Received Date:
19 February 2025 Accepted Date:
29 April 2025 Revised Date:
16 April 2025 Available Online:
15 April 2026
Abstract:
Hierarchical assembly provides a rational procedure to acquire complex supramolecular architectures from basic building blocks. In this work, a novel kind of double-stranded polyrotaxane motif was reported by Ag+-directed coordination-driven assembly following the preassembly of a four-connected pseudorotaxane (cpb)2CB8 linker. Moreover, supramolecular isomerism is observed in crystalline compounds based on double-stranded polyrotaxane motifs due to differences in lattice stacking mode. Interestingly, the resultant supramolecular isomers, cross-Ag-DSP-1 and para-Ag-DSP-1, show dual thermo- and anion-responsiveness. Benefiting from high crystallinity of these coordination assemblies, a combination of characterization techniques, especially X-ray diffraction, was used to unveil precise molecular mechanisms related to the inherent dynamic behavior of these assemblies, which can be attributed to remarkable lattice rearrangement and crystal transformations as temperature increases or after anion exchange, reflecting the adaptive adjustment ability of these supramolecular architectures in response to external stimuli. Based on the anion exchange capability, these two supramolecular materials show fast removal kinetics and high sorption capacity for perrhenate (ReO4-) anion, a surrogate of radioactive pertechnetate (TcO4-) in nuclear waste eluents. This work provides a feasible way to supramolecular assemblies with customized structures and stimuli-responsiveness, and is helpful to design and synthesize more functional supramolecular systems with complex structures and tailored functions.
Self-assembly is a spontaneous organization process of discrete subcomponents into large well-defined aggregates through a collection of weak noncovalent interactions such as hydrogen bonds, halogen bonds, aromatic π-π stacking, metal-ligand coordination [1-4]. Such processes are ubiquitous throughout biological systems, and contribute to the formation of a plethora of complex biological structures with diverse biochemical functions at molecular and cellular levels from relatively simple subunits [5-8]. Inspired by the principle of self-assembly in natural systems, supramolecular chemists also harness self-assembly for chemical synthesis to create a variety of intriguing artificial or biomimetic assembling structures, ranging from host-guest complexes driven by a mechanism of molecular recognition [9-13] to sophisticated chemical architectures [14-16] and high-order crystalline systems [17-20]. Especially, supramolecular architectures of varying complexity can be constructed through hierarchical assembly from the basic building blocks [21-24], which provides a rational procedure for assessing more complex superstructures.
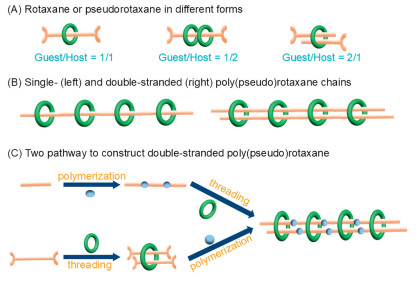
Crucial to hierarchical assembly is the designation and selection of functional chemical systems capable of facilitating the expected assembly process to form new aggregates through mutual recognition interactions between different subcomponents. Rotaxanes and pseudorotaxanes [9,25-28] are typical host-guest assembling systems in different mechanically-interlocked forms (Scheme 1A). Benefiting from non-covalent nature of host-guest complexing, (pseudo)rotaxane motifs exhibit good structural dynamics and can serve as good structural units or prototypes for further higher-order assembly of poly(pseudo)rotaxanes with different forms and functions, such as a single-stranded covalent-organic or metal-organic one-dimensional (1D) chain threaded by a collection of macrocyclic 'beads' (left diagram in Scheme 1B) [29-32], or a two- or three-dimensional network [33-37] with single-stranded linkers resembling them. A more complex variant of single-stranded poly(pseudo)rotaxane chain is a multi-stranded chain-based poly(pseudo)rotaxane with a set of polymer chains as main backbones tied by surrounding 'beads' (right diagram in Scheme 1B), resembling the topological structure of double-stranded chains of DNA in biological systems. Such kinds of double-stranded poly(pseudo)rotaxanes have been fabricated through polythreading of a couple of poly(ethylene glycol) [38,39] or poly(4-vinylpyridine) chains [40] by an array of macrocyclic hosts with large cavities with two possible pathways, i.e. polymerization followed by threading and prethreading prior to polymerization (Scheme 1C) [41-45], and the double-threaded crosslinker has been proven to be useful for improving the mechanical properties of polymer networks [40,46,47]. However, the reported poly(pseudo)rotaxane systems are all based on organic polymers without long-range order, while multi-stranded poly(pseudo)rotaxanes in crystalline state are still lacking, and desirable acquisition of precise molecular structures on these exquisite architectures remains elusive.
Scheme 1
Scheme 1.
Poly(pseudo)rotaxanes based on (pseudo)rotaxane units: (A) Rotaxanes and pseudorotaxanes in different mechanically-interlocked forms with varying host-guest ratios. (B) Schematic diagram of single-stranded and double-stranded poly(pseudo)rotaxane chains. (C) Two possible pathways, i.e., polymerization followed by threading and prethreading prior to polymerization, used to construct double-stranded poly(pseudo)rotaxane chains.
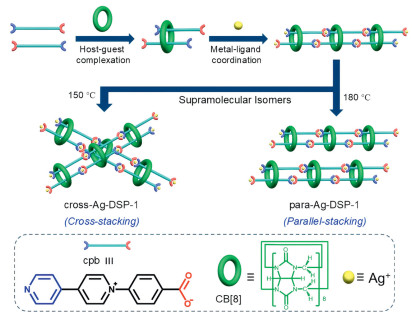
Here in, to construct double-stranded poly(pseudo)rotaxanes with high crystallinity, we propose a method of coordination-driven assembly [21,48-52], where a doubly threaded [3]pseudorotaxane linker (denoted as (cpb)2—CB8) composed of a pair of bifunctional guest components (1-(4-carboxyphenyl)-[4,4′-bipyridin]−1-ium, cpb, Scheme S1 and Figs. S1 and S2 in Supporting information) and a macrocyclic cucurbit[8]uril (CB8) host (Fig. S3 in Supporting information) that serves as a four-connected unit is linked by silver ion (Ag+) nodes by metal-ligand bonding. As expected, crystalline compounds consisting of silver-bridged double-stranded polyrotaxane chains were obtained successfully. More interestingly, on the basis of the same kind of double-stranded polyrotaxane chain, a pair of supramolecular isomers, cross-Ag-DSP-1 (cross-stacking) and para-Ag-DSP-1 (parallel-stacking), can be prepared from different conditions (Fig. 1), and both crystalline isomers show dual thermo- and anion-responsiveness. Benefiting from high crystallinity of these coordination assemblies, we could unveil precise molecular information related to inherent dynamic behavior of these assemblies by combined characterization techniques, especially X-ray diffraction, which are attributed to remarkable lattice rearrangement and crystal transformations as temperature increases or after anion exchange. Furthermore, based on the anion-responsive capability of these two supramolecular materials, their removal performance for perrhenate (ReO4-) anion, i.e., a surrogate of radioactive pertechnetate (TcO4-) in nuclear waste eluents, were also evaluated and discussed.
Figure 1
Figure 1.
Coordination-driven assembly of supramolecular isomers of double-stranded polyrotaxanes with different lattice stacking modes, cross-Ag-DSP-1 (cross-stacking) and para-Ag-DSP-1 (parallel-stacking) through metal-ligand coordination between a doubly threaded [3]pseudorotaxane linker, (cpb)2—CB8) and Ag+ ion.
According to the proposed coordination-driven assembly method for synthesizing double-stranded polyrotaxane chain, the design of four-connected doubly threaded [3]pseudorotaxane linker, (cpb)2—CB8, is the starting point for subsequent metal coordination (Fig. 1), and both host and guest components in (cpb)2—CB8 are crucial for the fulfillment of expected coordination capability. First, macrocyclic CB8 has a large internal cavity that can accommodate two guest molecules [35,53-55]. Isothermal titration calorimetry (ITC) experiment was conducted to verify stoichiometric ratios and the binding affinity of CB8 with cpb. The ITC data were well-fitted using a sequential two-site binding model, revealing a 1:2 host-guest stoichiometric ratio between CB8 and cpb. Thermodynamic analysis from ITC showed that the host-guest complexation is primarily enthalpy-driven. The first binding constant Ka1 was determined to be 3.075 × 105 L/mol and the second binding constant Ka2 was 1.520 × 104 L/mol, giving an overall binding constant of CB8 is 4.674 × 109 L/mol and indicating high stability of this ternary complex (Fig. S5 in Supporting information) [56-58]. Meanwhile, the other important component is the guest molecule, which must take both functions of macrocyclic inclusion and metal coordination. The cpb unit that is composed of both bipyridinium and 4-carboxyphenyl groups should be capable of conducting the above desirable functions: (i) The monovalent N-aryl bipyridinyl unit can be encapsulated in pairs in the macrocyclic cavity of CB8, which was proven by doubly threaded structure of [3]pseudorotaxane observed in crystal structure of (cpb)2—CB8 with a distance of ~3.46 Å between bipyridinyl units from the pair of cpb guest molecules encapsulated (Fig. S6 in Supporting information); (ii) The neutral pyridine moiety of monovalent N-aryl bipyridinyl unit and the 4-carboxyphenyl group at both ends of cpb retain their inherent metal-coordination ability, which thus provide a guarantee for further coordination assembly with Ag+ ion.
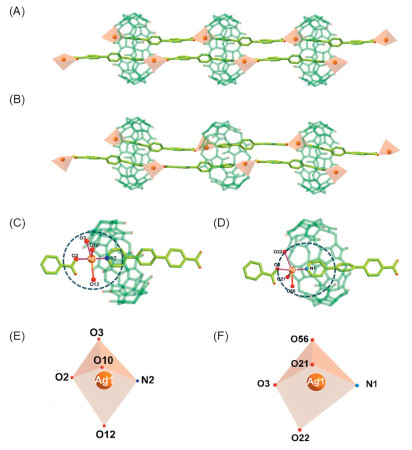
Following the identification of four-connected [3]pseudorotaxane ligand that forms based on cpb and CB8, Ag+ ion was further added to the mixture of cpb and CB8 under hydrothermal conditions, and two supramolecular isomers, cross-Ag-DSP-1 and para-Ag-DSP-1 were obtained at different reaction temperatures (Fig. S7 and Table S1 in Supporting information). It is found that, both cross-Ag-DSP-1 and para-Ag-DSP-1 contain the same primary structures of silver-coordination-directed double-stranded polyrotaxane chains (Figs. 2A and B), but have different stacking modes in the final crystal lattices. Attempts to synthesize double-stranded polyrotaxane compounds with other metal ions (Co2+, Ni2+, Cu2+, or Zn2+) instead of Ag+ ion proved to be unfeasible, illustrating the importance of Ag+ ion that might be more suitable for simultaneous coordination of cpb and CB8 ligands in (cpb)2—CB8. To further elucidate the role of Ag+ ion as a metal node for the formation of double stranded polyrotaxane chains, a direct comparison in assembly mode of such double-stranded polyrotaxane chains with the single-stranded polyrotaxane chain constructed by coordination assembly of the 1:2 host-guest complex, (bpy)2—CB8, with Cu2+ node (bpy, 4,4′-bipyridine) (Fig. S8 in Supporting information) [55] is conducted. Unlike the head-to-head coordination mode for [3]pseudorotaxane linker in single-stranded polyrotaxane chain, all the double-stranded polyrotaxane chains take a different head-to-tail pattern when coordinating to Ag+ node. Obviously, compared to bipyridine ligand for single-stranded polyrotaxane chain, the extended carbon chain at one end of the carboxylic acid allows the two adjacent macrocyclic CB8 molecules to remain far enough apart without spatial hindrance, while the high polarizability of Ag+ ions involved in the carboxyl coordination allows them to further coordinate with the pyridine N-donor embedded within the CB8 cavity (Fig. S9 in Supporting information), ensuring the formation of double-stranded chains.
Figure 2
Figure 2.
Double-stranded polyrotaxane structural units in cross-Ag-DSP-1 (A, C, E) and para-Ag-DSP-1 (B, D, F): molecular structures of double-stranded polyrotaxane chains (A, B), Coordination spheres (C, D) and geometric conformations (E, F) of Ag+ nodes (Ag1).
Besides a pyridine N-donor from the encapsulated tail part of one cpb molecule and a η1- or η2-carboxyl group from the protruding head part of another cpb molecule, the coordination sphere of Ag+ also contains one water molecule and two carbonyl groups from the adjacent CB8 macrocycle (Figs. 2C and D, and Fig. S10 in Supporting information), resulting in a distorted configuration of a quadrangular pyramid or pentagonal pyramid (Figs. 2E and F). It can be seen that Ag-N and Ag-O bonds between Ag+ and cpb linker are in the range of 2.187–2.238 Å (the longer carboxyl O atom of η2-carboxyl group is not included), which are much shorter than those between Ag+ and carbonyl groups of CB8 macrocycle or the only water molecule ligand (Table S2 in Supporting information). Based on the bonding feature shown above, it can be speculated that cpb linker that has higher affinity with Ag+ contributes mostly to the formation of double-stranded polyrotaxane chains, while CB8 macrocycle, together with the only water molecule ligand, plays an auxiliary role in Ag+ coordination, and helps to improve the structural stability of polyrotaxane chains.
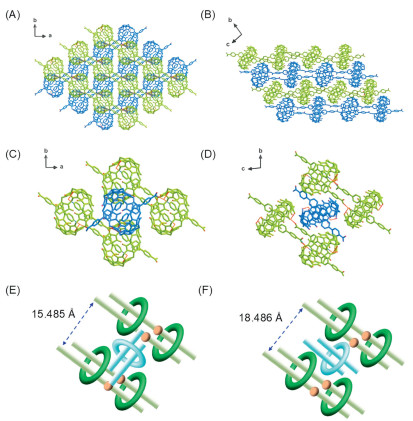
Another interesting phenomenon worth discussing is the occurrence of supramolecular isomerism [59], i.e., two different stacking modes (i.e., cross-stacking and parallel-stacking) based on the same double-stranded polyrotaxane chains as building units in cross-Ag-DSP-1 and para-Ag-DSP-1 (Figs. 3A–D and Fig. S11 in Supporting information), especially the cross-stacking mode, which is quite rare in cucurbituril-based metal-organic coordination poly(pseudo)rotaxanes. As indicated by the analysis result that the calculated lattice density of cross-Ag-DSP-1 in cross-stacking mode (~1.542 g/cm3) is slightly larger than that of its parallel-stacking counterpart para-Ag-DSP-1 (~1.385 g/cm3), cross-stacking should be more in line with the requirements of dense stacking. It is also reflected in microscopic lattice stacking by an observation that there is shorter spacing between two adjacent polyrotaxane chains within a layer in cross-Ag-DSP-1 (15.485 Å) than para-Ag-DSP-1 (18.486 Å) (Figs. 3E and F). The difference in cross-Ag-DSP-1 and para-Ag-DSP-1 suggests that the ordered arrangement will affect the density of lattice stacking, and the cross-stacking mode is more conducive to dense packing, which may be related to the fact that the cross-stacking mode can effectively reduce the steric hindrance caused by bulky macrocyclic CB8 molecules in the (cpb)2—CB8 (pseudo)rotaxane unit by taking a staggered orientation rather than a parallel one. From the perspective of synthesis, since it is not easy to form a denser structure at high temperature due to more intense molecular movement, it seems to be reasonable to obtain a loose structure at high temperature (180 ℃), and decreasing the reaction temperature (150 ℃) can help to promote the formation of a denser structure. Another kind of important components within the crystal lattices of cross-Ag-DSP-1 and para-Ag-DSP-1 are lattice water molecules, which occupy the free space between or within the polyrotaxane chains and help to stabilize the whole lattice (Figs. S12 and S13 in Supporting information). Although lattice water molecules are involved in the crystallization process and contribute to the lattice stability, their participation in the crystal lattice is premised on the formation of the supramolecular main backbone and should have little to do with the regulation of supramolecular isomers.
Figure 3
Figure 3.
Different stacking modes in cross-Ag-DSP-1 (A, C, E) and para-Ag-DSP-1 (B, D, F): crystal strucutures of cross-Ag-DSP-1 (A) and para-Ag-DSP-1 (B); local enlarged structures showing relative orientation and spatial relationship of adjcent double-stranded polyrotaxane chains (chains in green and blue denotes two different layers) in cross-Ag-DSP-1 (C) and para-Ag-DSP-1 (D); a comparision of spacing between two adjacent polyrotaxane chains within a layer in cross-Ag-DSP-1 (E) and para-Ag-DSP-1 (F).
Mechanically-interlocked molecules and related materials demonstrate significant potential in constructing complex dynamic molecular machines responsive to temperature and other stimuli [60-64]. The special lattice stacking modes of cross-Ag-DSP-1 and para-Ag-DSP-1 motivate us to explore their possible stimulus responsiveness, and the effect of temperature on their crystal structures was first studied. As indicated by variable-temperature single-crystal X-ray diffraction (VT-SCXRD) analysis at a series of preset temperature points, 150, 210, 230, 250, 260, 270, 280, 290 and 300 K, both compounds undergo interesting temperature-dependent SCSC transformation. Moreover, due to the difference in stacking mode, the structural transformation paths of these two supramolecular isomers are significantly different (Table S3 in Supporting information). To better understand this temperature-responsive process and differences between these two isomers, SCSC transformations of both compounds will be elaborated below.
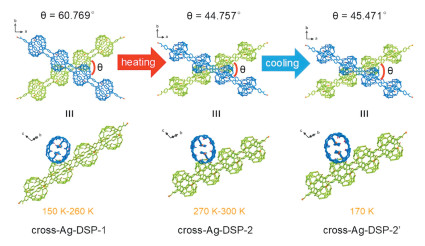
For cross-Ag-DSP-1, SCSC transformation to a new phase cross-Ag-DSP-2 is observed when the temperature increases to 270 K, and is always maintained within the range of 270–300 K (Fig. 4 and Table S3 in Supporting information). Although the space group does not change, all three axes, a, b and c, change significantly. Crystal structure analysis reveals that, there is little change in the lattice stacking mode, and specifically, the angle (θ) between two staggered chains undergoes a change from 60.769° to 44.757°. Further component analyses of crystal structures of cross-Ag-DSP-1 and cross-Ag-DSP-2 show that there is a loss of a large amount of lattice water in the heating process. Considering the dehydration of cross-Ag-DSP-1 will induce subsequent structural arrangement, the heating-triggered dehydration process is probably the origin of SCSC transformation observed here. Moreover, the SCSC transformation is an irreversible process, which is evidenced by the structure of cross-Ag-DSP-2′ that was collected by recooling the sample of cross-Ag-DSP-2 to 170 K. A comprehensive comparison between cross-Ag-DSP-2′ and cross-Ag-DSP-2 shows there is no significant change between these two compounds (θ = 45.471° in cross-Ag-DSP-2′ vs. θ = 44.757° in cross-Ag-DSP-2). Actually, the irreversibility of crystal transformation observed here is justified given the difficulty of regaining water after dehydration of cross-Ag-DSP-1.
Figure 4
Figure 4.
The heating-triggered irreversible SCSC transformation of cross-Ag-DSP-1: A transformation from cross-Ag-DSP-1 within 150–260 K to cross-Ag-DSP-2 at 270–300 K with a remarkable phase transition; foramtion of cross-Ag-DSP-2′ which has no signigicant change from cross-Ag-DSP-2 after recooling to 170 K.
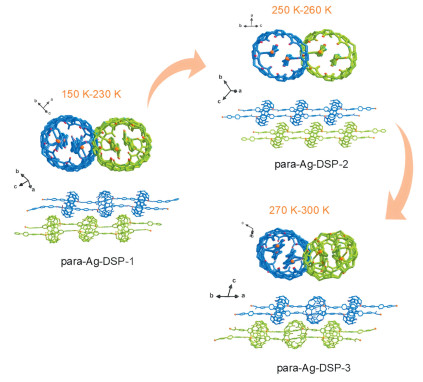
Similar temperature-triggered SCSC transformation can be seen for para-Ag-DSP-1, but the transformation process is more complex than that of cross-Ag-DSP-1 (Fig. 5). As the temperature increases from 150 K to 300 K, para-Ag-DSP-1 undergoes two sequential crystal transformation processes. The first one occurs at temperatures over 250 K, and the double-stranded polyrotaxane chains become more straight in the resultant intermediate compound, para-Ag-DSP-2. When temperature goes up over 270 K, a second transformation to a new phase para-Ag-DSP-3 is observed, where significant lattice shrinkage can be observed (cell volume decrease from 11,329 Å3 (150 K) to 9352 Å3 (290 K)), and the adjacent double-stranded polyrotaxane chains seem to be getting closer to each other. Detailed analysis and comparison of crystal structures suggest that, like the case in cross-Ag-DSP-1, the stepwise SCSC transformations observed here might be also attributed to the loss of lattice water induced by the increase of temperature, as there was a noticeable change in the amount of lattice water molecules before and after SCSC transformation.
Figure 5
Figure 5.
Stepwise SCSC transformation of para-Ag-DSP-1 within the temperature range of 150 K-300 K from para-Ag-DSP-1 (150 K-230 K) to para-Ag-DSP-2 (250 K-260 K), then to para-Ag-DSP-3 (270 K-300 K).
Furthermore, differential scanning calorimetry (DSC) analyses of cross-Ag-DSP-1 and para-Ag-DSP-1 were conducted within the temperature range from 173.15 K (i.e., −100 ℃) to 423.15 K (i.e., 150 ℃) to track the heat flow during crystal transformation (Fig. S14 in Supporting information), but the results show that significant heat flow can only be observed at the temperature range over room temperature (also corresponding to a dehydration process as suggested by the thermogravimetry analysis (TGA) result (Fig. S15 in Supporting information), but weight loss is significantly more than that for crystal transformation occurring below room temperature). The absence of observable heat flow in DSC diagram may be because, despite inducing SCSC transformations, the water loss process is not that violent and cannot be detected by DSC analyses, resembling the case observed in other thermo-responsive compounds [65,66].
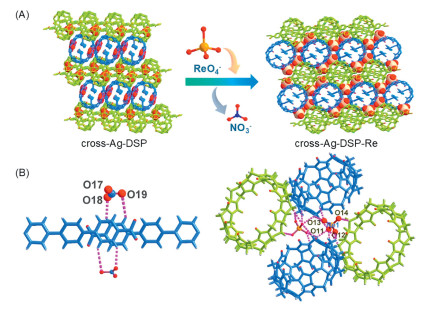
Meanwhile, it is found that, silver-directed double-stranded polyrotaxane chains and the resultant supramolecular networks in cross-stacking or parallel-stacking mode are both of cationic nature, accompanied by NO3- as counter anions in the final crystalline supramolecular materials, which make it possible for these materials to have potential ion exchange capacity. Moreover, the CH-rich outer surfaces of cucurbit[n]uril molecules like CB8 can interact with oxoanions through hydrogen bonds for stabilization of anions involved [55]. The above characteristics of cross-Ag-DSP and para-Ag-DSP make them promising candidate materials for separation of anions of interest through ion exchange [67-69]. Therefore, the anion exchange capability of these two cationic supramolecular networks is assessed. After immersing crystal samples into a solution of ReO4-, these samples underwent corresponding structure transformation triggered by an anion-exchange process, which were evidenced by single-crystal diffraction analyses of the resultant products, cross-Ag-DSP-Re and para-Ag-DSP-Re, from corresponding cationic supramolecular networks (Fig. 6 and Fig. S16 in Supporting information).
Figure 6
Figure 6.
Structure transformation from cross-Ag-DSP to cross-Ag-DSP-Re triggered by anion exchange between NO3- and ReO4-. (A) Lattice packing diagrams before and after anion exchange. Hydrogen interactions of NO3- (B) and ReO4- (C) with surrounding C—H groups.
As shown in Fig. 6A, NO3- anions in cross-stacking supramolecular networks of double-stranded polyrotaxane chains (denoted as cross-Ag-DSP, referring to this type of material in general without assigning specific temperature-dependent crystal phases) are all replaced by ReO4- to form a Re-containing cross-Ag-DSP-Re, among which ReO4- anions occupy different void sites than NO3- through enhanced hydrogen bonds with surrounding C—H groups from macrocyclic CB8 molecules (Figs. 6B and C, and Table S4 in Supporting information). In cross-Ag-DSP-Re, the angle (θ) between two staggered chains is 63.806° (Fig. S17 in Supporting information), which is similar to that of cross-Ag-DSP-1, but still shows a notable difference after replacing NO3- with ReO4- (Fig. S18 in Supporting information). Resembling the cross-Ag-DSP sample, the parallel-stacking supramolecular network (also denoted as para-Ag-DSP including all three temperature-dependent crystal phases) underwent a similar anion exchange process upon interacting with the aqueous solution of ReO4- (Fig. S16 in Supporting information). Interestingly, although the double-stranded polyrotaxane chains in the resultant compound para-Ag-DSP-Re are also a bit twisted in geometric structure like that observed in para-Ag-DSP-1, there are still significant differences in the relative orientation and packing patterns of macrocyclic molecules on adjacent chains for these two compounds (Fig. S19 in Supporting information). In all, the crystal transformation induced by ion exchange process observed here reflects the structural adaptability of supramolecular networks based on double-stranded polyrotaxane chains, which can take the corresponding structural adjustment according to different counter anions involved.
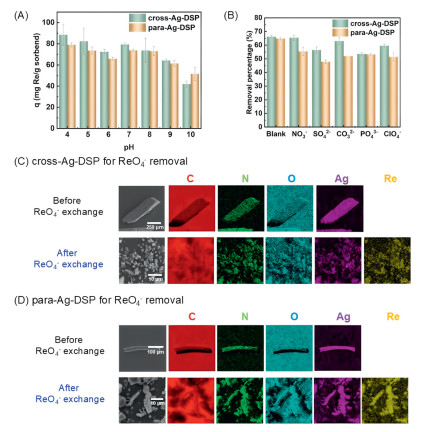
Inspired by structure transformation of cross-Ag-DSP and para-Ag-DSP samples triggered by ReO4- anion-exchange, we further assessed their performance as solid sorbents for the removal of ReO4-, a surrogate of radioactive TcO4- in the nuclear waste eluents. Stability test by soaking absorbents of cross-Ag-DSP and para-Ag-DSP in aqueous solutions of different pH ranges from 2–12 was first conducted, which suggests these two supramolecular materials are stable within the pH range of 4–10 (Fig. S20 in Supporting information). Batch experiment reveals that both materials reach adsorption equilibrium within 2 h, and over 80% of the saturated sorption capacity is achieved at 5 min (Fig. S21 in Supporting information). Between pH 4 and 8, the adsorption performances of the materials do not change much, and are the best at pH 4, which should be attributed to the difference in surface zeta potential for both materials (Fig. S22 in Supporting information). When pH is increased to 10, a significant decrease in removal capacity is observed (Fig. 7A). Sorption isotherm experiments suggest that the sorption behavior of both materials to ReO4- follows the Langmuir model, and the calculated maximum removal capacities are 174 and 200 mg Re per gram of sorbent (Figs. S23 and S24 in Supporting information), both of which are prior to that of previously-reported SCP-IHEP-1 (157 mg Re per gram of sorbent) [55] and UiO-66-NH3+ (119 mg Re per gram of sorbent) [70]. Experimental study of removal selectivity shows that, in the presence of an equal equivalent of typical competing oxoanions such as NO3-, SO42-, CO32-, PO43-, ClO4-, the removal performance of cross-Ag-DSP to ReO4- is largely unaffected, while the removal rate of para decreases from 64% to 50% (Fig. 7B). Moreover, competitive removal experiments involving other anions such as F-, Cl-, Br-, and I- were also performed (Fig. S25 in Supporting information). The results indicate that the adsorption performance of the materials remains essentially unchanged in the presence of F- and I-; however, with the presence of Cl- and Br-, the removal rate of ReO4- could only reach about half of the blank condition. This phenomenon may be related to the higher charge density of Cl- and Br- compared to I-, as their higher charge density makes it easier for them to occupy adsorption sites through electrostatic interactions. Despite F- having a higher charge density, its strong hydration capacity (approximately 515 kJ/mol) results in the formation of a stable hydration layer in aqueous solutions, making it difficult to approach the surface of the adsorbent. Competitive removal experiments in the presence of metal ions including Eu3+, Pr3+, Nd3+, Gd3+, Dy3+, Th4+, Ce4+, and Cs+ (Fig. S26 in Supporting information) show that, for most lanthanide metal ions, the competitive effects of cations could be neglected, except Th4+, Ce4+ and Cs+, which might interact directly with ReO4- and affect its adsorption. The cycle experiment shows that, due to incomplete desorption efficiency, the recyclability of these two materials was prevented (Fig. S27 in Supporting information).
Figure 7
Figure 7.
Performance of cross-Ag-DSP and para-Ag-DSP for ReO4- removal. (A) Effect of solution pH on the removal performance. (B) Removal rate of ReO4- in the presence of competing ion such as NO3-, SO42-, CO32-, PO43-, or ClO4-. EDS analyses before and after ReO4- exchange for both samples of cross-Ag-DSP (C) and para-Ag-DSP (D).
The sorption of ReO4- can be evidenced by further characterization of the materials after ReO4- exchange. In addition to single-crystal diffraction analysis mentioned above, analyses of cross-Ag-DSP and para-Ag-DSP before and after ReO4- exchange by powder X-ray diffraction (PXRD) (Fig. S28 in Supporting information), UV–vis absorption spectra (Fig. S29 in Supporting information), Fourier transform infrared spectroscopy (FTIR) (Fig. S30 in Supporting information) and energy dispersive X-ray spectroscopy (EDS) (Figs. 7C and D) were conducted. PXRD analyses suggest that the crystal phases of cross-Ag-DSP and para-Ag-DSP after ReO4- exchange are consistent with para-Ag-DSP-Re and cross-Ag-DSP-Re. Specifically, recognition of ReO₄- is achieved through synergistic interactions, which, in addition to electrostatic interactions, include multiple hydrogen bonding interactions to form specific recognition sites for ReO₄-. This enhances the binding capability and separation selectivity of ReO₄- after ion exchange. In this material, the CB8 macrocyclic cavity constructs the material through supramolecular inclusion, with its external alkyl side chains -CH/-CH₂ forming a hydrophobic microenvironment to recognize and accommodate ReO₄- ions. The Re-O vibration peak observed in FTIR confirms the successful inclusion of ReO4-. EDS analyses for both samples of cross-Ag-DSP and para-Ag-DSP indicate the presence of Re element after ReO4- exchange, accompanied by the shattering of block crystals into small pieces.
As for the thermodynamic driving force for ReO₄- ion exchange, it should be have the same origin as the case of ReO₄--adaptive CB8-based single-stranded polyrotaxane reported previously [55]. In detail, the difference of anions in hydration energy significantly influences the exchange selectivity. According to the Hofmeister effect [71], anions with lower hydration energy, due to their weak hydration shell characteristics, face smaller desolvation energy barriers during the ion exchange process, making them more easily selectively captured by hydrophobic solid-phase materials. Compared to anions like NO₃- (−306 kJ/mol) and SO₄²- (−1090 kJ/mol), ReO₄- (−215 kJ/mol) has lower hydration energy, and its hydrophobic nature gives it a competitive advantage for preferential adsorption.
In summary, by coordination-driven assembly between a four-connected pseudorotaxane (cpb)2—CB8 linker and Ag+ ion with high polarizability and deformability, a novel kind of crystalline double-stranded polyrotaxane compounds with supramolecular isomerism in lattice stacking mode were reported here. The resultant supramolecular isomers, cross-Ag-DSP-1 and para-Ag-DSP-1, show intriguing structure transformations when stimulated by temperature change and anion exchange, which reflects the adaptive adjustment ability of these supramolecular architectures in response to external stimuli. Based on the anion exchange capability of cross-Ag-DSP-1 and para-Ag-DSP-1, these two crystalline supramolecular materials show fast removal kinetics and high sorption amount to ReO4- ion, demonstrating their potentials as candidate sorbents for the removal of ReO4-/TcO4- ion. To achieve the widespread application of such materials in the field of radioactive anion separation, improving their recycling performance will be one of the key directions for future research. This work provides a feasible way to supramolecular assemblies with customized structures and stimuli-responsiveness, and will inspire the development and synthesis of more functional supramolecular systems with complex structures and tailored functions in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Y. Liu, Y. Yu, J. Gao, et al., Angew. Chem. Int. Ed. 49 (2010) 6576–6579. doi: 10.1002/anie.201002415
Scheme 1
Poly(pseudo)rotaxanes based on (pseudo)rotaxane units: (A) Rotaxanes and pseudorotaxanes in different mechanically-interlocked forms with varying host-guest ratios. (B) Schematic diagram of single-stranded and double-stranded poly(pseudo)rotaxane chains. (C) Two possible pathways, i.e., polymerization followed by threading and prethreading prior to polymerization, used to construct double-stranded poly(pseudo)rotaxane chains.
Figure 1
Coordination-driven assembly of supramolecular isomers of double-stranded polyrotaxanes with different lattice stacking modes, cross-Ag-DSP-1 (cross-stacking) and para-Ag-DSP-1 (parallel-stacking) through metal-ligand coordination between a doubly threaded [3]pseudorotaxane linker, (cpb)2—CB8) and Ag+ ion.
Figure 3
Different stacking modes in cross-Ag-DSP-1 (A, C, E) and para-Ag-DSP-1 (B, D, F): crystal strucutures of cross-Ag-DSP-1 (A) and para-Ag-DSP-1 (B); local enlarged structures showing relative orientation and spatial relationship of adjcent double-stranded polyrotaxane chains (chains in green and blue denotes two different layers) in cross-Ag-DSP-1 (C) and para-Ag-DSP-1 (D); a comparision of spacing between two adjacent polyrotaxane chains within a layer in cross-Ag-DSP-1 (E) and para-Ag-DSP-1 (F).
Figure 4
The heating-triggered irreversible SCSC transformation of cross-Ag-DSP-1: A transformation from cross-Ag-DSP-1 within 150–260 K to cross-Ag-DSP-2 at 270–300 K with a remarkable phase transition; foramtion of cross-Ag-DSP-2′ which has no signigicant change from cross-Ag-DSP-2 after recooling to 170 K.
Figure 5
Stepwise SCSC transformation of para-Ag-DSP-1 within the temperature range of 150 K-300 K from para-Ag-DSP-1 (150 K-230 K) to para-Ag-DSP-2 (250 K-260 K), then to para-Ag-DSP-3 (270 K-300 K).
Figure 6
Structure transformation from cross-Ag-DSP to cross-Ag-DSP-Re triggered by anion exchange between NO3- and ReO4-. (A) Lattice packing diagrams before and after anion exchange. Hydrogen interactions of NO3- (B) and ReO4- (C) with surrounding C—H groups.
Figure 7
Performance of cross-Ag-DSP and para-Ag-DSP for ReO4- removal. (A) Effect of solution pH on the removal performance. (B) Removal rate of ReO4- in the presence of competing ion such as NO3-, SO42-, CO32-, PO43-, or ClO4-. EDS analyses before and after ReO4- exchange for both samples of cross-Ag-DSP (C) and para-Ag-DSP (D).

 DownLoad:
DownLoad:

 下载:
下载:









 下载:
下载:
