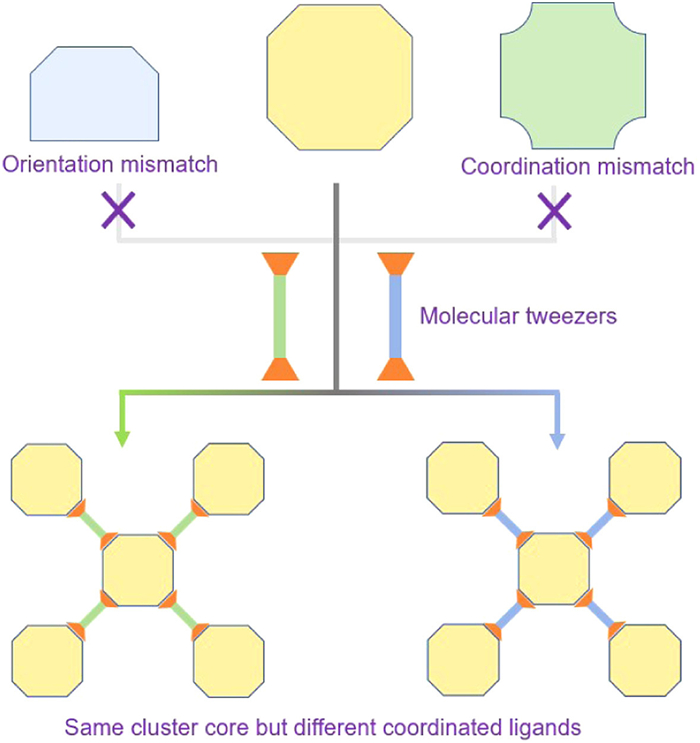
Scheme 1.
Illustration of molecular tweezers selecting metal clusters with identical cores.
Molecular tweezers in thiolate-protected Cu(Ⅰ) clusters: A framework platform for investigating ligand effects in cluster catalysis
Jiaojiao Zhang , Yifan Wu , Yifei Li , Fuxing Lin , Shengchang Xiang , Xi Fan , Zhangjing Zhang

Homogeneities and atomically precise coinage metal clusters (Cu, Ag, and Au) serve as precise platforms for bridging the gap between small metal-ligand molecules and metal nano-solids [1–5]. Consequently, they are selected as model catalysts due to their well-defined compositions and adjustable structures, establishing a robust foundation for correlating structures with properties [6–10]. Various ligands have been utilized to fabricate a range of coinage metal nanoclusters. It has been observed that surface ligands not only shield the metal core but also influence the catalytic properties, particularly the activity [11–15]. To comprehend the ligand effect involved in nanocluster catalysis, metal nanoclusters with fixed nuclearities but varying binding ligands serve as ideal model catalysts.
Owing to the weak metal-metal interactions and the diverse ligand-metal coordination, the structure of metal clusters exhibits considerable variability [16–18]. Generally, following the completion of a reaction, isolated single-crystalline metal clusters are not the exclusive synthesized product. They may be just more prevalent in yield or more readily form in saturated solutions, thereby facilitating crystallization. However, these conditions are notably sensitive, and even minor adjustments in the synthesis of metal clusters can lead to substantial alterations in the isolated cluster nuclei [19–23]. Moreover, guest solvents within the clusters tend to exhibit high volatility, resulting in the loss of crystallinity and presenting significant stability issues [24]. Consequently, only a limited number of investigations have focused on coinage metals possessing fixed cores but varying binding ligands in the context of catalysis [25–27].
Metal–organic frameworks constructed from novel cluster building blocks are fundamental aspects of reticular chemistry, representing naturally self-assembled superlattices of metal nanoparticles [28]. Both MOFs and coinage metal clusters have garnered extensive research attention. However, MOFs based on coinage metal clusters have received relatively scant attention, with only a handful of coinage metal clusters capable of serving as construction units for cluster-based frameworks [29]. Attaching clusters to adaptable bridging ligands enables their interconnection and the formation of robust cluster-based frameworks. These spatially separated clusters are likely to exhibit enhanced stability and unique properties compared to their monomeric counterparts. One crucial aspect of forming such supramolecular architectures is the use of effective building blocks, which provide ligand-accessible coordination sites with desired spatial orientations for assembly into architectures. In other words, the role of bridging ligands can also be viewed as acting like molecular tweezers, selectively picking preferred clusters and expediting their crystallization process. This eliminates the influence of factors such as inter-cluster interactions and solubility in solution on the resulting crystallized clusters. By employing tweezers with similar coordination preferences, it is possible to extract identical metal cluster building units, thereby enhancing stability and providing a platform to investigate the impact of ligands on cluster core catalytic performance (Scheme 1).
In this study, we successfully employed two types of carboxylic acid-based molecular tweezers to selectively capture two Cu6 clusters (Cu6-a and Cu6-b). They have identical cluster cores but different protected ligands, therefore provide accurate platform for investigating ligand effects in cluster catalysis. The incorporation of oxygen in OBB significantly enhances local spatial polarization, facilitating the charge separation and ROS generation efficiency of Cu6-b under visible-light irradiation. As a results, the oxygen-containing Cu6-b exhibits superior photocatalytic performance in the aerobic oxidation of sulfide, achieving both high yield and selectivity.
Solvothermal reaction of Cu(TFA)2·xH2O, 2-methyl-2-propanethiol (MPT) with 4,4′-biphenyl dicarboxylic acid (BPDC) in the present of Ph2SiH2 at 90 ℃ produced yellow needle-like crystals of Cu6-a. Single-crystal X-ray diffraction (SCXRD) analysis reveals that Cu6-a crystallizes in the monoclinic space group I2/a (Table S1 in Supporting information). The building units of Cu6-a resemble trigonal prismatic Cu6 clusters, stabilized by 4 MPT and 2 carboxylic acid ligands (Fig. 1a), indicating that Cu in Cu6-a exists as Cu(Ⅰ), and Cu6-a contains no free valence electron (6 – 4 – 2 = 0) (Fig. 1a). The Cu···Cu distances range from 2.725 Å to 3.067 Å (Fig. S1 in Supporting information), highlight the notable cuprophilic interactions within Cu6-a. 4 MPT ligands adopt two types of coordination modes with Cu atoms (
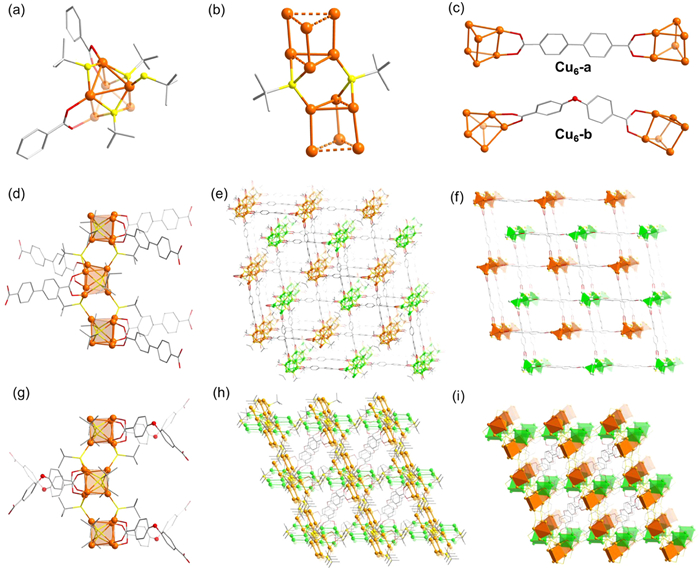
The less-than-optimal yield of Cu6-a indicates that Cu6 is clearly not the exclusive metal cluster product in this solvent reaction. BPDC can be likened to molecular tweezers, effectively selecting preferred Cu6 clusters and enhancing their crystallization rate. Therefore, employing another tweezer with similar coordination tendencies may aid in isolating identical Cu6 clusters. Analogously, substituting linear 4,4′-biphenyldicarboxylic acid ligands with Ⅴ-shaped 4,4′-oxybisbenzoic acid ligands (OBB) yields different crystals of Cu6-b. As expected, Cu6-b possess identical Cu6 building unit with Cu6-b, which are also linked into same 1D rods via MPT ligands (Figs. 1c and g). However, due to the Ⅴ-shaped OBB ligands, the rods propagate along the a- and b-axes in Cu6-b (Figs. 1h and i). It is noteworthy that one-directional straight rod SBUs, like those in Cu6-a, dominate the rod frameworks. While two-directional rod frameworks, where rods propagate along both the a- and b-axes, are rare [30]. Therefore, Cu6-b represents a significant addition to the scarce family of two-directional rod frameworks, which represents the first instance in coinage metal clusters-based MOFs.
This pair of isostructural Cu6 based rods frameworks provides us with a valuable opportunity to accurately study the ligand-oriented catalytic properties in Cu clusters. To evaluate the optical properties of Cu6-a and Cu6-b, UV–vis diffuse reflectance spectroscopy (DRS) measurements were performed. As shown in Fig. 2a, both materials exhibit absorption edges extending beyond 500 nm, indicating their partial capacity to absorb visible light. The slightly red-shift observed in Cu6-b compared to Cu6-a, suggesting that Cu6-b exhibits a stronger response to visible light. Furthermore, the optical bandgaps of Cu6-a and Cu6-b were investigated based on the Tauc plots. The Eg values for Cu6-a and Cu6-b were determined to be 2.55 eV and 2.50 eV, respectively (Fig. S7 in Supporting information). To ascertain the conduction band (CB) positions, Mott-Schottky (MS) electrochemical measurements were conducted (Fig. S8 in Supporting information). The ECB values were estimated to be −0.58 V and −0.62 V versus Ag/AgCl (−0.48 V and −0.52 V versus NHE). Consequently, the valence band (VB) positions were thus calculated to be 2.07 V (Cu6-a), and 1.98 V (Cu6-b) versus NHE by using the equation (Eg = EVB – ECB) (Fig. 2b). Photocurrent measurements corroborated enhanced charge separation efficiency in Cu6-b over Cu6-a, evidenced by significantly higher photocurrent intensity recorded for Cu6-b (Fig. 2c). Electrochemical impedance spectroscopy (EIS) plots demonstrated a gradual decrease in interfacial charge transfer resistance from Cu6-a to Cu6-b (Fig. 2d).
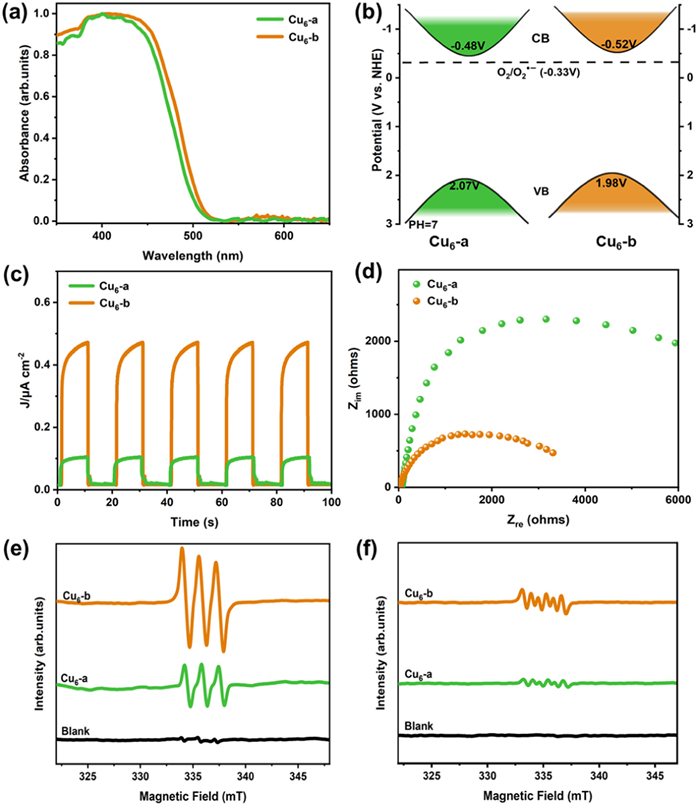
The CB positions of Cu6-a and Cu6-b possess more negative potentials than that of the O2/O2•- redox couple (−0.33 V versus NHE), indicating that the generation of reactive oxygen species (ROS) is thermodynamically favorable. In the presence of photocatalysts and molecular oxygen, two types of ROS, superoxide radical (O2•-) and singlet oxygen (1O2), are typically produced. Consequently, electron paramagnetic resonance (EPR) measurements were conducted to confirm the formation of both O2•− and 1O2. The presence of 1O2 was verified using 2,2,6,6-tetramethylpeperidine (TEMP) as trapping agent, while 5,5-dimethylpyrroline-N-oxide (DMPO) was employed for the detection of O2•−. The distinctive triplet signals of 1O2, characterized by the ratio of 1:1:1, are prominently observable in the EPR spectrum, with Cu6-b exhibiting a more pronounced signal for 1O2 in comparison to Cu6-a (Fig. 2e). Additionally, Cu6-a displayed weak six-line EPR signals indicative of O2•− formation; however, signal intensity observed with Cu6-b exceeded that seen with Cu6-a (Fig. 2f). The higher peak intensities suggest a greater efficiency in free radical generation. Consequently, Cu6-b demonstrates superior yields of both singlet oxygen and superoxide radicals compared to Cu6-a.
Given the differential capacities of Cu6-a and Cu6-b in generating reactive oxygen species, we further compared their photocatalytic potential in the oxidation of thioethers under visible light irradiation at room temperature. Specifically, Cu6-b demonstrated superior photocatalytic performance in the oxidation of thioalkyl methyl ether under blue light, achieving conversions exceeding 99% within 4 h (Table 1, Figs. S9 and S11 in Supporting information). In contrast, when Cu6-a was used as the photocatalyst under similar conditions, the conversion dropped to merely 8% even after 12 h, which correlates with its efficiency in generating reactive oxygen species (Table 1, Figs. S10 and S11 in Supporting information). The catalytic performance of Cu6-b remains consistent across three cycles and no significant structural alterations were observed as confirmed by PXRD and FTIR spectra (Figs. S12-S14 in Supporting information). Control experiments conducted in darkness, under a nitrogen atmosphere, or without Cu6-b showed negligible conversion rates, indicating that light, oxygen, and Cu6-b are all essential for this reaction (Table 1, entries 2–4). Furthermore, the photocatalytic efficacy of Cu6-b significantly decreased as the wavelength of the light source increased, suggesting that blue light energy is crucial for electronic excitation in this context (Table 1, entries 5 and 6).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Change from the “standard conditions” | Conv. (%)b | Sel. (2a:3a,%)b |
| 1 | None | 99 | 95 |
| 2 | No light | Trace | – |
| 3 | Under N2, instead of air | Trace | – |
| 4 | No Cu6-b | Trace | – |
| 5 | Green LED, instead of blue LED | Trace | – |
| 6 | Red LED, instead of blue LED | Trace | – |
| 7 | 3.0 mg Cu6-b | 90 | 95 |
| 8 | Cu6-a instead of Cu6-b | 8 | >99 |
| a Reaction conditions: 1a (0.20 mmol), Cu6-b (5.0 mg), acetonitrile (2.0 mL), H 2O (1 mL), r.t., 4 h, Cu6-a (5.0 mg), r.t., 12 h blue light-emitting diode (LED), in an O 2 atmosphere. b Determined by 1H NMR analysis of the crude reaction mixture. | |||
After optimizing the reaction conditions, we further investigated the substrate scope that could be applied in this reaction using Cu6-b as the catalyst (Table 2). The sulfones produced through the photocatalytic oxidation of substituted thioanisole derivatives exhibited remarkable yields and selectivity. A wide range of phenyl sulfide substrates, whether containing electron-withdrawing groups (-CHO) or electron-donating groups (-CH3, –OCH3, or -OH), were successfully converted into their corresponding sulfoxides. Importantly, there was no cooxidation observed with adjacent hydroxyl or carbonyl functionalities. This finding suggests a high degree of functional group tolerance during this gentle oxidation process (Figs. S9 and S15-S18 in Supporting information).
DownLoad:
CSV
 |
||||
| Entry | Substrate | Product | Yield (%)b | Sel. (2:3,%)b |
| 1 |  |
 |
99 | 95 |
| 2 |  |
 |
95 | 93 |
| 3 |  |
 |
98 | 93 |
| 4 |  |
 |
99 | 98 |
| 5 |  |
 |
99 | 94 |
| a Reaction conditions: 1a (0.20 mmol), Cu6-b (5.0 mg), acetonitrile (2.0 mL), H 2O (1 mL), r.t., 4 h, blue light-emitting diode (LED), in an O 2 atmosphere. b Determined by 1H NMR analysis of the crude reaction mixture. | ||||
In order to further investigate the photocatalytic oxidation of thioether, we conducted radical quenching experiments. The yield of sulfoxide significantly dropped when L-histidine (L-His) and p-benzoquinone (BQ) was employed as 1O2 and O2•− quenchers, respectively, with L-His causing a greater reduction (Figs. S19 and S20 in Supporting information). These results clearly indicate that both O2•− and 1O2 play significant roles in the synthesis of sulfoxides, and that 1O2 is the predominant species in the photocatalytic oxidation of thioethers. Additionally, the introduction of t-BuOH as a hydroxyl radical (·OH) scavenger did not result in a significant decrease in the yield of compound 2a. This observation excludes the participation of the ·OH pathway in the photocatalytic oxidation process of thioethers (Fig. S21 in Supporting information).
According to the data presented above, we propose analogous mechanisms for the photocatalytic oxidations. Initially, upon visible light irradiation, electrons in the HOMO of Cu6 are excited into the LUMO, generating free electrons and positively charged holes. Subsequently, O2 is activated into 1O2 and O2•- through energy transfer (ET) and singlet electron transfer (SET) processes, respectively. The ET process involves the formation of a phenyl sulfide radical cation via the interaction of 1O2 with phenyl sulfide. This radical cation then reacts with another phenyl sulfide to produce the desired product, phenyl sulfoxide. Additionally, photo-generated hole oxidizes the phenyl sulfide to its corresponding radical cation. In the SET process, the interaction between the phenyl sulfide radical cation and O2•− results in the formation of an intermediate peroxysulfoxide, which further reacts with additional phenyl sulfide molecules to generate target phenyl sulfoxide. Consequently, the synergistic effects of both ET and CT processes facilitate the production of this end productphenyl sulfoxide (Fig. 3a).

Polarization can be utilized to modulate the optical and electrical properties of solid-state materials, enhancing their charge transfer efficiency [31]. To gain a deeper understanding of the distinct optical and electrochemical properties between Cu6-a and Cu6-b, we calculated the dipole moments of the two component fragments depicted in the figure using the density functional theory (DFT) method with the M06–2X/def2tzvp basis set. The dipole moments of the two component fragments in the figure were calculated through the density functional theory (DFT) of the M06–2X/def2tzvp method. The results revealed that Cu6-b exhibits a dipole moment of 3.50 Debye, surpassing that of Cu6-a at 3.34 Debye (Figs. 3b and c). Therefore, the incorporation of oxygen in OBB significantly enhances local spatial polarization and favors the creation of a robust internal electric field, thereby accelerating in-plane charge transfer and facilitating efficient in-plane charge separation.
In summary, we successfully employed two types of carboxylic acid-based molecular tweezers to selectively capture two Cu6 clusters (Cu6-a and Cu6-b). A noteworthy aspect of Cu6-b is its unique two-directional rod framework, which represents the first example within coinage metal cluster-based MOFs. Both Cu6-a and Cu6-b share identical cluster cores but different protected ligands, therefore provide accurate platform for investigating ligand effects in cluster catalysis. The incorporation of oxygen in OBB significantly enhances local spatial polarization, facilitating the charge separation and ROS generation efficiency of Cu6-b under visible-light irradiation. As a result, the oxygen-containing Cu6-b exhibits superior photocatalytic performance in the aerobic oxidation of sulfide, achieving both high yield and selectivity. This work provides a valuable approach for precisely control the Cu clusters structures to regulate their properties.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiaojiao Zhang: Writing – original draft, Methodology, Formal analysis, Data curation. Yifan Wu: Formal analysis. Yifei Li: Formal analysis. Fuxing Lin: Resources. Shengchang Xiang: Resources. Xi Fan: Writing – review & editing, Writing – original draft, Supervision, Conceptualization. Zhangjing Zhang: Writing – review & editing, Conceptualization.
This work was supported by National Natural Science Foundation of China (Nos. 22101048, 22271046 and 22373015), the National Science Fund for Distinguished Young Scholars of China (No. 22425102), the Natural Science Foundation of Fujian Province (No. 2021J01150).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Deng, B.K. Teo, N. Zheng, J. Am. Chem. Soc. 143 (2021) 10214–10220. doi: 10.1021/jacs.1c03251
Y.L. Liu, S.Z. Zhan, J.X. Sun, et al., Angew. Chem. Int. Ed. 62 (2023) e202306748. doi: 10.1002/anie.202306748
W.D. Liu, J.Q. Wang, S.F. Yuan, et al., Angew. Chem. Int. Ed. 60 (2021) 11430–11435. doi: 10.1002/anie.202100972
Y.-P. Xie, Y.L. Shen, G.X. Duan, et al., Mater. Chem. Front. 4 (2020) 2205–2222. doi: 10.1039/d0qm00117a
X. Kang, M. Zhu, Chem. Soc. Rev. 48 (2019) 2422–2457.
S.F. Yuan, R.L. He, X.S. Han, et al., Angew. Chem. Int. Ed. 60 (2021) 14345–14349. doi: 10.1002/anie.202103060
X.K. Wan, J.Q. Wang, Q.M. Wang, Angew. Chem. Int. Ed. 60 (2021) 20748–20753. doi: 10.1002/anie.202108207
X. Du, R. Jin, ACS Nano 13 (2019) 7383–7387. doi: 10.1021/acsnano.9b04533
L.J. Liu, Z.Y. Wang, Z.Y. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202205626. doi: 10.1002/anie.202205626
X. Fan, J. Cheng, M. Qiu, et al., ACS Mater. Lett. 5 (2023) 1527–1531. doi: 10.1021/acsmaterialslett.3c00101
L. Chen, F. Sun, Q. Shen, et al., Nano Res. 15 (2022) 8908–8913. doi: 10.1007/s12274-022-4812-6
Q. Tang, G. Hu, V. Fung, et al., Acc. Chem. Res. 51 (2018) 2793–2802. doi: 10.1021/acs.accounts.8b00380
H. Liang, B.J. Liu, B. Tang, et al., ACS Catal. 12 (2022) 4216–4226. doi: 10.1021/acscatal.2c00841
Z.Y. Wang, M.Q. Wang, Y.L. Li, et al., J. Am. Chem. Soc. 140 (2018) 1069–1076. doi: 10.1021/jacs.7b11338
J. Wang, F. Xu, Z.Y. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202207492. doi: 10.1002/anie.202207492
Z. Lei, X.K. Wan, S.F. Yuan, et al., Acc. Chem. Res. 51 (2018) 2465–2474. doi: 10.1021/acs.accounts.8b00359
H.Y. Zhuo, H.F. Su, Z.Z. Cao, et al., Chem. Eur. J. 22 (2016) 17619–17626. doi: 10.1002/chem.201603797
S. Li, Z.Y. Wang, G.G. Gao, et al., Angew. Chem. Int. Ed. 57 (2018) 12775–12779. doi: 10.1002/anie.201807548
X. Fan, F. Yuan, J. Wang, et al., CCS Chem. 5 (2023) 350–360. doi: 10.31635/ccschem.022.202101741
X. Fan, F. Yuan, D. Li, et al., Angew. Chem. Int. Ed. 60 (2021) 12949–12954. doi: 10.1002/anie.202101664
B.L. Han, Z. Liu, L. Feng, et al., J. Am. Chem. Soc. 142 (2020) 5834–5841. doi: 10.1021/jacs.0c01053
S. Hossain, Y. Imai, D. Suzuki, et al., Nanoscale 11 (2019) 22089–22098. doi: 10.1039/c9nr07117b
X. Kang, M. Zhu, Chem. Soc. Rev. 48 (2019) 2422–2457.
R.W. Huang, Y.S. Wei, X.Y. Dong, et al., Nat. Chem. 9 (2017) 689–697. doi: 10.1038/nchem.2718
Q.J. Wu, D.H. Si, P.P. Sun, et al., Angew. Chem. Int. Ed. 62 (2023) e202306822. doi: 10.1002/anie.202306822
J.P. Dong, Y. Xu, X.G. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202313648. doi: 10.1002/anie.202313648
X.K. Wan, J.Q. Wang, Z.A. Nan, et al., Sci. Adv. 3 (2017) e1701823. doi: 10.1126/sciadv.1701823
X. Fan, K. Zuo, F. Yuan, et al., ACS Mater. Lett. 6 (2024) 1007–1011. doi: 10.1021/acsmaterialslett.3c01630
Y. Jin, C. Zhang, X.Y. Dong, et al., Chem. Soc. Rev. 50 (2021) 2297–2319. doi: 10.1039/d0cs01393e
Y.F. Zhang, Z.H. Zhang, L. Ritter, et al., J. Am. Chem. Soc. 143 (2021) 12202–12211. doi: 10.1021/jacs.1c04946
K. Wu, P.W. Cheng, X.Y. Liu, et al., Sci. China Chem. 67 (2024) 1000–1007. doi: 10.1007/s11426-023-1820-5

Scheme 1 Illustration of molecular tweezers selecting metal clusters with identical cores.

Figure 1 (a) The structure of the building block of Cu6 cluster. (b) Bridge linker MPT between the adjacent Cu6 clusters. (c) Carboxylic acid-based molecular tweezers capture two Cu6 clusters. (d) The chain, (e) the double-interpenetrated 3D framework and (f) its polyhedral model of Cu6-a. (g) The chain, (h) the crosswise 3D framework and (i) its polyhedral model of Cu6-b. Atom color codes: Cu, orange or green; O, red; C, gray; S, yellow. H atoms are omitted for clarity.

Figure 2 (a) The UV-visible diffuse reflectance spectrum (UV–vis DRS) of Cu6-a and Cu6-b. (b) Schematic illustration for the band structures of Cu6-a and Cu6-b. (c) Photocurrent of Cu6-a and Cu6-b under visible light irradiation. (d) The EIS plots of Cu6-a and Cu6-b. EPR spectra of Cu6-a and Cu6-b in the presence of (e) TEMP-1O2 and (f) DMPO-O2•− under visible light irradiation.

Figure 3 (a) Possible reaction mechanism of the photocatalytic thioether oxidation of Cu6-b. The molecular dipole of (b) Cu6-a and (c) Cu6-b.
Table 1. Visible-light-driven aerobic oxidation of thioanisole.a
|
|||
| Entry | Change from the “standard conditions” | Conv. (%)b | Sel. (2a:3a,%)b |
| 1 | None | 99 | 95 |
| 2 | No light | Trace | – |
| 3 | Under N2, instead of air | Trace | – |
| 4 | No Cu6-b | Trace | – |
| 5 | Green LED, instead of blue LED | Trace | – |
| 6 | Red LED, instead of blue LED | Trace | – |
| 7 | 3.0 mg Cu6-b | 90 | 95 |
| 8 | Cu6-a instead of Cu6-b | 8 | >99 |
| a Reaction conditions: 1a (0.20 mmol), Cu6-b (5.0 mg), acetonitrile (2.0 mL), H 2O (1 mL), r.t., 4 h, Cu6-a (5.0 mg), r.t., 12 h blue light-emitting diode (LED), in an O 2 atmosphere. b Determined by 1H NMR analysis of the crude reaction mixture. | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Visible-light-driven aerobic oxidation of sulfide derivatives.a
|
||||
| Entry | Substrate | Product | Yield (%)b | Sel. (2:3,%)b |
| 1 | |
|
99 | 95 |
| 2 | |
|
95 | 93 |
| 3 | |
|
98 | 93 |
| 4 | |
|
99 | 98 |
| 5 | |
|
99 | 94 |
| a Reaction conditions: 1a (0.20 mmol), Cu6-b (5.0 mg), acetonitrile (2.0 mL), H 2O (1 mL), r.t., 4 h, blue light-emitting diode (LED), in an O 2 atmosphere. b Determined by 1H NMR analysis of the crude reaction mixture. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: