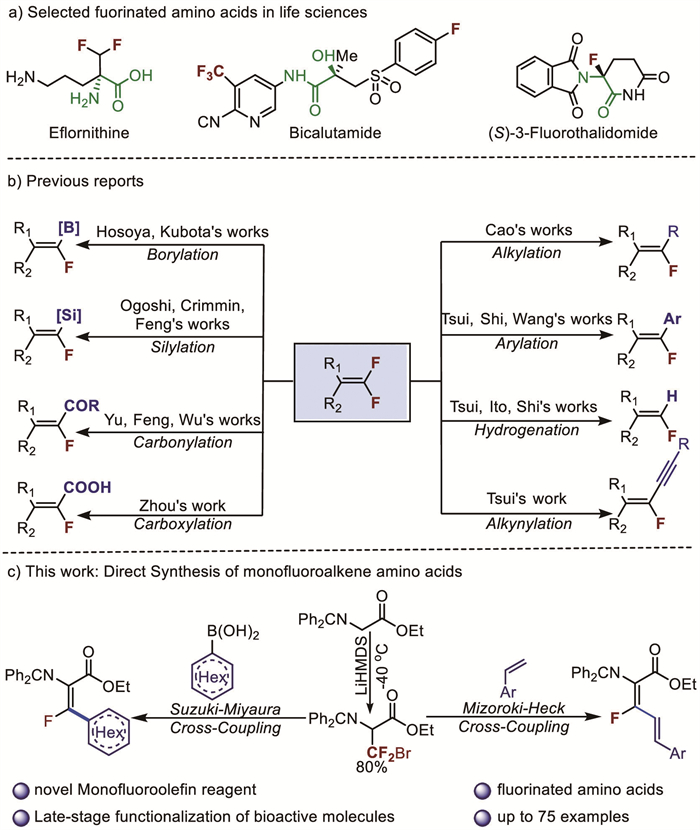
Figure 1.
Background and novel monofluoroalkene reagent.
A highly efficient approach to Z-monofluoroolefin-dehydroamino acid derivatives from gem-difluoroalkenes
Chuangchuang Liu , Yilian Song , Wenjie Ju , Xin Liu , Min Liang , Yingsheng Zhao

Organic fluorides have important applications in the pharmaceutical industry due to the ability of C-F bonds to alter the physical, chemical, and biological properties of small organic compounds, including solubility, metabolic stability, hydrogenbonding ability, lipophilicity, and chemical reactivity [1-4]. As a result, an increasing number of products of the pharmaceutical, agrochemical, and material science industries contain fluorine atoms as key components [5]. Nearly 20% of the top 200 drugs in 2023 contain a fluorine atom in their structure [6]. Monofluoroolefins are ideal amide-bond mimics in medicinal chemistry and hold great potential in isomer-based drug design [7], creating a growing need for efficient methods to synthesize various classes of monofluoroolefins. The alkylation [8], arylation [9-11], hydrogenation [12-14], alkynylation [15], borylation [16,17], silylation [18-20], carbonylation [21-23] and carboxylation [24] of C-F bonds in difluoroalkenes can be achieved through free radical [25-30] and metal catalysis strategies [31-48] (Fig. 1b). Although important progress has been achieved in the conversion of C-F bond to other functional groups, the application such strategy to monofluoro amino acid derivatives has not been disclosed. Here, we report a newly developed glycine containing fluoroalkylation reagent, which is directly applied to the synthesis of monofluorocarbon amino acids via a Pd-catalyzed cross-coupling reaction. A variety of arylboronic acids were well tolerated, yielding products with well-defined stereochemistry in moderate to high yields (Z:E > 20:1). Additionally, late-stage functionalization of bioactive compounds was also achieved. Furthermore, this new glycine-containing fluoroalkylation reagent was applied in Heck coupling reactions (Fig. 1c).
Amino acids are essential for the human body and each amino acid has unique functional properties. Among them, glycine is the smallest amino acid, which can be used as a raw material for the production of cephalosporins, intermediates of thiamphenicol, and intermediates for the synthesis of imidazoleacetic acid. The addition of fluorine atoms to amino acids has become a key synthetic strategy for the development of new drugs and pesticides, such as eflornithine, bicalutamide (Casodex) and (S)-3-fluorothalidomide (Fig. 1a). However, the approach of combining monofluoroolefins with small amino acids has not yet been reported till now. Therefore, it would be great importance and attractive to design a new gem-difluoroolefin precursor, using glycine as the backbone to construct the bank of α,β-dehydroamino acids which are non-natural amino acid with unique properties, such as planar conformation and the ability to resist degradation by peptidases [49]. The fluorinated -CF2Br block serves as a key reacting center for forming fluorinated products, a strategy that has been extensively explored by the groups of Zhang [50-53], Ackermann [54-56], and our group [57-63]. Introducing the -CF2Br group directly onto protected glycine may offer a novel approach to fluoroalkylation, allowing for the preparation of new framework compounds through well-established catalytic systems such as cross-coupling, C—H activation, photocatalytic radical processes, and electrocatalytic reactions (Fig. 1c).
At the beginning of the study, the 3-bromo-3,3-difluoroalanine Schiff base (1a) was easily prepared from protected glycine in good yield (8 g, 80%). We then explored its application in the Pd-catalyzed cross-coupling reaction to synthesize fluorinated amino acid derivatives. When 1a was treated with p-methoxyphenylboronic acid (2a), potassium carbonate, and water (150 µL) in the presence of PdCl2 (5 mol%) and PPh3 (10 mol%) in 1,4-dioxane at 100 ℃ for 2 h, the fluorinated dehydroamino acid 3a was obtained in 65% yield (Table 1, entry 1). Subsequently, we screened some phosphine ligands, including Davephos, Xantphos, dppf, and dppe, among which the 3a yield of the dppf ligand was slightly higher than that of the other ligands (Table 1, entries 1–5). We next employed Pd(dppf)Cl2 directly as the catalyst, giving the product 3a in 78% yield. We also screened different bases, including K3PO4, KHCO3, KOH, Et3N, and Cs2CO3, and obtained excellent yield of 3a when using Cs2CO3. In a control experiment using only Cs2CO3 no product was obtained, confirming that Pd(dppf)Cl2 was essential for this reaction (Table 1, entry 12).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | [Pd] | Ligand | Base | Yield (%)b |
| 1 | PdCl2 | PPh3 | K2CO3 | 65 |
| 2 | PdCl2 | Davephos | K2CO3 | 70 |
| 3 | PdCl2 | Xantphos | K2CO3 | 69 |
| 4 | PdCl2 | dppf | K2CO3 | 76 |
| 5 | PdCl2 | dppe | K2CO3 | 65 |
| 6 | Pd(dppf)Cl2 | None | K2CO3 | 78 |
| 7 | Pd(dppf)Cl2 | None | K3PO4 | 60 |
| 8 | Pd(dppf)Cl2 | None | KHCO3 | 65 |
| 9 | Pd(dppf)Cl2 | None | KOH | 35 |
| 10 | Pd(dppf)Cl2 | None | Et3N | 43 |
| 11 | Pd(dppf)Cl2 | None | Cs2CO3 | 89 |
| 12 | None | None | Cs2CO3 | 0 |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), base (3.0 equiv.), 150 µL H2O, [Pd] (5 mol%), 1,4-dioxane (0.5 mL), 100 ℃, under argon. b Solated yield. |
||||
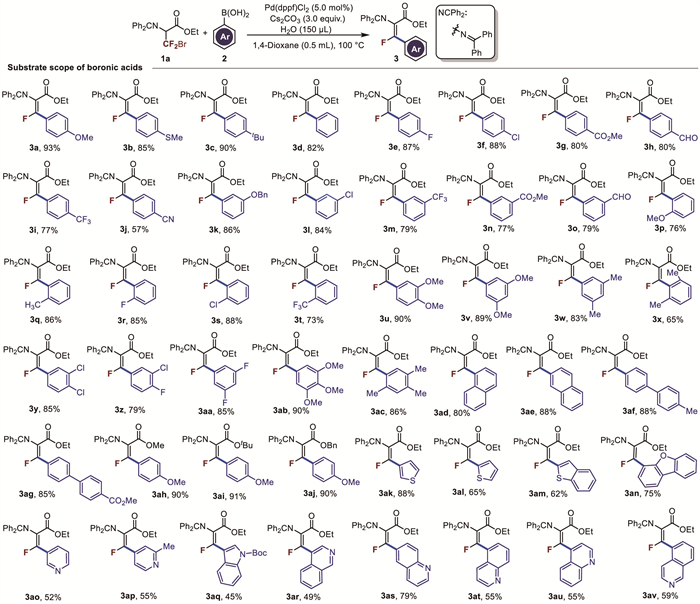
After optimizing the reaction conditions, the applicability of this reaction was further examined by evaluating the substrate scope of aryl boronic acids, as presented in Fig. 2. The reaction showed good tolerance to various functional groups, including electron-donating groups such as methoxy, methylthio, and tert-butyl, as well as electron-withdrawing groups like -F, -Cl, ester, formyl, and -CF3. For electron-donating substituents in the para position, such as -OMe, -SMe and -tBu, the target products were obtained in 93%, 85%, and 90% yields, respectively (3a-3c). With chlorine (3f) and fluorine (3e) substituents at the para position, the target products were obtained in 88% and 87% yields, respectively. The reaction yield was slightly decreased when electron-withdrawing substituents such as ester, formyl, and -CF3 were introduced on the aromatic ring (3g-3i). Notably, when cyano group was introduced in the para position, the target product was obtained in a lower yield (3j, 57%). The yields for meta and para-substituted phenylboronic acid were similar (3k-3o), but due to steric hindrance, the yield for ortho-substituted phenylboronic acid was lower compared with the para and meta-substituted ones (3p-3t). Polysubstituted phenylboronic acids were also examined, and they all provided the desired products in good to excellent yields (3u-3ac), except for ortho-disubstituted phenylboronic acids, which resulted in lower yields (3x). When naphthylboronic acids and biphenylboronic acidwere used, the corresponding products were isolated in excellent yields (3ad-3ag). The yield remained consistent when the ethyl ester was replaced with methyl ester, tert-butyl ester, or benzyl ester (3ah-3aj). Given the widespread presence of heterocycles in bioactive molecules, we further investigated heteroarylboronic acids. The yield of 3-thiopheneboronic acid was higher than that of 2-thiopheneboronic acid, with the target products obtained in 88% and 65% yields, respectively (3ak, 3al). The reaction also proceeded well with 2-benzothiopheneboronic acid, yielding 62% of the target product (3am). When other N-heterocyclic boric acids such as pyridine, quinoline, isoquinoline, and indole were used, the obtained yields were relatively poor (3ao-3av).
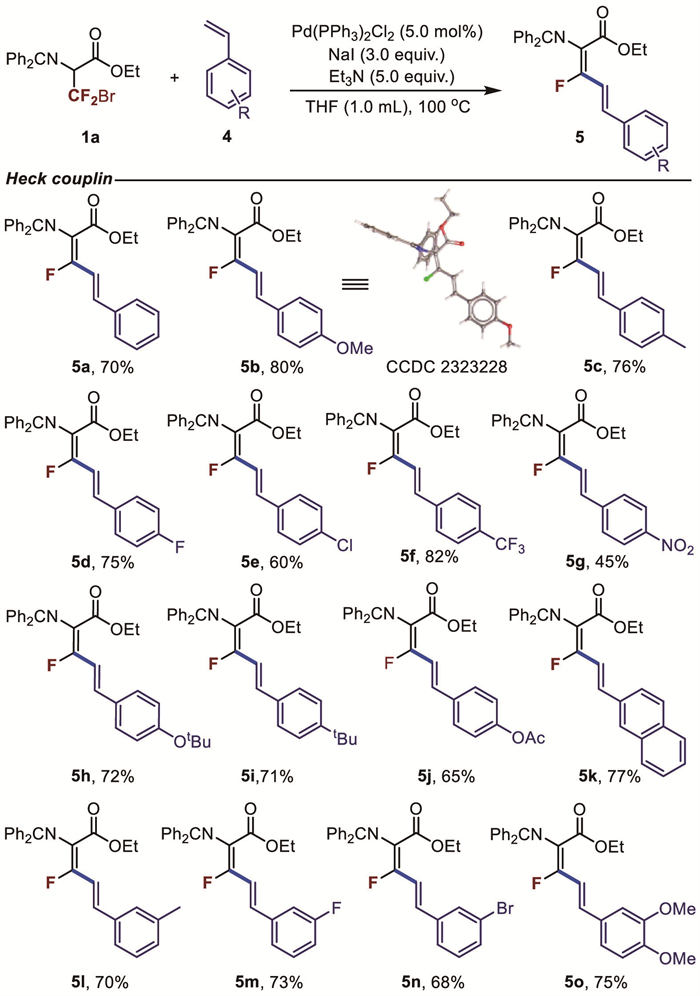
Subsequently, we found that this new glycine-containing fluoroalkylation reagent (1a) not only performed well in the Suzuki Cross-Coupling but also underwent Heck Cross-Coupling. Under optimal conditions, the scope of styrenes was investigated (Fig. 3, 5a-5o). Styrenes substituted with electron-donating and electron-withdrawing groups were all compatible, resulting in the corresponding products in moderate to good yields. Moreover, the stereoselectivity was excellent, favoring the (2Z,4E)-isomer regardless of the substituents. The product structure and the configuration of the two double bonds were confirmed by NMR and the X-ray crystal structure of compound (5b).
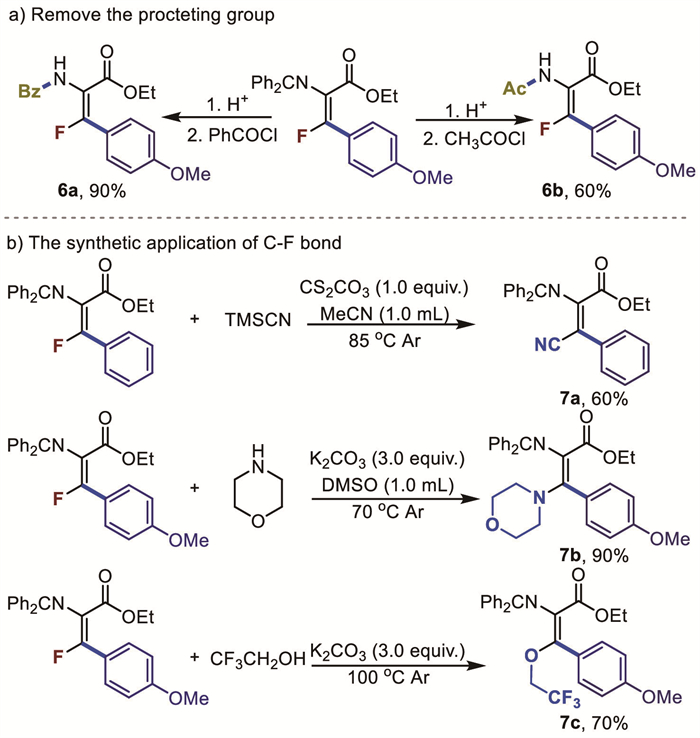
Moreover, to further demonstrate the practical aspects of the present method, several transformations involving the amino and fluoride group of 3a were conducted (Fig. 4). Treatment of 3a with a catalytic amount of acid in water led to a deamination product, which could be further modified with acyl chloride or benzoyl chloride, yielding amide products (6a, 6b) in good yields. Fluorinated olefins can be diversified on the C(sp2)-F bonds through various nucleophilic substitution reactions. To this end, we performed a series of follow-up transformations with different nucleophiles. For instance, 3d or 3a reacted directly with trimethylsilyl cyanide, morpholine, and trifluoroethanol under mild conditions, yielding the corresponding products in good to excellent yields (7a-7c).
Given the essential role of amino acids in biological systems, site-selective transformations of amino acids, peptides, and proteins have become a prominent topic in organic synthesis, medicinal chemistry, and chemical biology. Among various methods developed, the selective introduction of fluorinated functional groups into amino acids and peptides has emerged as an effective strategy for modifying their physicochemical and biological properties. However, the synthesis of fluorinated reported to date typically involves multiple synthetic steps. In a previous study, the synthesis of peptides containing 2-amino-3-fluoroacrylic acid required 7 steps, with a yield of only 16% [49]. By using our strategy with glycine, 60% of the target product (8a) was obtained in just 2 steps (Fig. 5a). The late-stage functionalization of well-known bioactive organic molecules provide a novel, highly efficient approach in this field, which can facilitate the discovery of new drugs. Till now, there were no studies on the direct introduction of the 2-amino-3-fluoroacrylic acid moiety into complex molecules. In this work, we demonstrated that estrone (a female hormone), clofibrate (a lipid-lowering agent), and paclitaxel (an anticancer agent) were successfully coupled with 2-amino-3-fluoropropenoic acid in excellent yields (83%–86%) (Fig. 5b, 8b-8d). Moreover, our method enabled the synthesis of fluorodehydroborated L-phenylalanine (8e), a highly effective anti-cancer drug, as well as derivatives of the tryptophan framework (8f). Notably, when 1a reacted with hydroxyphenylboronic acid, an F-aminocoumarin derivative (8g) is obtained under standard reaction conditions with a 69% yield. This derivative was then treated with 1 mol/L hydrochloric acid, resulting in F-aminocoumarin (8h) in a good yield (50%) (Fig. 5c).
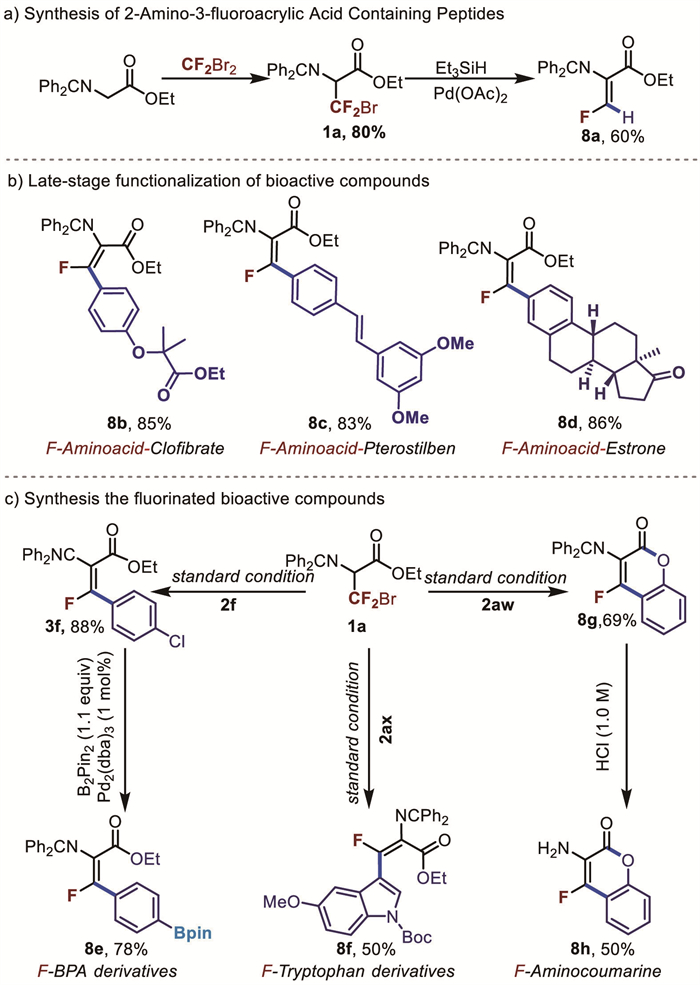
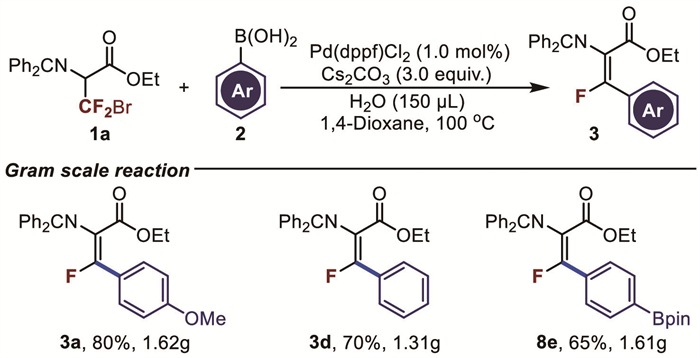
Following the successful small-scale reactions, the reaction was scaled up to evaluate its applicability at the gram scale (Fig. 6). Reactions between 5 mmol of 3-bromo-3,3-difluoropropanoic acidethyl ester Schiff base and 15 mmol of 4-methoxyphenylboronic acid or phenylboronic acid yielded the target products 3a (80%) and 3d (70%) with excellent yields. Additionally, scale-up experiments for the antitumor drug derivative (F-BPA) (8e) were conducted, achieving a 65% yield of the target product. It is worth mentioning that these reactions only require a low catalytic amount of palladium (1 mol%).
Several control experiments were conducted to explore the reaction pathway and the origin of stereoselectivity (Scheme 1). Initially, substrate 9 was synthesized and used instead of 1a. However, the desired product 9a was not observed, along with 9 being recovered. Based on these results, we believe that direct oxidative addition between Pd(0) and RCF2Br does not occur in the catalytic cycle (Scheme 1a). According to previous reports [64,65], we speculate that difluoroalkene amino acids (10) may be formed during the reaction under basic conditions. As expected, difluoroalkene amino acid (10) was easily isolated by treatment with triethylamine (Scheme 1b-1). However, it was unstable and readily transformed into other compounds. When 10 was used as the substrate instead of 1a, the product 3a was isolated in 87% yield (Scheme 1b-2), suggesting that 10 is the key precursor in this newly developed Pd-catalyzed cross-coupling reaction. Due to the rapid progression of the Suzuki coupling reaction, we decided to employ the Heck coupling to monitor the formation of the difluorodehydroalanine intermediate. The reaction progress was monitored at 0.5, 1.0, and 2.0 h using 19F NMR spectroscopy.
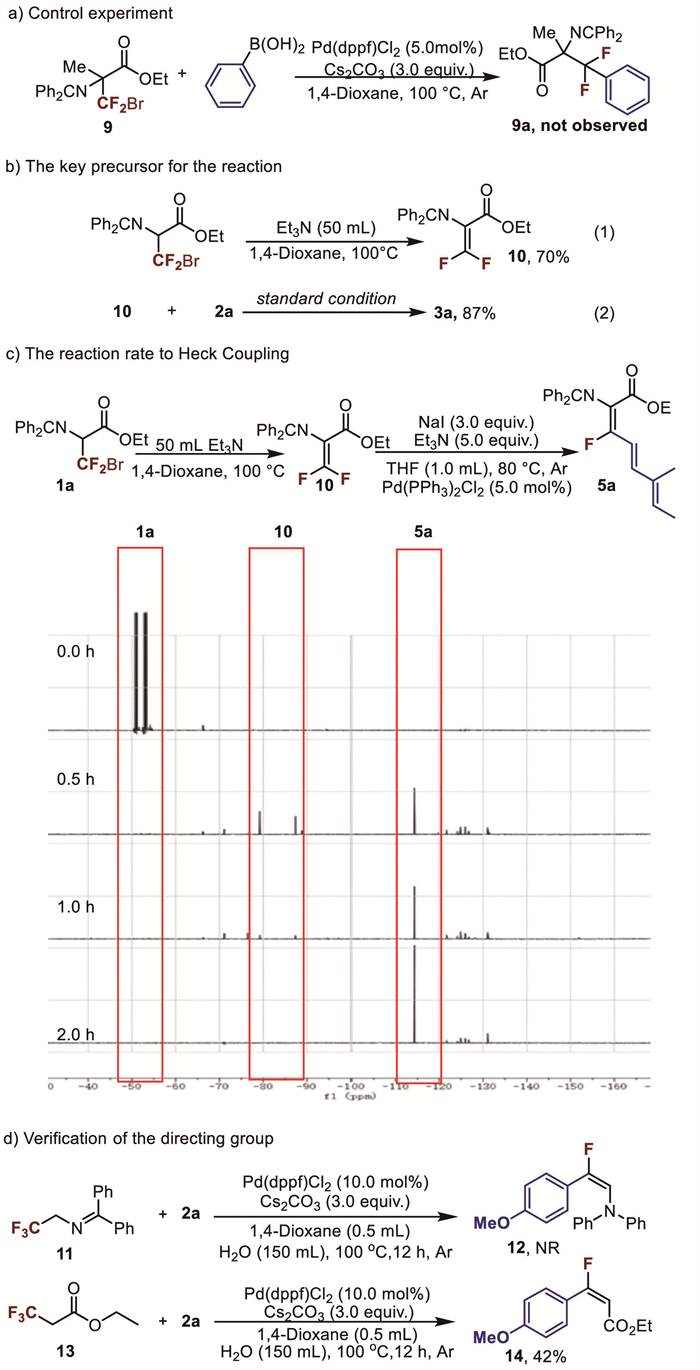
The results showed that difluoroalkene 10 formed rapidly, followed by its trans formation into the product (Scheme 1c). To further investigate the reaction pathway, substrates 11 and 13 were prepared. When they were used instead of 1a, product 12 was not obtained, whereas product 14 was isolated in 42% yield (Scheme 1d) [66]. These findings suggest that the oxidative addition step was assisted by the ester group rather than the nitrogen atom in substrate 1a, which contributed to high selectivity toward Z-isomer.
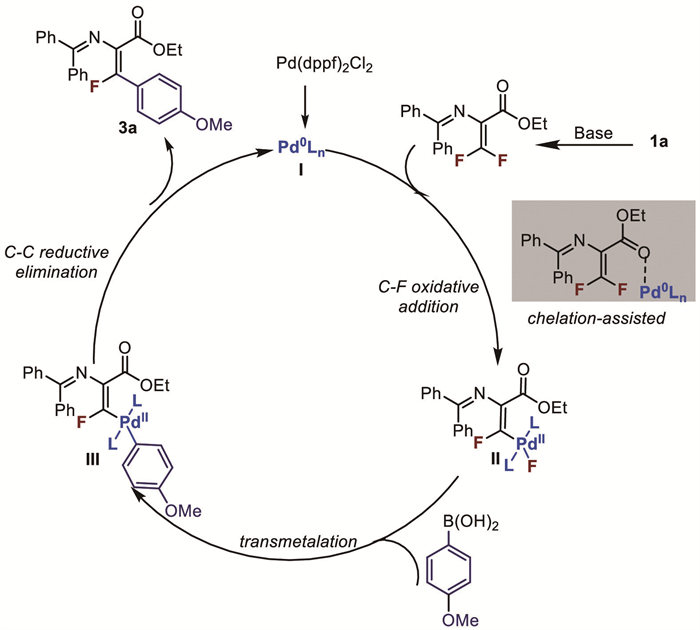
Based on the above mechanistic verification and previous reports, we propose the following catalytic cycle (Scheme 2): First, 2-amino-3-fluoropropenoic acid undergoes HBr elimination under basic conditions to produce difluorodehydroalanine 10. Pd(dppf)Cl₂ is reduced to Pd(0) by boric acid, and the Pd(0) complex is selectively added to the C-F bond of the difluoroolefin amino acids with the assistance of ester groups to form complex Ⅱ. This is followed by transmetalation and reductive elimination, yielding the product 3a, with the Pd complex being reduced back to Pd(0).
In summary, we have demonstrated that ethyl 3-bromo-2-((diphenylmethylene)amino)-3,3-difluoropropanoate is an effective gem-difluoroalkenes precursor, applicable in Suzuki and Heck couplings with palladium catalysts. This reaction provides a straightforward approach for constructing a library of fluorinated amino acids. The novel gem-difluoroalkenes precursor not only offer an efficient method for introducing fluorine-containing amino acid moieties into biologically important compounds such as clofibrate, pterostilbene, estrone, and tryptophan but also facilitates the synthesis of fluorinated BPA and aminocoumarin derivatives, further underscoring the synthetic potential of this approach.
The authors declare that they have no known competing financial interestsor personal relationships that could have appeared to influence the work reported in this paper.
Chuangchuang Liu: Writing – original draft, Validation, Resources, Methodology, Data curation. Yilian Song: Validation, Data curation. Wenjie Ju: Data curation. Xin Liu: Validation. Min Liang: Validation. Yingsheng Zhao: Writing – review & editing, Funding acquisition, Conceptualization.
This work was supported by the Natural Science Foundation of China (No. 22171197), the Major Basic Research Project of the Natural Science Foundation of Jiangsu Higher Education Institutions (No. 21KJA150002), the National Local Joint Engineering Laboratory to Functional Adsorption Material Technology for Environmental Protection (No. SDGC2121), and the PAPD Project.
Supplementary material associated with this article can be found, in the online version, at doi:
M. Hird, Chem. Soc. Rev. 36 (2007) 2070–2095. doi: 10.1039/b610738a
K. Müller, C. Faeh, F. Diederich, Science 317 (2017) 1881–1886.
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
R. Smits, C.D. Cadicamo, K. Burger, B. Koksch, Chem. Soc. Rev. 37 (2008) 1727–1739. doi: 10.1039/b800310f
Y. Yu, A. Liu, G. Dhawan, et al., Chin. Chem. Lett. 32 (2021) 42–52.
A. Papapetropoulos, S. Topouzis, S. Alexander, et al., British J. Pharma. 181 (2024) 1553–1575. doi: 10.1111/bph.16337
M. Drouin, J. Paquin, Beilstein J. Org. Chem. 13 (2017) 2637–2658. doi: 10.3762/bjoc.13.262
W. Dai, H. Shi, X. Zhao, S. Cao, Org. Lett. 18 (2016) 4284–4287. doi: 10.1021/acs.orglett.6b02026
Y. Wang, X. Qi, Q. Ma, P. Liu, G.C. Tsui, ACS Catal. 11 (2021) 4799–4809. doi: 10.1021/acscatal.0c05141
J.Q. Wu, S.S. Zhang, H. Gao, et al., J. Am. Chem. Soc. 139 (2017) 3537–3542. doi: 10.1021/jacs.7b00118
Z. Zhu, L. Lin, J. Xiao, Z. Shi, Angew. Chem. Int. Ed. 61 (2022) e202113209. doi: 10.1002/anie.202113209
R. Kojima, K. Kubota, H. Ito, Chem. Commun. 53 (2017) 10688–10691. doi: 10.1039/C7CC05225A
J. Hu, X. Han, Y. Yuan, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 13342–13346. doi: 10.1002/anie.201708224
Q. Ma, C. Liu, G.C. Tsui, Org. Lett. 22 (2020) 5193–5197. doi: 10.1021/acs.orglett.0c01813
Q. Ma, Y. Wang, G.C. Tsui, Angew. Chem. Int. Ed. 59 (2020) 11293–11297. doi: 10.1002/anie.202002219
H. Sakaguchi, Y. Uetake, M. Ohashi, et al., J. Am. Chem. Soc. 139 (2017) 12855–12862. doi: 10.1021/jacs.7b08343
H. Ito, T. Seo, R. Kojima, K. Kubota, Chem. Lett. 47 (2018) 1330–1332. doi: 10.1246/cl.180656
H. Sakaguchi, M. Ohashi, S. Ogoshi, Angew. Chem. Int. Ed. 57 (2018) 328–332. doi: 10.1002/anie.201710866
G. Coates, H.Y. Tan, C. Kalff, A.J.P. White, M.R. Crimmin, Angew. Chem. Int. Ed. 58 (2019) 12514–12518. doi: 10.1002/anie.201906825
H. Zhang, E. Wang, S. Geng, et al., Angew. Chem. Int. Ed. 60 (2021) 10211–10218. doi: 10.1002/anie.202100049
S.S. Yan, D.S. Wu, J.H. Ye, et al., ACS Catal. 9 (2019) 6987–6992. doi: 10.1021/acscatal.9b02351
C. Zhu, Y.F. Zhang, Z.Y. Liu, et al., Chem. Sci. 10 (2019) 6721–672.
F.P. Wu, Y. Yuan, J. Liu, X.F. Wu, Angew. Chem. Int. Ed. 60 (2021) 8818–8822. doi: 10.1002/anie.202017365
S. Xie, X. Gao, H. Wu, F. Zhou, J. Zhou, Org. Lett. 22 (2020) 8424–8429. doi: 10.1021/acs.orglett.0c03051
J. Xie, J. Yu, M. Rudolph, F. Rominger, A.S.K. Hashmi, Angew. Chem. Int. Ed. 55 (2016) 9416–9421. doi: 10.1002/anie.201602347
J. Li, Q. Lefebvre, H. Yang, Y. Zhao, H. Fu, Chem. Commun. 53 (2017) 10299–10302. doi: 10.1039/C7CC05758J
H. Yang, C. Tian, D. Qiu, et al., Org. Chem. Front. 6 (2019) 2365–2370. doi: 10.1039/c9qo00495e
J. Wang, B. Huang, C. Yang, W. Xia, Chem. Commun. 55 (2019) 11103–11106. doi: 10.1039/c9cc05293c
C. Zhu, Y. Zhang, Z. Liu, et al., Chem. Sci. 10 (2019) 6721–6726. doi: 10.1039/c9sc01336a
S. Xie, X. Gao, H. Wu, F. Zhou, J. Zhou, Org. Lett. 22 (2020) 8424–8429. doi: 10.1021/acs.orglett.0c03051
X. Lu, Y. Wang, B. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 12632–12637. doi: 10.1021/jacs.7b06469
X. Qi, Q. Ma, P. Liu, G.C. Tsui, ACS Catal. 11 (2021) 4799–4809. doi: 10.1021/acscatal.0c05141
Y. Wang, G.C. Tsui, Org. Lett. 25 (2023) 6217–6221. doi: 10.1021/acs.orglett.3c02452
S. Porey, Y. Bairagi, S. Guin, X. Zhang, D. Maiti, ACS Catal. 13 (2023) 14000–14011. doi: 10.1021/acscatal.3c02975
S.S. An, D.S. Wu, J.H. Ye, et al., ACS Catal. 9 (2019) 6987–6992. doi: 10.1021/acscatal.9b02351
D.H. Tan, E. Lin, W.W. Ji, et al., Adv. Synth. Catal. 360 (2018) 1032–1037. doi: 10.1002/adsc.201701497
J. Hu, X. Han, Y. Yuan, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 13342–13346. doi: 10.1002/anie.201708224
W. Dai, H. Shi, X. Zhao, S. Cao, Org. Lett. 18 (2016) 4284–4287. doi: 10.1021/acs.orglett.6b02026
T. Ma, Y. Chen, Y. Li, Y. Ping, W. Kong, ACS Catal. 9 (2019) 9127–9133. doi: 10.1021/acscatal.9b03172
L. Kong, B. Liu, X. Zhou, F. Wang, X. Li, Chem. Commun. 53 (2017) 10326–10329. doi: 10.1039/C7CC06048C
J.Q. Wu, S.S. Zhang, H. Gao, et al., J. Am. Chem. Soc. 139 (2017) 3537–3545. doi: 10.1021/jacs.7b00118
P. Tian, C. Feng, T.P. Loh, Nat. Commun. 6 (2015) 7472–7479. doi: 10.1038/ncomms8472
L. Kong, X. Zhou, X. Li, Org. Lett. 18 (2016) 6320–6323. doi: 10.1021/acs.orglett.6b03203
D. Zell, U. Dhawa, V. Müller, et al., ACS Catal. 7 (2017) 4209–4213. doi: 10.1021/acscatal.7b01208
N. Li, J. Chang, L. Kong, X. Li, Org. Chem. Front. 5 (2018) 1978–1982. doi: 10.1039/c8qo00297e
R.T. Thornbury, F.D. Toste, Angew. Chem. Int. Ed. 55 (2016) 11629–11632. doi: 10.1002/anie.201605651
L. Yang, W.W. Ji, E. Lin, et al., Org. Lett. 20 (2018) 1924–1927. doi: 10.1021/acs.orglett.8b00471
Y. Wang, G.C. Tsui, Org. Lett. 26 (2024) 5822–5826. doi: 10.1021/acs.orglett.4c02112
H. Zhou, W.A. van der Donk, Org. Lett. 3 (2001) 593–596. doi: 10.1021/ol006997s
M.K. Wang, Y.C. Luo, H.Y. Zhao, et al., ACS Catal. 13 (2023) 14090–14102. doi: 10.1021/acscatal.3c03578
C. Xu, R. Cheng, Y.C. Luo, M.K. Wang, X.G. Zhang, Angew. Chem. Int. Ed. 59 (2020) 18741–18747. doi: 10.1002/anie.202008498
C. Xu, Z.F. Yang, L. An, X.G. Zhang, ACS Catal. 9 (2019) 8224–8229. doi: 10.1021/acscatal.9b02488
L.C. Yu, J.W. Gu, S. Zhang, X.G. Zhang, J. Org. Chem. 82 (2017) 3943–3949. doi: 10.1021/acs.joc.7b00111
S. Chen, B.B. Yuan, Y.L. Wang, L. Ackermann, Angew. Chem. Int. Ed. 62 (2023) e202301168. doi: 10.1002/anie.202301168
S.N. Han, S.D. Liu, L. Liu, L. Ackermann, J. Li, Org. Lett. 21 (2019) 5387–5391. doi: 10.1021/acs.orglett.9b01400
Z.X. Ruan, S.K. Zhang, C.J. Zhu, et al., Angew. Chem. Int. Ed. 56 (2017) 2045–2049. doi: 10.1002/anie.201611595
X.P. Jiang, Y.Q.Q. Jiang, Q.Q. Liu, et al., J. Org. Chem. 87 (2022) 3546–3554. doi: 10.1021/acs.joc.1c03095
W.T. Fan, Y.T. Li, D.J. Wang, S.J. Ji, Y.S. Zhao, J. Am. Chem. Soc. 14 (2020) 20524–20530. doi: 10.1021/jacs.0c09545
X. Xu, N. Tao, W.T. Fan, et al., J. Org. Chem. 85 (2020) 13868–13876. doi: 10.1021/acs.joc.0c01909
G.L. Tu, D.J. Wang, C.C. Yuan, J.Y. Zhang, Y.S. Zhao, J. Org. Chem. 85 (2020) 10740–10749. doi: 10.1021/acs.joc.0c01257
G.L. Tu, C.C. Yuan, Y.T. Li, J.Y. Zhang, Y.S. Zhao, Angew. Chem. Int. Ed. 57 (2019) 15597–15601.
C.C. Yuan, L. Zhu, C.P. Chen, et al., Nat. Commun. 9 (2018) 1189–1199. doi: 10.1038/s41467-018-03341-6
C.C. Yuan, L. Zhu, R.S. Zeng, Y. Lan, Y.S. Zhao, Angew. Chem. Int. Ed. 57 (2018) 1277–1281. doi: 10.1002/anie.201711221
J. He, K. Yang, J. Zhao, S. Cao, Org. Lett. 21 (2019) 9714–9718. doi: 10.1021/acs.orglett.9b03815
W.H. Chen, X.J. Pei, X.X. Li, Y.S. Feng, Synth. Commun. 50 (2020) 3212–3220. doi: 10.1080/00397911.2020.1797813
T.L. Andersen, S.D. Friis, H. Audrain, et al., J. Am. Chem. Soc. 137 (2015) 1548–1555. doi: 10.1021/ja511441u

Figure 2 Substrate scope of Suzuki-Miyaura cross coupling. Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Cs2CO3 (3.0 equiv.), 150 µL H2O, Pd(dppf)2Cl2 (5 mol%), 1,4-dioxane (0.5 mL), 100 ℃, under argon, isolated yield.

Figure 3 Substrate scope of Mizoroki-Heck cross coupling. Reaction conditions: 1a (0.1 mmol), 4 (0.3 mmol), Et3N (5.0 equiv.), NaI (3.0 equiv.), Pd(PPh3)2Cl2 (5.0 mol%), THF (1.0 mL), at 100 ℃ for 2 h, under argon, isolated yield.
Table 1. Optimization of reaction conditions.a
|
||||
| Entry | [Pd] | Ligand | Base | Yield (%)b |
| 1 | PdCl2 | PPh3 | K2CO3 | 65 |
| 2 | PdCl2 | Davephos | K2CO3 | 70 |
| 3 | PdCl2 | Xantphos | K2CO3 | 69 |
| 4 | PdCl2 | dppf | K2CO3 | 76 |
| 5 | PdCl2 | dppe | K2CO3 | 65 |
| 6 | Pd(dppf)Cl2 | None | K2CO3 | 78 |
| 7 | Pd(dppf)Cl2 | None | K3PO4 | 60 |
| 8 | Pd(dppf)Cl2 | None | KHCO3 | 65 |
| 9 | Pd(dppf)Cl2 | None | KOH | 35 |
| 10 | Pd(dppf)Cl2 | None | Et3N | 43 |
| 11 | Pd(dppf)Cl2 | None | Cs2CO3 | 89 |
| 12 | None | None | Cs2CO3 | 0 |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), base (3.0 equiv.), 150 µL H2O, [Pd] (5 mol%), 1,4-dioxane (0.5 mL), 100 ℃, under argon. b Solated yield. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们






 下载:
下载: