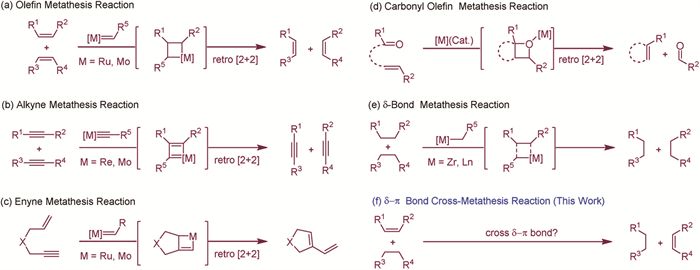
Scheme 1.
Different types of metathesis reactions.
A σ-π bond interchange assisted σ-π cross-metathesis reaction for nonoxidative conversion of methyl ketones to carboxylic acids
Huidan Geng , Jiuwei Nie , Wei Chen , Chong Zhao , Zikun Yao , Hui Wang , Jiquan Zhang , Lei Tang , Yuanyong Yang

The alkene metathesis reaction is one of the most powerful C—C bond-forming reactions in organic chemistry [1,2], which has been applied extensively in the total synthesis of natural products and pharmaceuticals due to its ability to form key carbon-carbon bonds in a predictable and reliable manner. Beyond alkene metathesis, various other types such as alkyne metathesis [3,4], enyne metathesis [5,6], σ-bond metathesis [7,8], and carbonyl-olefin metathesis [9,10], they are good complements to the alkene metathesis reaction and have garnered extensive research interest due to their ability to exchange reactive partners in a simple and efficient manner, thereby greatly expanding the synthetic toolkit available to chemists (Scheme 1). These metathesis reactions typically rely on transition metal catalysis, operating through a well-established metallacyclobutane intermediate [1-10]. Given the central role of transition metal catalysts, the development of metathesis reactions under transition metal-free conditions poses a significant challenge. Consequently, only a few examples of transition metal-free metathesis have been reported, predominantly within the context of carbonyl-olefin metathesis [11,12]. Notably, cross-metathesis reactions strictly occur between similar types of covalent bonds, specifically σ-σ or π-π bonds. In contrast, cross-metathesis reactions involving σ and π bonds are exceptionally rare (Scheme 1f), which is a missing gap waiting to be fulfilled, and a σ-π cross-metathesis reaction under transition metal-free conditions is even more elusive. Recently, Garrec and Kaïm reported an elegant Phospha-Brook/Smiles cascade reaction leading to diarylacetamide derivatives, suggesting a σ-π metathesis mechanism to explain the observed transformations [13]. However, solid experimental evidence for a σ-π metathesis reaction is still lacking. The rarity of such reactions can be attributed to the intrinsic nature of metathesis, which typically proceeds via a [2 + 2] cycloaddition to form a four-membered cyclic intermediate, followed by a [2 + 2] cycloreversion to facilitate partner exchange. However, the participation of both a σ-bond and a π-bond in a [2 + 2] cycloaddition to form the requisite four-atom cyclic intermediate is highly improbable. These current limitations present significant obstacles to the development of cross-metathesis reactions between σ and π bonds. Overcoming these challenges and successfully developing a σ-π cross-metathesis reaction would not only deepen our understanding of metathesis mechanisms but also expand the boundaries of metathesis chemistry by introducing a new class of reactions into the synthetic repertoire.
Current methods for catalytic C—C single bond activation, especially with ketone substrates, often rely on strategies such as the extrusion of functional fragments, the release of ring strain, or the use of directing groups to facilitate the reaction [14-16]. While effective, these approaches are limited by the need for specialized substrate classes such as ring-strained compounds or substrates with preinstalled directing groups, thus restricting their broader applicability. An emerging strategy in this field involves molecular editing through the cleavage of inactivated C(=O)-C bonds, enabling the conversion of ketones into various value-added products such as esters [17,18], amides [19,20], and carboxylic acids [21-30]. Among these, carboxylic acids have garnered particular interest due to their potential for further transformation into a range of valuable compounds [31-33]. The conversion of ketones to carboxylic acids is usually carried out under oxidative conditions, often using sustainable dioxygen or the classical haloform reaction to selectively cleave the C(=O)-C bond and introduce a C(=O)-O bond [34-42]. Since its discovery in 1822, the haloform reaction has remained the most widely practiced method for converting ketones to carboxylic acids [43]. However, this protocol is not suitable for molecules containing secondary alcohols, phenols, nitro groups, alkenes, etc., due to its oxidative nature [43,44]. Given the importance of the oxidant, current methods are generally incompatible with substrates containing oxidation-sensitive functional groups. Additionally, these oxidative processes often require high reaction temperatures and transitional metals as catalysts, which can limit functional group tolerance and lead to the formation of overoxidation byproducts [34-41]. Consequently, finding alternative oxidation strategies to facilitate C—O redox processes has become a critical area of focus in green chemistry research [45]. There is still a strong need for a general and efficient method to achieve ketone-to-carboxylic acid conversion without compromising sensitive functional groups.
To date, there is one type of reaction that closely resembles the concept of σ-π cross-metathesis reaction: The fragmentation reaction of a 1,2-dioxetan-3-olate intermediate generated from the hydroperoxidation of a trisubstituted unsymmetrical ketone (Scheme 2a) [35,42]. However, this is not a real σ-π cross-metathesis reaction owing to two main reasons. First, the hybrid orbital of oxygen is not a simple π-bond but consists of a σ-bond and a three-electron π-bond [46]. Second, reactions involving oxygen are generally classified as oxidation reactions due to their high redox potential [47]. Nevertheless, this reaction provides a clue: if a carbon variant intermediate, oxetan-2-olate, could be generated, it might be utilized in similar fragmentation reactions as shown in Scheme 2b. It is important to note, however, that the existence of an oxetan-2-olate has not been verified yet, and the precursor likely to generate the oxetan-2-olate intermediate is known to be prone to Aldol dehydration, resulting in a more thermally stable product [48]. Additionally, the formation of a four-membered ring is not kinetically favorable. As a result, the formation of the proposed but unproven oxetan-2-olate is neither thermodynamically nor kinetically favored in this context. While the concept of steering the reaction towards the formation of an oxetan-2-olate intermediate is intriguing, substantial investigation may be required to identify effective strategies to overcome the kinetic and thermodynamic barriers associated with the classical Aldol condensation pathway.
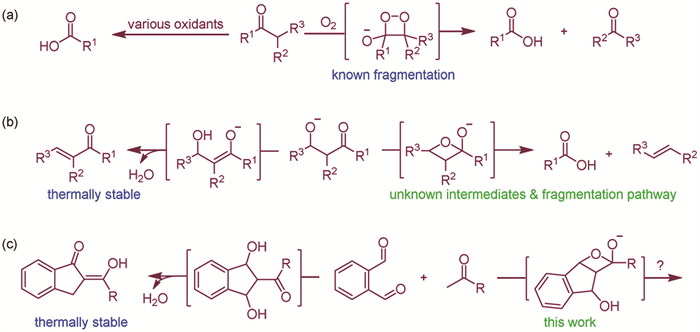
On a different note, it has been reported that the reaction of o-phthalaldehyde with aryl-substituted methyl ketones under basic conditions afforded carbonyl-substituted 2-benzylidene-1-indanones in high yields (Scheme 2c) [49]. We envision that it may be possible to direct the double Aldol addition reaction between o-phthalaldehyde and methyl ketones towards the formation of oxetan-2-olate. By carefully adjusting the reaction conditions, it may be possible to induce a retro-[2 + 2] cycloaddition reaction, taking advantage of the fused ring strain in this crucial intermediate (Scheme 2c). In this context, a proof-of-concept experiment was conducted, leading to the discovery of a novel σ-π cross-metathesis reaction. This reaction enabled a nonoxidative ketone-to-carboxylic acid conversion and facilitated the efficient formation of 11H-benzo[b]fluoren-11-one derivatives. Specifically, the carbon atom of the methyl group was selectively replaced with an oxygen atom through a redox-neutral pathway, with the carbon atom precisely incorporated into the product. This resulted in a new type of single-carbon atom doping (SCAD) reaction [50].
In our initial attempt to investigate the reaction between acetophenone 1 and o-phthalaldehyde 2, a small amount of a pure product as a yellow solid was obtained when using a polar aprotic solvent. However, NMR spectrum analysis revealed that the product was not the previously reported 2-benzylidene-1-indanone [49]. Further characterization, including X-ray diffraction, confirmed that the product was 11H-benzo[b]fluoren-11-one 3. A literature review indicated that this type of compound has potential applications in white organic light-emitting diodes (WOLEDs) [51], and also privileged structural motifs with important pharmacological activities in medicinal studies [52-54]. Notably, the previously reported synthesis of this core structure typically requires multiple steps and transition metal catalysis [55]. This unexpected result led to further investigation into all possible products formed in this reaction. Upon acidification of the reaction mixture, benzoic acid 4 was identified, with a yield comparable to that of compound 3, suggesting these were the two major products. Inspired by this finding, we conducted extensive optimization of reaction conditions, examining factors such as base, solvent, temperature, and water content, which were found to significantly influence the reaction outcome (Table 1, entries 1–7). Ultimately, the optimal conditions were determined as follows: Cs2CO3 as the base, a mixture of DMF and water (1:1, v/v) as the solvent, and a reaction temperature of 60 ℃ for 24 h. Remarkably, this reaction could also be carried out at room temperature, still yielding favorable results. This highlights the advantage of the current methodology, as many aerobic oxidative methods often require additional heating to achieve high yields [34-42].
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Variation from the standard condition | Yield (%) | |
| 3 | 4 | ||
| 1 | None | 88 | 86 |
| 2 | In the absence of water | 55 | 35 |
| 3 | THF instead of DMF | 41 | 33 |
| 4 | K3PO4 instead of Cs2CO3 | 38 | 19 |
| 5 | KOH instead of Cs2CO3 | 42 | 10 |
| 6 | rt instead of 60 ℃ | 75 | 73 |
| 7 | 1.2 equiv. of Cs2CO3 | 83 | 81 |
| a Reaction conditions A: The reaction was carried out with 0.2 mmol of acetophenone, 0.42 mmol of o-phthalaldehyde, and 0.3 mmol of Cs2CO3 in 0.2 mL of solvent (DMF:H2O 1:1, v/v) at 60 ℃ for 24 h. All reported yields are isolated yields. | |||
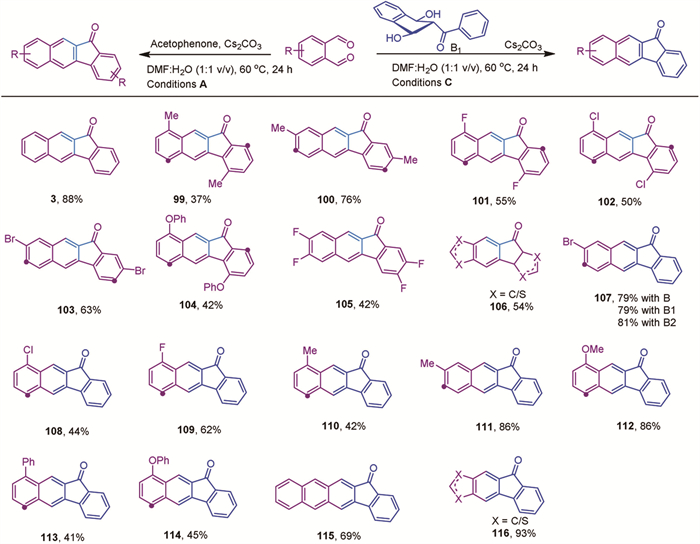
With the optimized reaction conditions established, we then explored the substrate scope of this reaction. We first tested aryl methyl ketones and found that the reaction conditions were compatible with a wide range of substrates, as summarized in Scheme 3. Both electron-rich and electron-deficient aryl substrates, including those with ethers, esters, amides, sulfonamides, alkyl groups, and halogens, were well tolerated, yielding products in 57% to 98% (Scheme 3, 4–47). Notably, the presence of ortho-substituents did not significantly impact the yields (Scheme 3, 24–33), suggesting that the reaction is not particularly sensitive to steric hindrance.
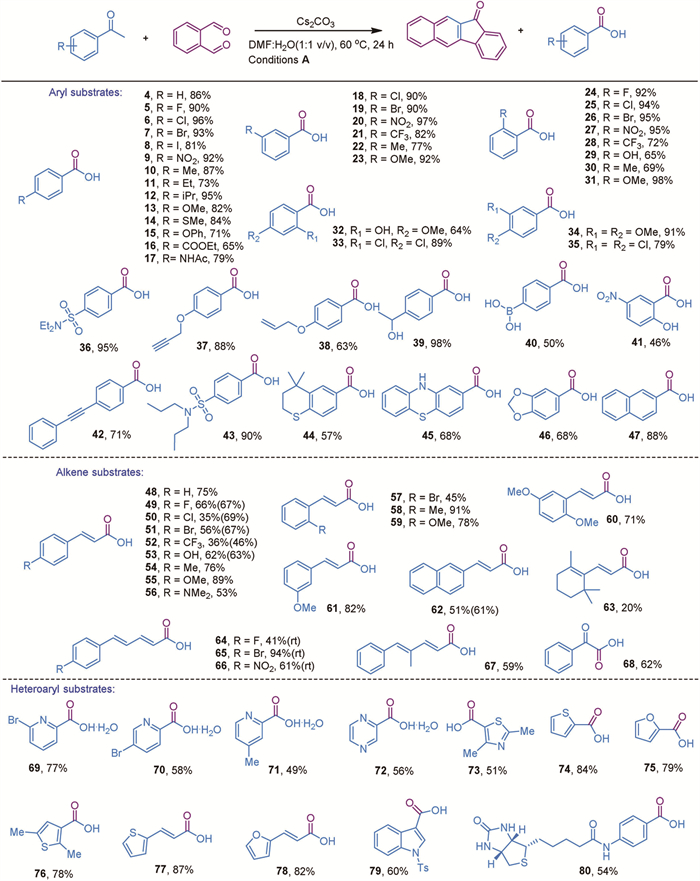
As mentioned earlier, conventional oxidation methods for converting ketones to carboxylic acids are often incompatible with oxidation-sensitive functional groups. Therefore, we focused on examining substrates containing oxidation-sensitive groups. Encouragingly, this protocol demonstrated high compatibility with a variety of oxidation-sensitive functional groups, such as iodo, ether, benzyl, sulfide, phenol, hydroxyl, amino, boronic acid, double bonds, and triple bonds (Schemes 3, 8, 12, 13, 14, 29, 32, 37–45) with 64%−98% yields. This distinguishes our method from conventional approaches, including the classical haloform reaction. Additionally, the oxidation of alkenyl-methyl ketones and diene-containing ketones to carboxylic acids is typically challenging due to the risk of overoxidation [40]. However, our method achieved moderate-to-excellent yields for majority of substrates (Scheme 3, 48–68), with some reactions proceeding even at room temperature to yield more favorable results (Scheme 3, 49–53 and 62), demonstrating the mild nature of the current methodology. Furthermore, the method was verified with heteroaryl methyl ketones, accommodating both electron-rich and electron-deficient substrates, and achieved moderate-to-high yields, including for a biotin-derived compound (Scheme 3, 69–80).
Furthermore, we were eager to explore the scope of alkyl methyl ketones, as these substrates are challenging for conventional oxidative methods [37-39]. According to the literature, C—C cleavage typically occurs on both sides adjacent to the carbonyl group, often leading to further decarboxylation and resulting in a mixture of carboxylic acids with shortened carbon chains [38,39]. Under our reaction conditions, the overoxidation could be minimized. However, the linear alkyl substrate yielded product 81 in low amounts due to the formation of a seven-membered ring byproduct 82 under standard reaction conditions [56]. To solve this problem, we modified the reaction conditions by using a mixture of DMI (1,3-dimethyl-2-imidazolidinone) and water in a 1:1 volume ratio, Na2CO3 as the base, and increased the reaction temperature to 140 ℃. This adjustment helped suppress the formation of the seven-membered ring byproduct to some extent, resulting in moderate yields. This modification enabled the successful conversion of various alkyl substrates into the desired alkyl carboxylic acids with moderate-to-high yields for majority of substrates (Scheme 4, 83–93). Even for the sterically hindered substrate 1-acetyladamantane, the desired product 88 was obtained in 65% yield under these modified reaction conditions. Moreover, the yield of compound 63 improved from 20% to 62%, suggesting that alkyl-substituted alkene substrates require higher energy input to achieve satisfactory conversion.
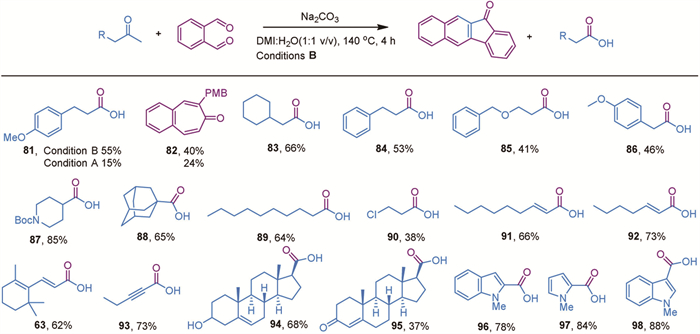
Furthermore, this methodology can be extended to steroid functionalization, achieving low to moderate yields (Scheme 4, 94 and 95). Notably, N-methyl-2-acetyl indole was converted to the corresponding carboxylic acid 96 in 78% yield. It is important to note that the transformation of protected 2-acetyl indole into the corresponding carboxylic acids has not been achieved so far. Accordingly, N-methyl-3-acetyl indole and N-methyl-3-acetyl pyrrole could be converted into the corresponding acid 97 and 98 in 88% and 84% yield respectively, and the transformation of protected 2-acetyl pyrrole into the corresponding carboxylic acids are generally achieved with poor yields [57,58].
After testing the ketone substrates, we proceeded to examine the scope of o-phthalaldehyde as the reaction partner. The results are summarized in Scheme 5. The unsubstituted o-phthalaldehyde gave product 3 in pure form with high yield (Scheme 5). However, when unsymmetric 3-methylphthalaldehyde was used, the reaction resulted in an inseparable mixture of four isomers, corresponding to different positions of the methyl group, with a decreased yield. This suggests that steric hindrance negatively affects the yield (Scheme 5, 99 and 100). The relative ratios of these isomers could not be determined due to overlapping NMR signals. This phenomenon was observed for all unsymmetric o-phthalaldehydes, resulting in only moderate yields (Scheme 5, 101–105). These results provide insights into the reaction mechanism, suggesting that the Aldol addition and proton shift processes are nonregioselective.
To gain a better understanding of the reaction process, we focused on the separation and characterization of the key intermediates (Scheme 6). To distinguish the aromatic region, acetophenone was replaced with 4-chloroacetophenone. This allowed us to successfully isolate intermediates A, B, and C. The structure of intermediate A was confirmed by X-ray diffraction analysis. The structure of intermediate B was determined based on its high symmetry observed in NMR studies. The primary difference between intermediates A and B is the positioning of the hydroxyl group: In intermediate A, one hydroxyl group is in the axial position, whereas in intermediate B, all the two hydroxyl groups are in equatorial positions. We then investigated the reaction rates of these two intermediates. The results showed that intermediate A reacted more rapidly in the first 30 min, but the rates became similar after 1 h (Fig. 1, left). Because the Aldol reaction is reversible, intermediates A and B are expected to interconvert under basic conditions. Next, intermediate A was subjected to standard reaction conditions in the presence or absence of o-phthalaldehyde. The results indicated that in the absence of o-phthalaldehyde, the reaction still proceeded to form the products, but with significantly lower yields. This finding suggests that aromatization leading to the formation of compound 3 may be a driving force for the reaction, and that intermediate A could revert to o-phthalaldehyde to produce compound 3. Additionally, when o-phthalaldehyde was absent, the yield of the carboxylic acid product was higher than that of 11H-benzo[b]fluoren-11-one 3. This suggests that aromatization is not the sole driving force for the reaction, as the formation of the carboxylic acid may occur prior to the formation of 11H-benzo[b]fluoren-11-one 3.
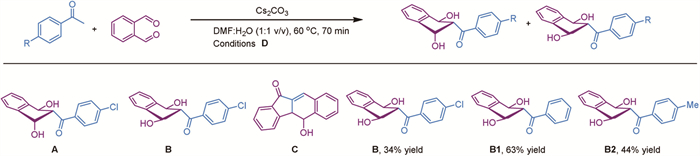
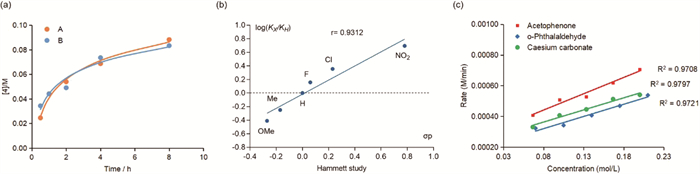
Based on the reaction of compound A with o-phthalaldehyde, we postulate that isolating intermediate A or B and subsequently reacting it with various substituted o-phthalaldehydes could lead to a wider range of 11H-benzo[b]fluoren-11-one derivatives. A preliminary study was conducted to determine the optimal reaction time for isolating intermediate B1, and it was found that a yield of ~63% could be achieved by quenching the reaction at 70 min under lower concentration (0.05 mol/L) conditions. Using these optimized reaction conditions, intermediates B and B2 were prepared and then reacted with 4-bromophthalaldehyde, resulting in comparable yields for compound 107. Subsequently, B1 was reacted with various substituted o-phthalaldehydes, affording diverse 11H-benzo[b]fluoren-11-one derivatives, each in two isomers (Scheme 5, 108–116). Interestingly, this approach yielded better results compared to using the ketone as the substrate. This strategy allows for the introduction of various functional groups and structural modifications in the final products, expanding their potential applications. After establishing the substrate scope, we investigated the reaction mechanism (Fig. 1). First, the Hammett equation was applied to evaluate the effect of acetophenone substitution on the reaction rate. A linear relationship was observed, with electron-deficient aryl substrates reacting faster, suggesting a possible anionic pathway for the reaction. Second, reaction order was determined for each reactant, revealing that the reaction is first order with respect to acetophenone, o-phthalaldehyde, and Cs2CO3. Based on the reaction kinetics for o-phthalaldehyde, it appears that the initial reaction of acetophenone with the first o-phthalaldehyde is significantly faster than the subsequent reaction of intermediate A with the second o-phthalaldehyde. This conclusion is supported by the successful isolation of B1 in moderate yield.
Additionally, 13C-labeled acetophenone was reacted with o-phthalaldehyde, and the isolated product, 11H-benzo[b]fluoren-11-one 3, was found to retain the 13C label with an 86% yield, as shown in Scheme 7a. This indicates that the bridgehead carbon in 11H-benzo[b]fluoren-11-one 3 originates from acetophenone, confirming that this reaction is a new SCAD reaction involving the formation of four new C—C bonds in a single step. Furthermore, H218O was used in place of normal water, resulting in the formation of 18O-labeled 11H-benzo[b]fluoren-11-one 3 and benzoic acid (Scheme 7b). Given that oxygen exchange between aldehyde and water is known [59], this result does not rule out the possibility of 18O exchange after the reaction. To clarify this, we prepared 18O-labeled o-phthalaldehyde (~55% 18O by NMR analysis) through exchange with 18O-labeled water and subjected it to the standard reactions in the absence of water and under a nitrogen atmosphere. Because 18O causes an upfield shift in the attached carbon signal in NMR spectroscopy, its presence and amount could be clearly identified and quantified based on carbon integration [60]. Benzoic acid was obtained with 55% 18O incorporation and in a 46% yield (Scheme 7c); the lower yield is mainly due to the absence of water. Thus, the cleavage of the C(=O)-C bond and the C=O bond, leading to carbon-oxygen exchange, was confirmed, validating the occurrence of a σ-π cross-metathesis reaction. Additionally, compound A was reacted with 18O-labeled o-phthalaldehyde (~63% 18O by NMR analysis) in the absence of water. The resulting acid was isolated with a 55% yield and with ~42% 18O incorporation (Scheme 7d). Combined with the results obtained without o-phthalaldehyde (Scheme 7e), it can be concluded that the carbon-oxygen exchange reaction primarily occurs between compound A and the second o-phthalaldehyde. Furthermore, we were unable to isolate 2,3-dihydro-1H-inden-1-one 118 from the reaction mixture in the absence of o-phthalaldehyde (Scheme 7e); only compound 3 was isolated in relatively low yield, suggesting that the intermediate can partially convert back into o-phthalaldehyde and react with A to produce the final product. This also explains the low yields of compounds 3 and 6, as intermediate A serves as two components. Although we did not isolate compound 117 from the reaction mixture (possibly due to its low concentration), the formation of 117 from acetophenone and o-phthalaldehyde is known [49]. We synthesized this compound and subjected it to the standard reaction conditions. In the absence of o-phthalaldehyde, 2,3-dihydro-1H-inden-1-one and benzoic acid were isolated in 36% and 44% yields, respectively (Scheme 7f), suggesting that a retro-Claisen rearrangement occurred. This result rules out the possibility that intermediate A is converted into 117 before forming the final product; they likely proceed through different pathways. Additionally, in the presence of o-phthalaldehyde, the yield of compound 3 is lower (64% vs. 88%) than that of compound 4 (Scheme 7f), indicating that the formation of compound 3 from 2,3-dihydro-1H-inden-1-one is not straightforward, as it is an intermolecular reaction. This provides a clue that the comparable yields of compounds 3 and 4 under standard reaction conditions may derive from an intramolecular reaction.
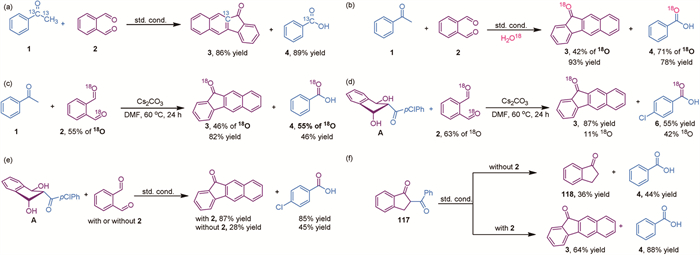
Based on the mechanistic study and the isolated intermediates A, B, and C, two possible reaction pathways are proposed in Scheme 8. The enolate generated from acetophenone in the presence of a base attacks o-phthalaldehyde, resulting in an initial Aldol addition reaction to form intermediate D. As the formation of a five-membered ring is kinetically favored, the reaction proceeds with a second Aldol addition rather than dehydration, leading to the formation of intermediates A and B, which can interconvert. Intermediate A, with a cis‑hydroxyl group relative to the benzoyl group, is prone to attack the carbonyl group, forming a four-membered oxetan-2-olate E. This intermediate then undergoes a [2 + 2] cycloreversion, releasing the benzoate group and generating intermediate F, which then undergoes a base-catalyzed 1,3-proton shift followed by a double Aldol condensation, resulting in the formation of product 3 [61]. Alternatively, intermediate B, which lacks an axial hydroxyl group, attacks a second molecule of o-phthalaldehyde from the less hindered face, leading to the formation of intermediate K. The hydroxyl group on the outer ring then attacks the carbonyl group, forming another four-membered oxetan-2-olate L. This intermediate undergoes a [2 + 2] cycloreversion, releasing the benzoate group and generating intermediate M. Under basic conditions, a 1,3-proton shift occurs [62], producing intermediate N, which then undergoes a series of dehydration, 1,3-proton shift, and additional dehydration reactions, ultimately forming the final product 3.
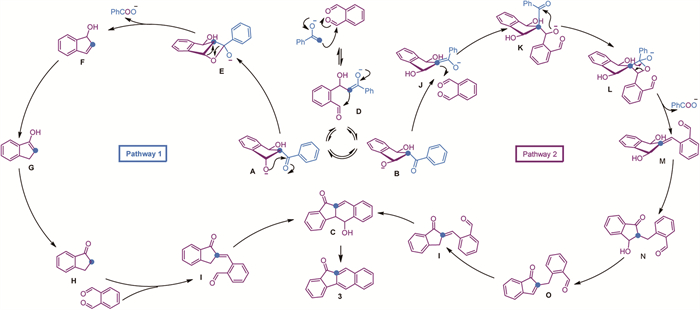
In the proposed mechanism, the C(=O)-C σ bond is selectively transformed into a C=O π bond, while the original C=O π bond is converted into a C—OH σ bond. This interchange is essential for the generation of oxetan-2-olate and its subsequent [2 + 2] cycloreversion reaction, facilitating the exchange of reaction partners. Moreover, intermediates H or/and O can undergo sequential reactions to achieve complete aromatization, which further accelerates the conversion to product 3. Aromatization is a commonly employed strategy in organic transformations to drive reactions [63,64]. Overall, the combination of aromatization and benzoate extrusion enables the ketone-to-carboxylic acid conversion even at room temperature without the need for an oxidant, highlighting the mildness of this transformation.
In this reaction, the nucleophilicity of the α-carbon and the electrophilicity of the carbonyl group are effectively harnessed to work together, a concept originally demonstrated using hexaphenylcarbodiphosphorane [65]. In other words, the methyl ketone in this protocol serves as a practical ylide equivalent with significantly higher atom efficiency (59% vs. 29%). From a green chemistry perspective, developing Wittig chemistry alternatives without Ph3PO and finding alternatives for oxidations in C—O redox processes are important research areas [45]. The protocol described here could provide a promising approach to achieving these goals.
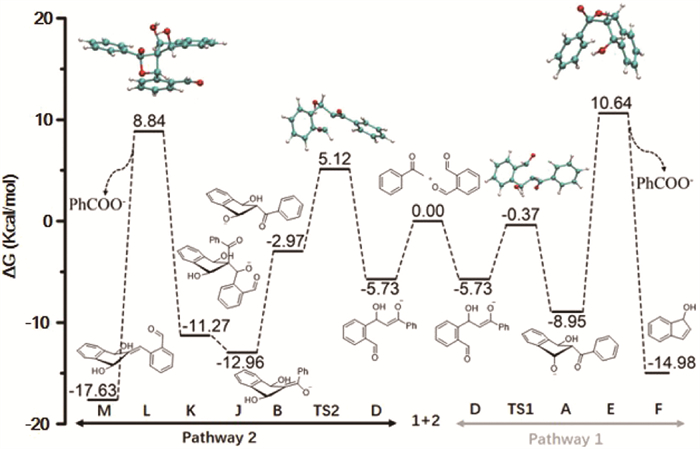
To gain a better understanding of the reaction pathway, DFT calculations were performed for the two proposed reaction cycles using acetophenone 1 and o-phthalaldehyde 2 as molding substrates, as shown in Fig. 2. Because both pathways lead to the same final product, only the relative energies of key transition states (TS) were calculated to minimize the computational costs. The second Aldol addition favors the formation of intermediate A (ΔG = −8.95 kcal/mol) over intermediate B (ΔG = −2.97 kcal/mol), as the carbonyl group must rotate out of the aryl plane in the transition state (TS2) to form intermediate B. However, intermediate B can be smoothly converted to dihydroxy enolate J, followed by a third Aldol addition to form intermediate K. With the assistance of two water molecules, both pathways 1 and 2 proceed with relatively low energy barriers in their transition states, with pathway 2 being slightly lower. This aligns with our observation that, in the absence of second o-phthalaldehyde, the reaction does not proceed to completion and yields are significantly lower, suggesting that the subsequent aromatization process to generate product 3 is crucial for the reaction’s success. Based on the DFT calculations, the formation of oxetan-2-olate (E and L) and their subsequent [2 + 2] cycloreversion reactions appear to be the rate-determining steps. Although kinetic studies from Scheme 3 indicate that the reaction is first order with respect to acetophenone, o-phthalaldehyde, and Cs2CO3 in the transition state, the reaction with the first o-phthalaldehyde is so fast that it can be considered pseudo-first order. Combining the mechanistic study and DFT calculations, this reaction seems to primarily proceed through pathway 2.
A base-promoted σ-π cross-metathesis reaction was developed, avoiding the classical Aldol dehydration pathway. Unlike conventional oxidative C(=O)-C bond cleavage methods, this protocol proceeds at lower reaction temperatures and in the absence of harsh oxidants, which even enables the transformations of sensitive substrates like protected pyrrole or indole into the corresponding carboxylic acids, broadening the substrate scope by tolerancing wider functional groups such as benzyl, sulfide, phenol, hydroxyl, alkenes, and alkynes. This unique transformation was elucidated through detailed mechanistic studies and DFT calculations, revealing that the reaction proceeds via a key four-membered oxetan-2-olate intermediate to achieve the carbon-oxygen exchange. Synthetically, it provides a method for nonoxidative ketone-to-carboxylic acid transformation as well as offers a new synthetic strategy for constructing medicinal significant 11H-benzo[b]fluoren-11-one derivatives using readily available starting material. Theoretically, this mechanism maybe extended to the activation of other types of carbonyl-containing compounds in a redox neutral way. Besides, the principles learned from this study may inspire the design of new catalysts or reaction conditions for other challenging bond activations based on σ-π cross-metathesis concept.
Huidan Geng: Investigation, Formal analysis, Data curation. Jiuwei Nie: Validation, Methodology, Investigation. Wei Chen: Methodology, Investigation, Data curation. Chong Zhao: Visualization, Validation, Software. Zikun Yao: Investigation. Hui Wang: Investigation. Jiquan Zhang: Visualization. Lei Tang: Resources, Project administration. Yuanyong Yang: Writing – review & editing, Writing – original draft, Supervision, Funding acquisition, Conceptualization.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are thankful for financial support from the National Natural Science Foundation of China (No. 22061012), the Guizhou Provincial Excellent Young Talents Plan (No. YQK[2023]030), the Excellent Young Talents Plan of Guizhou Medical University (Nos. [2023]105, [2022]102), GMU Training Program of National Natural Science Foundation of China (No. 22NSFCP13), and the support from National-Local Joint Engineering Research Center for Innovative & Generic Chemical Drug & Guizhou Province Innovation Base of Common Major Chronic Disease Pathogenesis and Drug Development and Application (No. [2021]4029).
G.C. Vougioukalakis, R.H. Grubbs, Chem. Rev. 110 (2010) 1746–1787. doi: 10.1021/cr9002424
R.R. Schrock, A.H. Hoveyda, Angew. Chem. Int. Ed. 42 (2003) 4592–4633. doi: 10.1002/anie.200300576
S. Huang, Z. Lei, Y. Jin, W. Zhang, Chem. Sci. 12 (2021) 9591–9606. doi: 10.1039/d1sc01881g
A. Fürstner, P.W. Davies, Chem. Commun. 18 (2005) 2307–2320. doi: 10.1039/b419143a
S.T. Diver, A.J. Giesser, Chem. Rev. 104 (2004) 1317–1382. doi: 10.1021/cr020009e
H. Villar, M. Frings, C. Bolm, Chem. Soc. Rev. 36 (2007) 55–66. doi: 10.1039/B508899M
R. Waterman, Organometallics 32 (2013) 7249–7263. doi: 10.1021/om400760k
Z. Lian, B.N. Bhawal, P. Yu, B. Morandi, Science 356 (2017) 1059–1063. doi: 10.1126/science.aam9041
H. Albright, A.J. Davis, J.L. Gomez-Lopez, et al., Chem. Rev. 121 (2021) 9359–9406. doi: 10.1021/acs.chemrev.0c01096
J.R. Ludwig, P.M. Zimmerman, J.B. Gianino, C.S. Schindler, Nature 533 (2016) 374–379. doi: 10.1038/nature17432
A. Soicke, N. Slavov, J.M. Neudörfl, H.G. Schmalz, Synlett 17 (2011) 2487–2490.
A.K. Griffith, C.M. Vanos, T.H. Lambert, J. Am. Chem. Soc. 134 (2012) 18581–18584. doi: 10.1021/ja309650u
C. Cheibas, N. Fincias, N. Casaretto, J. Garrec, L.E. Kaïm, Angew. Chem. Int. Ed. 61 (2022) e202116249. doi: 10.1002/anie.202116249
W. Schutyser, T. Renders, S.V. Bosch, et al., Chem. Soc. Rev. 47 (2018) 852–908. doi: 10.1039/c7cs00566k
Y. Xia, G. Dong, Nat. Rev. Chem. 4 (2020) 600–614. doi: 10.1038/s41570-020-0218-8
X.Y. Yu, J.R. Chen, W.J. Xiao, Chem. Rev. 121 (2021) 506–561. doi: 10.1021/acs.chemrev.0c00030
X. Huang, X. Li, M. Zou, et al., J. Am. Chem. Soc. 136 (2014) 14858–14865. doi: 10.1021/ja5073004
M. Liu, Z. Zhang, J. Yan, et al., Chem 6 (2020) 3288–3296. doi: 10.1016/j.chempr.2020.09.006
C. Tang, N. Jiao, Angew. Chem. Int. Ed. 53 (2014) 6528–6532. doi: 10.1002/anie.201403528
W. Ding, Q. Song, Org. Chem. Front. 2 (2015) 765–770. doi: 10.1039/C5QO00101C
J.T. Minor, C.A. Vanderwerf, J. Org. Chem. 17 (1952) 1425–1430. doi: 10.1021/jo50011a001
W. Liu, P. Liu, L. Lv, C.J. Li, Angew. Chem. Int. Ed. 57 (2018) 13499–13503. doi: 10.1002/anie.201807181
C. Wang, Z. Li, Y. Ju, S. Koo, Eur. J. Org. Chem. 2012 (2012) 6976–6985. doi: 10.1002/ejoc.201201194
L. Xu, S. Wang, B. Chen, et al., Synlett 29 (2018) 1505–1509. doi: 10.1055/s-0037-1609751
A. Roy, K.R. Reddy, P.K. Mohanta, H. Ila, H. Junjappa, Synthetic Commu. 29 (1999) 3781–3791. doi: 10.1080/00397919908086017
S.I. Hirashima, T. Nobuta, N. Tada, A. Itoh, Synlett 12 (2009) 2017–2019.
J.N. Capilato, P.J. Pellegrinelli, J. Bernard, et al., Org. Biomol. Chem. 19 (2021) 5298–5302. doi: 10.1039/d1ob00786f
K.A.A. Kumar, V. Venkateswarlu, R.A. Vishwakarma, S.D.A. Sawant, Synthesis 47 (2015) 3161–3168. doi: 10.1055/s-0034-1381026
A.M. Vanarendonk, M.E. Cupery, J. Am. Chem. Soc. 53 (1931) 3184–3186. doi: 10.1021/ja01359a506
S. Zhu, Y. Lu, J. Jin, J. Yu, W. Lu, Dyes Pigments 196 (2021) 109783. doi: 10.1016/j.dyepig.2021.109783
F. Penteado, E.F. Lopes, D. Alves, et al., Chem. Rev. 119 (2019) 7113–7278. doi: 10.1021/acs.chemrev.8b00782
Y. Wei, P. Hu, M. Zhang, W. Su, Chem. Rev. 117 (2017) 8864–8907. doi: 10.1021/acs.chemrev.6b00516
A. Varenikov, E. Shapiro, M. Gandelman, Chem. Rev. 121 (2021) 412–484. doi: 10.1021/acs.chemrev.0c00813
M. Wang, J. Lu, X. Zhang, et al., ACS Catal. 6 (2016) 6086–6090. doi: 10.1021/acscatal.6b02049
X. Wang, R.X. Chen, Z.F. Wei, et al., J. Org. Chem. 81 (2016) 238–249. doi: 10.1021/acs.joc.5b02506
R. Nakamura, Y. Obora, Y. Ishii, Adv. Synth. Catal. 351 (2009) 1677–1684. doi: 10.1002/adsc.200900099
H. Liu, M. Wang, H. Li, et al., J. Catal. 346 (2017) 170–179. doi: 10.1016/j.jcat.2016.12.016
T. Hattori, H. Okami, T. Ichikawa, et al., Adv. Synth. Catal. 359 (2017) 3490–3495. doi: 10.1002/adsc.201700774
P. Sathyanarayana, O. Ravi, P.R. Muktapuram, S.R. Bathula, Org. Biomol. Chem. 13 (2015) 9681–9685. doi: 10.1039/C5OB01569C
P. Sathyanarayana, A. Upare, O. Ravi, P.R. Muktapuram, S.R. Bathula, RSC Adv. 6 (2016) 22749–22753. doi: 10.1039/C6RA02962K
L. Xu, Y. Chen, Z. Shen, Y. Wang, M. Li, Tetrahedron Lett. 59 (2018) 4349–4354. doi: 10.1016/j.tetlet.2018.10.060
A.S.K. Tsang, A. Kapat, F. Schoenebeck, J. Am. Chem. Soc. 138 (2016) 518–526. doi: 10.1021/jacs.5b08347
R.C. Fuson, B.A. Bull, Chem. Rev. 15 (1934) 275–309. doi: 10.1021/cr60052a001
A.M. VanArendonk, M.E. Cupery, J. Am. Chem. Soc. 53 (1931) 3184–3186. doi: 10.1021/ja01359a506
M.C. Bryan, P.J. Dunn, D. Entwistle, et al., Green Chem. 20 (2018) 5082–5103. doi: 10.1039/c8gc01276h
W.T. Borden, R. Hoffmann, T. Stuyver, B. Chen, J. Am. Chem. Soc. 139 (2017) 9010–9018. doi: 10.1021/jacs.7b04232
J. Wilshire, D.T. Sawyer, Acc. Chem. Res. 12 (1979) 105–110. doi: 10.1021/ar50135a005
C.L. Perrin, K.L. Chang, J. Org. Chem. 81 (2016) 5631–5635. doi: 10.1021/acs.joc.6b00959
M..L. González, M.E. Sánchez-Vergara, J.R. ‘Alvarez-Bada, et al., J. Mater. Chem. C 2 (2014) 5607–5614. doi: 10.1039/C4TC00599F
M. Kamitani, B. Nakayasu, H. Fujimoto, et al., Science 379 (2023) 484–488. doi: 10.1126/science.ade5110
W. Yang, X. Chen, Phys. Chem. Chem. Phys. 16 (2014) 4242–4250. doi: 10.1039/c3cp54462a
Q.F. Hu, B. Zhou, J.M. Huang, et al., J. Nat. Prod. 76 (2013) 292–296. doi: 10.1021/np300727f
G. Wang, J. Qiu, X. Xiao, A. Cao, F. Zhou, Bioorg. Chem. 76 (2018) 249–257. doi: 10.1016/j.bioorg.2017.11.017
B. Wang, F. Guo, J. Ren, et al., Nat. Commun. 6 (2015) 7674–7678. doi: 10.1038/ncomms8674
S. Zhu, D. Wang, S. Liu, et al., ACS Catal. 13 (2023) 8402–8412. doi: 10.1021/acscatal.3c00857
D. Mukherjee, Y.D. Wu, F.R. Fronczek, K.N. Houk, J. Am. Chem. Soc. 110 (1988) 3328–3330. doi: 10.1021/ja00218a069
M. Chatzopoulou, I.D. Bonovolias, I. Nicolaou, et al., Eur. J. Med. Chem. 50 (2012) 75–80. doi: 10.1016/j.ejmech.2012.01.041
A.C. Rowett, S.G. Sweeting, D.M. Heard, A.J.J. Lennox, Angew. Chem. Int. Ed. 63 (2024) e202400570. doi: 10.1002/anie.202400570
Y.C. Hsu, J.H. Lai, R.S. Liu, Chem. Commun. 53 (2017) 6009–6012. doi: 10.1039/C7CC03421K
J.C. Vederas, J. Am. Chem. Soc. 102 (1980) 374–376. doi: 10.1021/ja00521a063
J.J. Eisch, J.E. Galle, J. Org. Chem. 55 (1990) 4835–4840. doi: 10.1021/jo00303a015
K. Wang, S. Niu, X. Guo, et al., J. Org. Chem. 87 (2022) 3804–3809. doi: 10.1021/acs.joc.1c02916
X.Y. Lv, R. Abrams, R. Martin, Angew. Chem. Int. Ed. 62 (2023) e202217386. doi: 10.1002/anie.202217386
Y. Xu, X. Qi, P. Zheng, et al., Nature 567 (2019) 373–378. doi: 10.1038/s41586-019-0926-8
A. Streitwieser Jr., S.M. Brown, J. Org. Chem. 53 (1988) 904–906. doi: 10.1021/jo00239a050

Scheme 3 Substrate scope of the cross-metathesis reaction. Reaction conditions A: The reaction was conducted with 0.2 mmol of acetophenone, 0.42 mmol of o-phthalaldehyde, and 0.3 mmol of Cs2CO3 in 0.2 mL of solvent (DMF:H2O, 1:1, v/v) at 60 ℃ for 24 h unless otherwise noted. All yields are isolated yields, with yields in parentheses indicating those obtained at room temperature.

Scheme 4 Substrate scope for the cross-metathesis reaction. Reaction conditions B: The reaction was conducted with 0.2 mmol of acetophenone, 0.42 mmol of o-phthalaldehyde, and 0.3 mmol of Na2CO3 in 2 mL of solvent (DMI:H2O, 1:1, v/v) at 140 ℃ for 4 h. All reported yields are isolated yields.

Scheme 5 Scope of o-phthalaldehyde. Reaction conditions A: The reaction was conducted with 0.2 mmol of acetophenone, 0.42 mmol of o-phthalaldehyde, and 0.3 mmol of base in 0.2 mL of solvent (DMF:H2O, 1:1, v/v) at 60 ℃ for 24 h. All reported yields are isolated yields. Reaction conditions C: The reaction was conducted with 0.2 mmol of intermediate A, 0.22 mmol of o-phthalaldehyde, and 0.22 mmol of base in 0.2 mL of solvent (DMF:H2O, 1:1, v/v) at 60 ℃ for 24 h. All reported yields are isolated yields.

Scheme 6 Preparation of intermedates. Reaction conditions D: Methyl aryl ketone (1 mmol), o-phthalaldehyde (2.1 mmol, 2.1 equiv.), Cs2CO3 (1.5 mmol, 1.5 equiv.), and DMF/H2O (1/1 v/v, 20 mL) were added to the reaction flask and reacted at 60 ℃ for 70 min.

Figure 1 Proposed mechanistic study results. (a) Product concentrations of A and B over time. (b) Hammett study. (c) Reaction order study.

Figure 2 DFT calculations (Cs2HCO3+ and water are omitted for clarity; relative energies are given in kcal/mol).
Table 1. Summary of key results from reaction condition optimization.a
|
|||
| Entry | Variation from the standard condition | Yield (%) | |
| 3 | 4 | ||
| 1 | None | 88 | 86 |
| 2 | In the absence of water | 55 | 35 |
| 3 | THF instead of DMF | 41 | 33 |
| 4 | K3PO4 instead of Cs2CO3 | 38 | 19 |
| 5 | KOH instead of Cs2CO3 | 42 | 10 |
| 6 | rt instead of 60 ℃ | 75 | 73 |
| 7 | 1.2 equiv. of Cs2CO3 | 83 | 81 |
| a Reaction conditions A: The reaction was carried out with 0.2 mmol of acetophenone, 0.42 mmol of o-phthalaldehyde, and 0.3 mmol of Cs2CO3 in 0.2 mL of solvent (DMF:H2O 1:1, v/v) at 60 ℃ for 24 h. All reported yields are isolated yields. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: