.D. |
| 5 | P(4-OMeC6H4)3 as a ligand | 45 |
| 6 | PCy3 as a ligand | N.D. |
| 7 | DMSO as solvent | 25 |
| 8 | DMAc as solvent | 34 |
| 9 | K2CO3 as base | 61 |
| 10 | without ligand | 43 |
| 11 | without ZnCl2 | 59 |
| 12 | without Cs2CO3 | 48 |
| 13 | S1:S2 = 1:1 | 52 |
| 14 | no light, up to 160 ℃ | N.D. |
| 15 | without Pd(PPh3)4 | 0 |
| a Reaction conditions: S1 (0.2 mmol, 1.0 equiv.), S2 (0.4 mmol, 2.0 equiv.), Pd(PPh 3) 4 (10 mol%), Cs 2CO 3 (0.4 mmol, 2.0 equiv.), ZnCl 2 (0.4 mmol, 2.0 equiv.), HMPA (2.0 mL), irradiation with 10 W 455 nm blue LEDs at 40–50 ℃ under CO 2 (1 atm) for 16 h, and then MeI (0.6 mmol, 3.0 equiv.) was added to react at room temperature for 4 h. N.D. = Not detected. DMSO = Dimethyl sulfoxide. DMAc = N, N-dimethylacetamide. HMPA = Hexamethylphosphoramide. b Yields determined by GC analysis. Isolated yields in the parentheses. | ||
With the optimized conditions in hand, the scope of electron-deficient alkenes was examined first (Scheme 2A). Electron-deficient alkenes with different substitutions were identified as suitable substrates for this transformation. Thus, a diverse array of terminal acrylates substrates (2–7), including the free hydroxyl group, exhibited excellent reactivity, providing the desired alkenylcarboxylation products in good to high yields. Similarly, acrylamides (8 and 9) were also well tolerated, and give the desired carboxylic acids. Acrylonitrile afforded the three-component coupling product (10) in moderate yield under mild reaction conditions. In addition, 1,1-disubstituted electron-deficient alkenes, such as acrylates and α, β-unsaturated lactones, efficiently participated in this reaction, providing various alkenylcarboxylation products (11–16) in good yields with excellent regio- & chemoselectivity. To our delight, methyl 2-acetamidoacrylate was well performed to generate (±) aspartic acid derivatives (12), thereby proving that this protocol has the potential to synthesize functionalized amino acid derivatives. However, the ester exchange product (14) was obtained when a precursor with an allylic alcohol portion was used. Notably, even more challenging internal olefins, such as ethyl crotonate and crotononitrile, successfully underwent alkenylcarboxylation with vinyl arenes (17–19). Moreover, trisubstituted endo- and exocyclic acrylates (20 and 21) were found to be capable substrates for this alkenylcarboxylation reaction, which efficiently construct highly complex dicarboxylic acid derivatives with alkenyl functional groups and quaternary carbon centers.
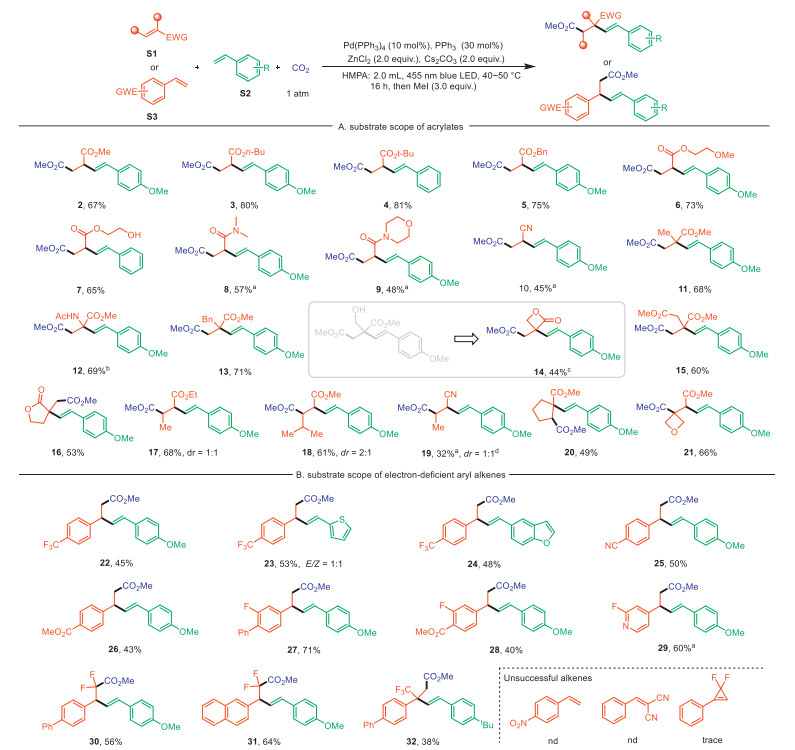
We found that the reaction was not limited to acrylates substrates, as electron-deficient vinyl aromatics (heteroarenes) can also be reduced by excited-state Pd(0) species and successfully underwent the alkenylcarboxylation to furnish monocarboxylic acid derivatives (Scheme 2B). A series of vinyl arenes with electron-withdrawing group, such as trifluoromethyl (22–24), nitrilea (25), ester (26) and fluoro (27 and 28), underwent alkenylcarboxylation efficiently, yielding the desired products in moderate yields. Additionally, electron-deficient heteroarenes (29) proved to be compatible with the light-driven alkenylcarboxylation reaction. Building on these encouraging findings, we shifted our focus to exploring more challenging substrates. Instead of relying on transition metal-catalyzed defluorination to produce monofluoroalkenes [77], this robust protocol facilitated the efficient carboxylation of gem‑difluoroalkenes, providing a straightforward route to difluorocarboxylic acid products (30 and 31). Besides these, α-trifluoromethylstyrene was also successfully subjected to the reaction conditions, generating the desired product (32) in a reasonable yield. Unfortunately, this methodology was proved to be incompatible with several alkenes, such as 4-nitrostyrene, benzalmalononitrile, and gem‑difluorocyclopropene.
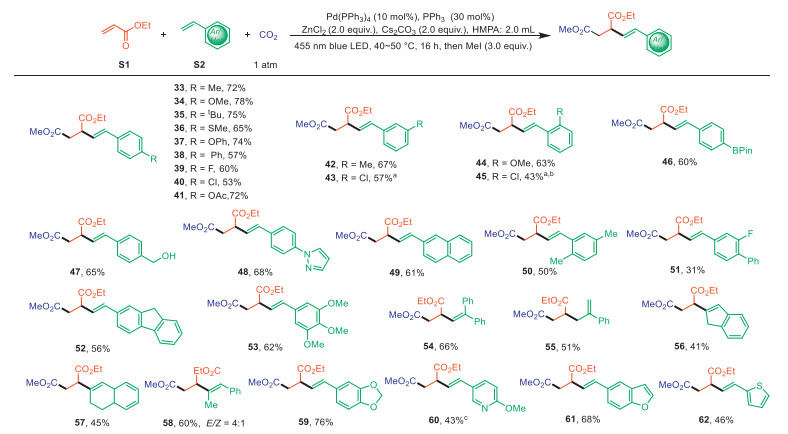
We next became interested in the scope of another alkenyl conjugations (Scheme 3). Such as, a wide variety of electron-neutral, electron-donating (e.g., OMe, t-Bu, OAc) or electron-withdrawing (e.g., F, Cl, ) groups were tolerant at the para (33–41), meta (42 and 43), and ortho positions (44 and 45) of the vinyl arenes moiety, affording the corresponding products in moderate to good yields. Notably, this method demonstrates good tolerance for reactive functional groups on the vinyl arenes, including boronate (46), free benzyl alcohol (47), and pyrazole (48). Disubstituted and trisubstituted styrene derivatives were also efficiently transformed into the corresponding desired products (49–53). Besides monosubstituted alkenes, 1,1-disubstituted alkenes (54 and 55) were exhibited good reactivity under the standard conditions. To our surprise, when α-methylstyrene was used as a radical acceptor, the alkene isomerization product (55) was obtained. Remarkably, more challenging internal alkenes, such as indene and 1,2-dihydronaphthalene, were successfully delivered alkenylcarboxylation products (56–58) in moderate to good yields. We were delighted to find that the method tolerated a range of heterocyclic styrene derivatives, including 1,3-benzodioxole (59), pyridine (60), benzofuran (61), and thiophene (62). However, unactivated alkyl alkenes proved incompatible with these carboxylation conditions, generating only trace amounts of the corresponding carboxylated products (for other unsuccessful examples, see Supporting information).
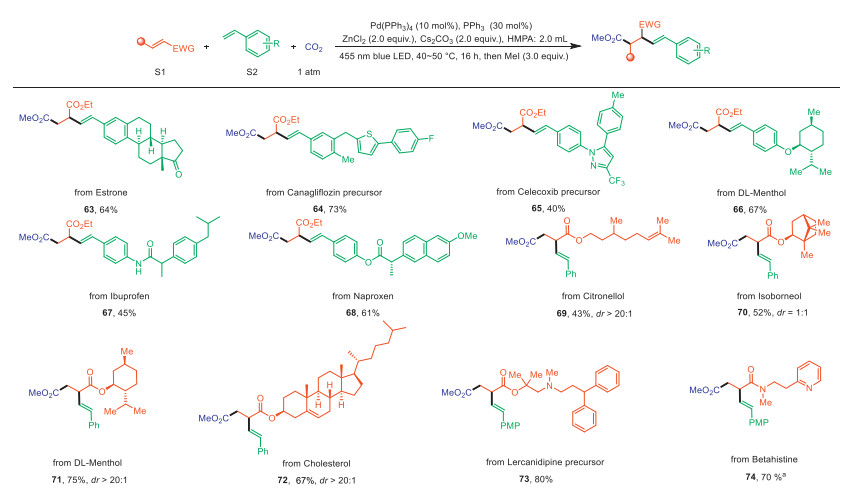
Then, to prove the application potential of this new synthetic strategy, the late-stage functionalization derived from drugs and natural products was carried out, as shown in Scheme 4. Gratifyingly, vinyl arenes containing estrone, canagliflozin intermediate, celecoxib intermediate, dl-menthol, ibuprofen and naproxen derivatives furnished target products (63–68) in moderate to high yields. Furthermore, acrylates derived from terpenes (70 and 71), terpenoids (69 and 73), and cholesterol (72) exhibited excellent compatibility in this reaction. Analogously, betahistine derivative (74) was also tolerated in this method. This approach offers a novel synthetic pathway for building diverse complex molecules to generate diverse chemical libraries, accelerating the discovery and development of new drugs.
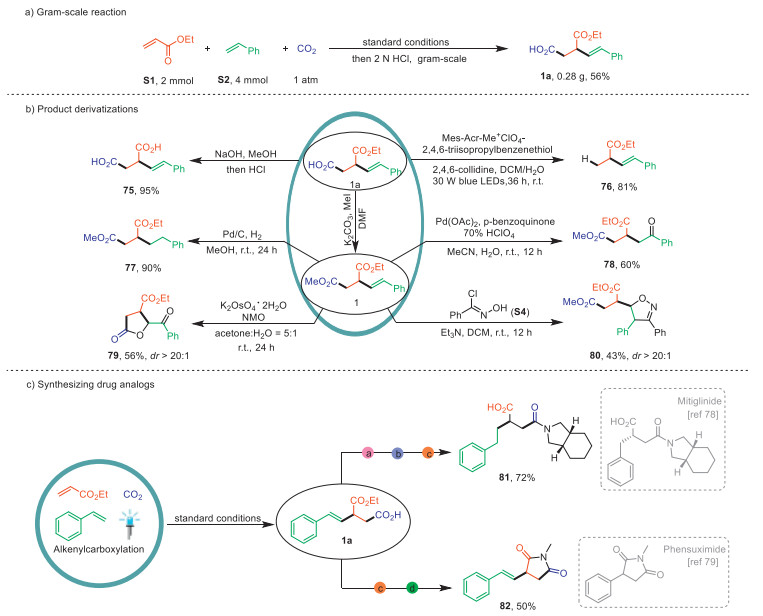
In order to further demonstrate the utility of this approach, we performed a gram-scale reaction and product derivatizations, as shown in Fig. 1 (For details, see Supporting information). Firstly, the reaction was performed on a 2 mmol scale under standard conditions, generating product 1a in moderate yield. Then, we performed facile derivatization of the products to demonstrate their potential synthetic applications. Hydrolysis of product 1a by using NaOH produced the diacid 75 in 95% yield. A highly efficient decarboxylation of primary carboxylic acid 1a was accomplished through synergistic photoredox and HAT catalysis, delivering compound 76 in excellent yields. Additionally, the reduction of 1 under the a H2 atmosphere, catalyzed by Pd/C, successfully produced the compound 77 in 90% yield. The double bond in 1 was oxidized using the Wacker process, affording the corresponding ketone 78. Surprisingly, performing dihydroxylation on compound 1 results in intramolecular cyclization, yielding the lactones 79. Moreover, treatment of compound 1 with N-hydroxybenzimidoyl chloride and triethylamine led to the formation of desired dihydroisoxazole 80. Notably, further transformation studies highlighted the practical value of the monoethyl succinate product. For example, we successfully synthesized an analogue 81 of mitiglinide [78], a drug for treatment of diabetes, in just three steps. Furthermore, succinimide, an analog 82 of the nervous system drug phensuximide [79], was also synthesized via a two-step procedure. All these outcomes underscore the outstanding practical value of our highly modular approach to the synthesis of structurally diverse molecules from abundant feedstocks, thus reinforcing their potential application in the rapid and efficient synthesis of drug molecule analogues.
To gain mechanistic insight into this photoinduced palladium catalysis process, a series of experiments were carried out (Fig. 2). Firstly, when 2,2,6,6-tetramethyl-1-piperidiny-1-oxy (TEMPO) or diphenyldiselenide (PhSeSePh) was added as a radical inhibitor under standard conditions, the formation of product 1 was completely inhibited, which indicated that a radical intermediate was involved in this transformation (Fig. 2a). In the radical probe experiment, the cross-alkenylcarboxylation of acrylate S1 with cyclopropyl-substituted styrene S5 produced dehydronaphthalene 83, seemingly through a radical rearrangement cascade (Fig. 2b). After that, TEMPO completely suppressed the alkenylcarboxylation reaction of 4-(trifluoromethyl)styrene S3, with HRMS analysis revealing the formation of a possible alkyl-TEMPO adducts 84′ (Fig. 2c, see Supporting information for more details). Secondly, when D2O was added as the hydrogen source in the model reaction of ethyl acrylate and styrene under N2, the deuterated product 85 was obtained in 39% yield with 90% deuterium incorporation at the β-position of the acrylate moiety (Fig. 2d). This result indicates that the electron-deficient alkenes undergo SET reduction to form radical anions, which subsequently undergo protonation at the β-carbon [80]. Furthermore, neither formate (HCO2−) nor oxalate (C2O4)2− was detected under the reaction conditions without alkenes, suggesting that the reduction of CO2 to CO2•− is less favored (Fig. 2e). Finally, Stern-Volmer quenching experiments (Fig. 2f) revealed that the excited-state of Pd(0) catalyst was mainly quenched by the electron-deficient acrylate S1 rather than styrene derivatives S2.
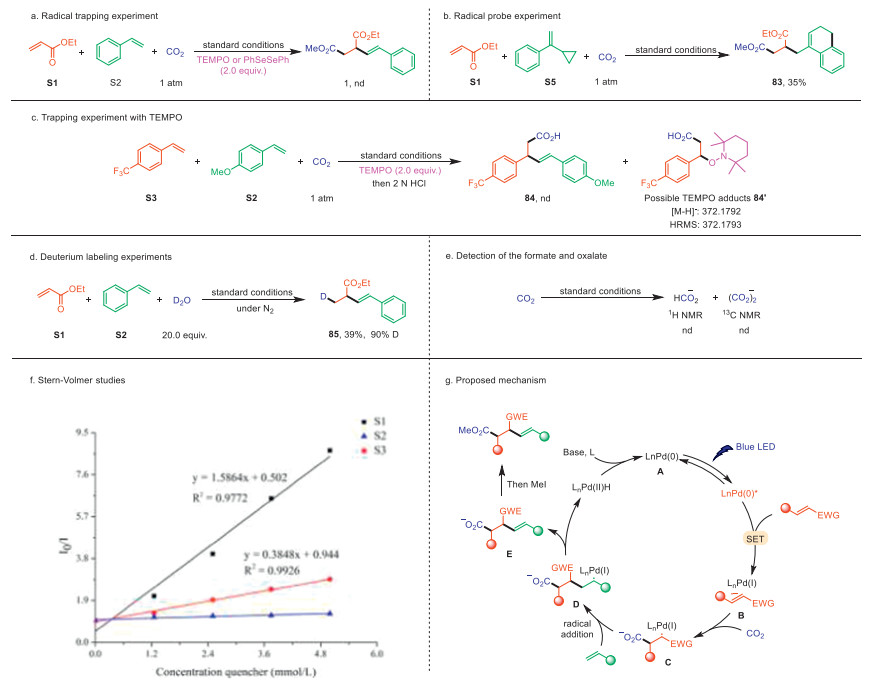
Based on the above mechanistic investigations and previous reports [10,41,42,80-82], we propose a possible reaction mechanism (Fig. 2g). Initially, upon exposure to blue LED irradiation, the excited-state Pd(0) can selectively reduce electron-deficient alkenes via SET, to form hybrid Pd(Ⅰ) alkyl radical anion species B. It could react with CO2 to generate the intermediate C. The intermediate D is then formed by the addition of the carbon radical to vinyl aromatics (heteroarenes), which subsequently undergo β-H elimination to produce the Pd(Ⅱ)-H species and carboxylated product E. Finally, methylation of the carboxylated product E furnishes the target product, and the Pd(Ⅱ)-H react with the base to regenerates the Pd(0) catalyst A.
In summary, we have developed the three-component coupling of two distinct alkenes with CO2 via visible-light induced palladium-catalyzed process. The present method features high regio- & chemoselectivity, broad substrate versatility and compatibility with complex molecules. The key to the success of reaction is that the excited-state Pd(0) species can selectively facilitate the reductive activation of electron-deficient alkenes, leading to the formation of hybrid Pd(Ⅰ) alkyl radical anion intermediates. Overall, this methodology represents the first example of selective activation of two distinct alkenes via photoexcited Pd(0) catalysis, which effectively suppresses the undesired two-component alkenes coupling and enables the simultaneous construction of multiple C–C bonds in a one-pot operation. This operationally simple and modular strategy provides a versatile platform for synthesizing structurally diverse molecules.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jie Zhou: Writing – original draft, Methodology, Investigation, Data curation. Luping Hu: Methodology, Investigation, Data curation. Rui Wang: Methodology, Data curation. Ruijie Wang: Methodology, Data curation. Jun Xu: Writing – review & editing, Methodology, Investigation. Huajian Xu: Writing – review & editing, Methodology, Investigation.
We gratefully acknowledge the financial support from the Fundamental Research Funds for the Central Universities (No. PA2020GDKC0021), the National Natural Science Foundation of China (No. 21971051) and the Hefei University Talent Research Fund (No. 24RC31).
Supplementary material associated with this article can be und, in the online version, at doi:
S.H. Cho, J.Y. Kim, J. Kwak, S. Chang, Chem. Soc. Rev. 40 (2011) 5068–5083. doi: 10.1039/c1cs15082k
C.J. Li, Acc. Chem. Res. 42 (2009) 335–344. doi: 10.1021/ar800164n
T. Koike, M. Akita, Org. Chem. Front. 3 (2016) 1345–1349. doi: 10.1039/C6QO00139D
G. Yin, X. Mu, G.S. Liu, Acc. Chem. Res. 49 (2016) 2413–2423. doi: 10.1021/acs.accounts.6b00328
W. Ge, J.X. Wang, M.C. Fu, Y. Fu, Chin. J. Chem. (2024) 1203–120842. doi: 10.1002/cjoc.202300740
A.H. Hoveyda, A.R. Zhugralin, Nature 450 (2007) 243–251. doi: 10.1038/nature06351
O.M. Ogba, N.C. Warner, D.J. O’Leary, R.H. Grubbs, Chem. Soc. Rev. 47 (2018) 4510–4544. doi: 10.1039/c8cs00027a
C.Y. Ho, C.W. Chan, L. He, Angew. Chem. Int. Ed. 54 (2015) 4512–4516. doi: 10.1002/anie.201411882
T.V. RajanBabu, Chem. Rev. 103 (2003) 2845–2860. doi: 10.1021/cr020040g
S. Sarkar, S. Ghosh, D. Kurandina, Y. Noffel, V. Gevorgyan, J. Am. Chem. Soc. 145 (2023) 12224–12232. doi: 10.1021/jacs.3c02410
Z.K. Wen, Y.H. Xu, T.P. Loh, Chem. Sci. 4 (2013) 4520–4524. doi: 10.1039/c3sc52275j
Q.J. Liang, C. Yang, F.F. Meng, et al., Angew. Chem. Int. Ed. 56 (2017) 5091–5095. doi: 10.1002/anie.201700559
B. Jiang, M. Zhao, S.S. Li, Y.H. Xu, T.P. Loh, Angew. Chem. Int. Ed. 57 (2018) 555–559. doi: 10.1002/anie.201710601
K. Meng, Y. Sun, J. Zhang, et al., Org. Lett. 21 (2019) 8219–8224. doi: 10.1021/acs.orglett.9b02935
J.C. Lo, J. Gui, Y. Yabe, C.M. Pan, P.S. Baran, Nature 516 (2014) 343–348. doi: 10.1038/nature14006
J. Streuff, Chem. Eur. J. 17 (2011) 5507–5510. doi: 10.1002/chem.201100501
M. Kong, Y. Tan, X. Zhao, et al., J. Am. Chem. Soc. 143 (2021) 4024–4031. doi: 10.1021/jacs.1c01073
Z.G. Wu, M.Y. Wu, K. Zhu, J. Wu, Y.X. Lu, Chem 9 (2023) 978–988. doi: 10.1016/j.chempr.2022.12.013
B.H. Rotstein, S. Zaretsky, V. Rai, A.K. Yudin, Chem. Rev. 114 (2014) 8323–8359. doi: 10.1021/cr400615v
A. Dömling, W. Wang, K. Wang, Chem. Rev. 112 (2012) 3083–3135. doi: 10.1021/cr100233r
G. Tan, F. Paulus, A. Petti, et al., Chem. Sci. 14 (2023) 2447–2454. doi: 10.1039/d2sc06497a
F. Paulus, C. Stein, C. Heusel, et al., J. Am. Chem. Soc. 145 (2023) 23814–23823. doi: 10.1021/jacs.3c08898
D. Wu, W. Kong, Y. Bao, et al., Nat. Catal. 6 (2023) 1030–1041. doi: 10.1038/s41929-023-01032-0
Y. Chen, J. Wu, Y. Ding, H.M. Huang, Org. Chem. Front. 12 (2025) 85–89. doi: 10.1039/D4QO01312C
Y. Zhang, K.D. Li, C.Q. Zhou, Z.X. Xing, H.M. Huang, Green Chem. 26 (2024) 10434–10440. doi: 10.1039/d4gc02879a
X.B. Lu, W.M. Ren, G.P. Wu, Acc. Chem. Res. 45 (2012) 1721–1735. doi: 10.1021/ar300035z
Q. Liu, L.P. Wu, R. Beller, M. Jackstell, Nat. Commun. 6 (2015) 5933. doi: 10.1038/ncomms6933
M.Y. He, Y.H. Sun, B.X. Han, Angew. Chem. Int. Ed. 61 (2022) e202112835. doi: 10.1002/anie.202112835
Q.Y. Meng, S. Wang, G.S. Huff, B. König, J. Am. Chem. Soc. 140 (2018) 3198–3201. doi: 10.1021/jacs.7b13448
H. Huang, J.H. Ye, D.G. Yu, et al., C–C S Chem. 3 (2021) 1746–1756. doi: 10.31635/ccschem.020.202000374
Y. Jin, J. Caner, S. Nishikawa, N. Toriumi, N. Iwasawa, Nat. Commun. 13 (2022) 7584. doi: 10.1038/s41467-022-35293-3
B. Yu, Y. Liu, H.Z. Xiao, et al., Chem 10 (2024) 938–951. doi: 10.1016/j.chempr.2023.12.005
V.R. Yatham, Y.Y. Shen, R. Martin, Angew. Chem. Int. Ed. 56 (2017) 10915–10919. doi: 10.1002/anie.201706263
H. Wang, Y.Z. Gao, C.L. Zhou, G. Li, J. Am. Chem. Soc. 142 (2020) 8122–8129. doi: 10.1021/jacs.0c03144
L.L. Liao, G.M. Cao, Y.X. Jiang, et al., J. Am. Chem. Soc. 143 (2021) 2812–2821. doi: 10.1021/jacs.0c11896
J. Hou, A. Ee, H. Cao, et al., Angew. Chem. Int. Ed. 57 (2018) 17220–17224. doi: 10.1002/anie.201811266
J.H. Ye, M. Miao, H. Huang, et al., Angew. Chem. Int. Ed. 56 (2017) 15416–15420. doi: 10.1002/anie.201707862
Q. Fu, Z.Y. Bo, J.H. Ye, et al., Nat. Commun. 10 (2019) 3592. doi: 10.1038/s41467-019-11528-8
W. Zhang, Z. Chen, Y.X. Jiang, et al., Nat. Commun. 14 (2023) 3529. doi: 10.1038/s41467-023-39240-8
T. Ju, Y.Q. Zhou, K.G. Cao, et al., Nat. Catal. 4 (2021) 304–311. doi: 10.1038/s41929-021-00594-1
J.P. Yue, J.C. Xu, H.T. Luo, et al., Nat. Catal. 6 (2023) 959–968. doi: 10.1038/s41929-023-01029-9
B. Zhang, T.T. Li, Z.C. Mao, et al., J. Am. Chem. Soc. 146 (2024) 1410–1422. doi: 10.1021/jacs.3c10439
J.C. Xu, J.P. Yue, M. Pan, et al., Nat. Commun. 16 (2025) 1850. doi: 10.1038/s41467-025-57060-w
S. Sarkar, K.P.S. Cheung, V. Gevorgyan, Angew. Chem. Int. Ed. 63 (2024) e202311972. doi: 10.1002/anie.202311972
M. Parasram, P. Chuentragool, D. Sarkar, V. Gevorgyan, J. Am. Chem. Soc. 138 (2016) 6340–6343. doi: 10.1021/jacs.6b01628
Y.C. Luo, F.F. Tong, Y. Zhang, C.Y. He, X.G. Zhang, J. Am. Chem. Soc. 143 (2021) 13971–13979. doi: 10.1021/jacs.1c07459
Y. Liang, T.C. Bian, K. Yadav, et al., ACS Cent. Sci. 10 (2024) 1191–1200. doi: 10.1021/acscentsci.4c00094
N. Oku, M. Murakami, T. Miura, Org. Lett. 24 (2022) 1616–1619. doi: 10.1021/acs.orglett.2c00121
H. Xin, L.N. Guo, M. Yang, et al., Org. Chem. Front. 10 (2023) 1147–1152. doi: 10.1039/d2qo02001g
J. Wang, Q.X. Zhou, L.J. Zhou, Z.X. Zhang, ACS Catal. 14 (2024) 18499–18506. doi: 10.1021/acscatal.4c06842
K. Yamada, K.P. Cheung, V. Gevorgyan, J. Am. Chem. Soc. 146 (2024) 18218–18223. doi: 10.1021/jacs.4c06421
N. Kvasovs, J. Fang, F. Kliuev, V. Gevorgyan, J. Am. Chem. Soc. 145 (2023) 18497–18505. doi: 10.1021/jacs.3c04968
X.Y. Ruan, D.X. Wu, W.A. Li, et al., J. Am. Chem. Soc. 146 (2024) 12053–12062. doi: 10.1021/jacs.4c01690
Y. Cai, G. Gaurav, T. Ritter, Angew. Chem. Int. Ed. 63 (2024) e202311250. doi: 10.1002/anie.202311250
S. Maiti, P. Ghosh, D. Raja, et al., Nat. Catal. 7 (2024) 285–294. doi: 10.1038/s41929-024-01109-4
P.S.K. Cheung, J. Fang, K. Mukherjee, A. Mihranyan, V. Gevorgyan, Science 378 (2022) 1207–1213. doi: 10.1126/science.abq1274
H.H. Han, W.Q. Yi, S.J. Ding, X.Y. Ren, B.G. Zhao, Angew. Chem. Int. Ed. 64 (2025) e202418910. doi: 10.1002/anie.202418910
Z.L. Liu, Z.P. Ye, Z.H. Liao, et al., ACS Catal. 14 (2024) 3725–3732. doi: 10.1021/acscatal.4c00470
H. Yu, Q.L. Zhang, W.W. Zi, Angew. Chem. Int. Ed. 61 (2022) e202208411. doi: 10.1002/anie.202208411
G.Z. Wang, R. Shang, W.M. Cheng, Y. Fu, J. Am. Chem. Soc. 139 (2017) 18307–18312. doi: 10.1021/jacs.7b10009
W. Yao, G.Y. Zhao, Y. Wu, et al., J. Am. Chem. Soc. 144 (2022) 3353–3359. doi: 10.1021/jacs.1c13299
D. Kurandina, M. Parasram, V. Gevorgyan, Angew. Chem. Int. Ed. 56 (2017) 14212–14216. doi: 10.1002/anie.201706554
T.Z. Zhang, M.Q. Shen, Q. Zhang, M.C. Fu, Org. Lett. 26 (2024) 8890–8898. doi: 10.1021/acs.orglett.4c03343
Y.J. Du, X.X. Sheng, J.H. Li, et al., Chem. Sci. 14 (2023) 3580–3586. doi: 10.1039/d2sc06852d
P. Chuentragool, D. Yadagiri, T. Morita, et al., Angew. Chem. Int. Ed. 58 (2019) 1794–1798. doi: 10.1002/anie.201812398
K.P.S. Cheung, D. Kurandina, T. Yata, V. Gevorgyan, J. Am. Chem. Soc. 142 (2020) 9932–9937. doi: 10.1021/jacs.0c03993
M. Ratushnyy, N. Kvasovs, S. Sarkar, V. Gevorgyan, Angew. Chem. Int. Ed. 59 (2020) 10316–10320. doi: 10.1002/anie.201915962
S. Yang, S.S. Cai, J.H. Li, M. Chen, J. Org. Chem. 89 (2024) 7243–7254. doi: 10.1021/acs.joc.4c00703
W.W. Ding, Y. Zhou, S. Song, Z.Y. Han, Org. Lett. 24 (2022) 7350–7354. doi: 10.1021/acs.orglett.2c02877
X.X. Sheng, Y.J. Du, J.H. Li, Q.Q. Teng, M. Chen, Org. Lett. 25 (2023) 3664–3669. doi: 10.1021/acs.orglett.3c01030
W.W. Jin, S.Y. Yu, Org. Lett. 23 (2021) 6931–6935. doi: 10.1021/acs.orglett.1c02509
Z.Y. Zhang, N. Kvasovs, A. Dubrovina, V. Gevorgyan, Angew. Chem. Int. Ed. 61 (2022) e202110924. doi: 10.1002/anie.202110924
Z.L. Liu, J.L. Yan, K. Chen, H.Y. Xiang, H. Yang, Org. Lett. 26 (2024) 8762–8767. doi: 10.1021/acs.orglett.4c03080
N. Kvasovs, V. Gevorgyan, Org. Lett. 24 (2022) 4176–4181. doi: 10.1021/acs.orglett.2c01409
H.M. Huang, M. Koy, E. Serrano, et al., Nat. Catal. 3 (2020) 393–400. doi: 10.1038/s41929-020-0434-0
K. Ⅲ. Tanaka, ACS Catal. 14 (2024) 5269–5274. doi: 10.1021/acscatal.4c00510
M.Z. Lu, J. Goh, M. Maraswami, et al., Chem. Rev. 122 (2022) 17479–17646. doi: 10.1021/acs.chemrev.2c00032
T.U. Sastry, K.N. Rao, T.A. Reddy, P. Gandhi, Asian J. Org. Chem. 26 (2014) 2417–2421. doi: 10.14233/ajchem.2014.16137
C.A. Miller, L.M. Long, J. Am. Chem. Soc. 73 (1951) 4895–4898. doi: 10.1021/ja01154a126
W. Zhou, I.A. Dmitriev, P. Melchiorre, J. Am. Chem. Soc. 145 (2023) 25098–25102. doi: 10.1021/jacs.3c11285
C.H. Song, X.H. Bai, B. Li, Y.F. Dang, S.Y. Yu, J. Am. Chem. Soc. 146 (2024) 21137–21146. doi: 10.1021/jacs.4c07126
C.H. Song, S.Y. Yu, ACS Catal. 14 (2024) 15997–16002. doi: 10.1021/acscatal.4c04956

Scheme 2 Substrate scope of electron-deficient alkenes. Reaction conditions: S1 or S3 (0.2 mmol), S2 (0.4 mmol), Pd(PPh3)4 (10 mol%), PPh3 (30 mol%), Cs2CO3 (2.0 equiv.), ZnCl2 (2.0 equiv.), HMPA (2.0 mL), irradiation with 10 W 455 nm blue LEDs at 40–50 ℃ under CO2 (1 atm) for 16 h, and then MeI (3.0 equiv.) was added to react at room temperature for 4 h. Isolated yields. Unless otherwise stated, only E products were observed. a Pd(tBu3P)2 (10 mol%) was used as a catalyst, P(4-OMeC6H4)3 (15 mol%) was used as a ligand and 2.0 equiv. of NH4Cl was used. b Cs2CO3 (1.0 equiv.) was used. c The ester exchange product. d Diastereoselectivity was determined through 1H NMR.

Scheme 3 Substrate scope of vinyl aromatics (heteroarenes). Reaction conditions: S1 (0.2 mmol), S2 (0.4 mmol), Pd(PPh3)4 (10 mol%), PPh3 (30 mol%), Cs2CO3 (2.0 equiv.), ZnCl2 (2.0 equiv.), HMPA (2.0 mL), irradiation with 10 W 455 nm blue LEDs at 40–50 ℃ under CO2 (1 atm) for 16 h, and then MeI (3.0 equiv.) was added to react at room temperature for 4 h. Isolated yields. Unless otherwise stated, only E products were observed. a Pd(PPh3)4 (5 mol%) was used and 12 h. b A mixture composed of the target product and a dechlorination byproduct (for details, see Supporting information). c Pd(tBu3P)2 (10 mol%) was used as a catalyst, P(4-OMeC6H4)3 (15 mol%) was used as a ligand and 2.0 equiv. of NH4Cl was used.

Scheme 4 Late-stage modification of natural products and drug derivatives. Reaction conditions: S1 (0.2 mmol), S2 (0.4 mmol), Pd(PPh3)4 (10 mol%), PPh3 (30 mol%), Cs2CO3 (2.0 equiv.), ZnCl2 (2.0 equiv.), HMPA (2.0 mL), irradiation with 10 W 455 nm blue LEDs at 40–50 ℃ under CO2 (1 atm) for 16 h, and then MeI (3.0 equiv.) was added to react at room temperature for 4 h. Isolated yields. Unless otherwise stated, only E products were observed. a Pd(tBu3P)2 (10 mol%) was used as a catalyst, P(4-OMeC6H4)3 (15 mol%) was used as a ligand and 2.0 equiv. of NH4Cl was used.

Figure 1 Synthetic applications. Reaction conditions see Supporting information for experimental details. (a) cis-Octahydro-1H-isoindole, EDCI, HOBt, DMF, 70 ℃, 8 h; (b) Pd/C, H2 (1 atm), MeOH, r.t., 24 h; (c) NaOH, H2O, MeOH, 45 ℃, 4 h; (d) N-hydroxybenzimidoyl chloride, Et3N, toluene, 4 Å molecular sieves, reflux, 24 h.
Table 1. Optimization of reaction conditions.a
|
|
||
| Entry | Variations from standard conditions | Yield (%) |
| 1 | None | 78 (70)b |
| 2 | Pd(PPh3)4 (5 mol%), PPh3 (15 mol%) | 71 |
| 3 | Pd(OAc)2 as Pd catalyst | 32 |
| 4 | Xantphos as a ligand | N.D. |
| 5 | P(4-OMeC6H4)3 as a ligand | 45 |
| 6 | PCy3 as a ligand | N.D. |
| 7 | DMSO as solvent | 25 |
| 8 | DMAc as solvent | 34 |
| 9 | K2CO3 as base | 61 |
| 10 | without ligand | 43 |
| 11 | without ZnCl2 | 59 |
| 12 | without Cs2CO3 | 48 |
| 13 | S1:S2 = 1:1 | 52 |
| 14 | no light, up to 160 ℃ | N.D. |
| 15 | without Pd(PPh3)4 | 0 |
| a Reaction conditions: S1 (0.2 mmol, 1.0 equiv.), S2 (0.4 mmol, 2.0 equiv.), Pd(PPh 3) 4 (10 mol%), Cs 2CO 3 (0.4 mmol, 2.0 equiv.), ZnCl 2 (0.4 mmol, 2.0 equiv.), HMPA (2.0 mL), irradiation with 10 W 455 nm blue LEDs at 40–50 ℃ under CO 2 (1 atm) for 16 h, and then MeI (0.6 mmol, 3.0 equiv.) was added to react at room temperature for 4 h. N.D. = Not detected. DMSO = Dimethyl sulfoxide. DMAc = N, N-dimethylacetamide. HMPA = Hexamethylphosphoramide. b Yields determined by GC analysis. Isolated yields in the parentheses. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: