Shenzhen Key Laboratory of Polymer Science and Technology, Guangdong Research Center for Interfacial Engineering of Functional Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China
b.
CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China
c.
Institute of Critical Materials for Integrated Circuits, Shenzhen Polytechnic University, Shenzhen 518055, China
zengqd@nanoctr.cn (Q.-D. Zeng). 1 These authors contributed equally to this work.
Received Date:
01 November 2023 Accepted Date:
28 January 2024 Revised Date:
10 January 2024 Available Online:
15 October 2024
Abstract:
The topology of conjugated macrocycles had significant impacts on their photo-physical and photo-chemical properties. Herein, a series of π-conjugated macrocycles with diverse topology were synthesized via intramolecular McMurry coupling. Their chemical structure and macrocyclic topology were unambiguously confirmed via NMR, MALDI-TOF mass spectra, crystal analysis and scanning tunneling microscopy (STM). Depending on the structural topology and structural rigidity, these cyclic compounds display obviously distinctive emission behavior and photochemical reactions in the solution and in the solid state. Monocyclic phenylene vinylene macrocycle (denoted as MST) exhibiting aggregation-induced emission behavior, was more vulnerable to photo-cyclization in solution and triplet sensitizer promoted photo-dimerization due to lower strain and more flourishing intramolecular motions. After UV light irradiation, relatively more flexible MST could yield the anti-dimer via triplet excimer on the HOPG surface confirmed by STM investigation. By contrast, highly constrained bicyclic analogue (named as DMTPE) with central tetraphenylethene core, displayed high emission quantum yields of 68% both in solution and in the solid state, and was relatively inert to photochemical reactions and yield syn-dimer on the surface via singlet excimer involved [2 + 2] photo-dimerization. Based on the solution-mediated photo-polymerization of MST moiety, multicyclic porous carbon-rich ribbon connected with four-membered ring was successfully constructed and validated via STM imaging.
The exploration of structural diversities, complexities and topologies of molecules and frameworks is always the core task for chemists and material scientists [1]. Among them, π-conjugated macrocyclic molecules have attracted extensive attentions owing to their unique chemical structures and novel physical properties [2–4]. For pursuing the structural and topological boundary and complexity of conjugated macrocycle, tremendous efforts have been devoted to their synthetic exploration. Owing to the inherent voids within the cyclic skeleton, conjugated macrocycles could be regarded as the supramolecular host for small molecules, like fullerene and aromatic compounds [5,6]. In addition, these cyclic carbon-rich compounds was also represented as the nano-sized substructures and infinite lattices for nanoporous carbon allotropes and related materials [7], such as the relationship between cycloparaphenylenes (CPP) with carbon nanotube [8,9] and dehydro[n]annulene with graphdiyne [10]. This structural similarity would also shed clues on the fabrications of nano-porous carbon materials [11]. Parallel with the flourishing synthetic adventure to-ward novel conjugated macrocycles, their applications in diverse fields had also emerged in the past decade [4,12]. Originated from their infinite π-conjugated backbone, conjugated macrocycles had proven their unique electronic properties and performance in organic photonics and electronics, ranging from emitters in organic light-emitting diodes [13,14], bio-imaging fluorophores [15–17] and sensors [18–21], to organic semiconductors in photodetectors [22], field effect transistor [23], and organic photovoltaic [24].
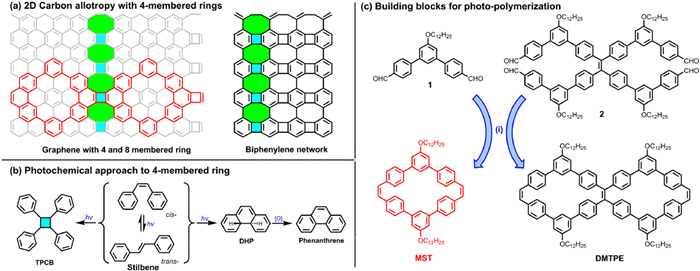
Among those reported conjugated macrocycles with diverse structure, phenylene vinylene macrocycles (PVMs) [25–28], had shared similar rigid and planar cyclic conjugated backbone which endow them controllable tendency for self-aggregation and guest binging [29]. As the main components in PVMs, diarylethene moieties were well known for their complicated photophysical and photochemical processes [30]. And thus, PVMs composed by benzene rings and ethylene linkages were also rendered the possibility to undergo diverse photo-pumped process, which had rarely been explored for PVMs before. Beside the photophysical process like fluorescence and phosphorescence, diarylethene molecules and its analogues could mainly undergo three types of photochemical reactions (Scheme 1a): (i) Fast unimolecular cis-trans isomerization; (ii) slow electrocyclic rearrangement of cis-S1 state to an isomeric 4a, 4b-dihydrophenanthrene (DHP) and subsequent conversion to phenanthrene in presence of oxidants; (iii) very slow bimolecular [2 + 2] dimerization between an excited stilbene and a ground state stilbene to yield 1,2,3,4-tetraphenylcyclobutane (TPCB). These non-radiative processes of diarylethene molecules were competitive with each other and other radiative emissive process under the impacts of inherent chemical and electronic structures, and exterior environments. Although the [2 + 2] dimerization toward four-membered ring was featured with low yield due to the bimolecular mechanism, the dimerization reaction of alkene derivatives had recently been proven to be a reliable approach to construct photo-responsive systems [31], shape memory materials [32] and crystal-to-crystal transformation [33]. However, the photo-chemical process for PVMs, especially the impacts of cyclic topology on photochemical reactions, was rarely studied. Considering the reliable [2 + 2] photo-dimerization of alkenes, we envisioned the possibility of building-up non-benzenoid nanoporous carbon-rich materials with PVM as the elementary building blocks and [2 + 2] dimerization as the polymerization routes.
Scheme 1
Scheme 1.
(a) Photochemical reactions related to stilbene: (i) cis-trans isomerization; (ii) photo-cylization; (iii) [2 + 2] photo-dimerization. (b) Representative structure of four-membered ring containing carbon networks. (c) Synthetic routes toward π-conjugated macrocycles with diverse topology. Condition: TiCl4, Zn, Pyridine, THF, refluxing, 24.7% for MST, 38.8% for DMTPE.
For carbon allotropes and related carbon-rich materials, six-membered benzenoid ring was the dominating building blocks, whereas the introduction of non-six membered ring into carbon network would dramatically impact their electronic structure and performance [34–36]. Among reported non-benzenoid carbon-rich structures, four-membered ring was rarely explored owing to the lacking of efficient approach to construct four-membered ring. Recently, the biphenylene network, a planar sp2-hybridized carbon allotropes with periodically arranged four-, six-, and eight-membered rings, were fabricated through an on-surface interpolymer dehydrofluorination (HF-zipping) reaction (Scheme 1b) [37]. The metallic and multiradical nature of this new allotrope had made biphenylene network a promising candidate for conducting future electronic and spintronic devices [38]. However, the lacking of efficient reaction toward four-membered ring had severely hampered the solution-mediated synthesis of carbon-rich structures containing four-member rings, and consequently limited their further structural exploration and derivatization.
Herein, we had presented the synthesis of two π-conjugated PVMs (MST and DMTPE in Scheme 1c) with diverse topology, and explored their photo-physical and photo-chemical properties. Topology of these cyclic building blocks was revealed to be of great significance for their inherent photo-physical performance and photochemical reactions. Relatively flexible monocyclic MST exhibited aggregation-induced emission behavior, whereas bicyclic DMTPE was verified to be a dual-phase emitter owing to the more rigid skeleton constrained by bicyclic topology and consequently restricted intramolecular motions [39]. On the other hand, the cyclic topology of PVMs also plays a critical role in their photochemical process. Both PVMs underwent on-surface bimolecular [2 + 2] photo-dimerization confirmed by STM, however, monocyclic MST yield the anti-dimer from its triplet excimer and bicyclic DMTPE give out the syn-dimer from its singlet excimer. In the solution, monocyclic MST could readily undergo photocyclization and photo-dimerization, whereas bicyclic DMTPE was relatively inert to photochemical reactions due to the restraining bicyclic topology. Then, solution-mediated photo-polymerization of monocyclic MST toward multicyclic chain connected via four-membered ring was accomplished and confirmed via STM imaging.
The synthesis toward bicyclic DMTPE was firstly attempted via ring closure olefin metathesis following the classical procedures toward PVMs (Scheme S1 in Supporting information) [27], in which the AIE-active tetraphenylethene (TPE) core could act as a template to facilitate the cyclization. However, GPC curves of crude products had indicated the yield of oligomers instead of cyclic targets even after elongating the reaction time and raising the reaction temperature (Table S1 in Supporting information). This was probably owing to the relative higher strain within the bis-cyclic skeleton, which disfavored the yield of DMTPE. Hence, irreversible McMurry coupling reaction was applied to build-up the bis-cyclic DMTPE and monocyclic analogue MST from the aldehyde-terminted precursor 2 and 3 (Schemes S1 and S2 in Supporting information) [40,41]. Fortunately, two targeted MST and DMTPE were successfully obtained with a satisfactory yield of 24.7% and 38.8%, respectively.
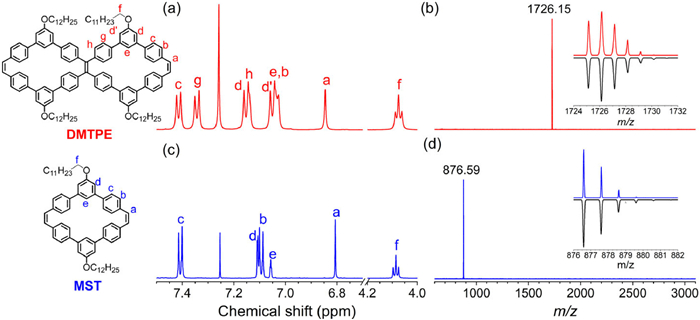
The chemical identities of these two cyclic compounds were unambiguously verified by 1H nuclear magnetic resonance (NMR) spectroscopy and matrix-assisted laser ionization time-of-flight (MALDI-TOF) mass spectra (Fig. 1). 1H NMR spectra together with 2D correlation spectroscopy (COSY) and nuclear Overhauser enhancement spectroscopy (NOESY) provided further structural proof for the PVMs, where every proton signal could be unambiguously assigned (Figs. 1a and c). The MALDI-TOF MS spectra of DMTPE and MST both indicated the presence of a single species with m/z = 1726.15 and 876.59, which were consistent with their desired molar mass. In addition, the experimental isotopic distributions were also in perfect agreement with their simulated patterns (Figs. 1b and d).
Figure 1
Figure 1.
Partial 1H NMR and MALDI-TOF MS spectra of π-conjugated macrocycles DMTPE (a, b) and MST (c, d).
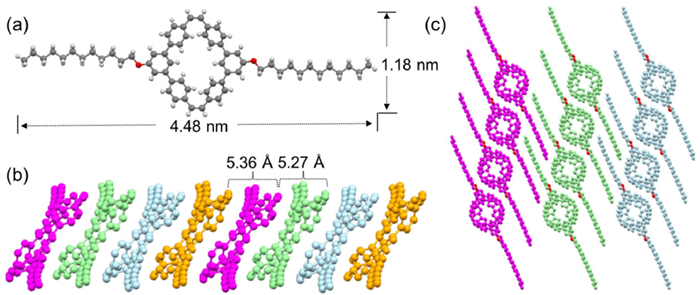
To further confirm the cyclic structure of these conjugated macrocycles, the single crystals of the two PVMs were grown via slowly evaporation of their solution in DCM. Fortunately, crystal of monocyclic MST (CCDC: 2291479) was obtained and resolved (Fig. 2a). As shown in Fig. 2b, the cyclic MST was tilted and further arrayed into a column with the voids connecting into a nano-sized channel. In the arrayed column, the conjugated ring was parallel with each other whereas the distance between closest alkene moieties of neighboring rings was estimated to be approximately 5.36 and 5.27 Å. This large distance between alkene bonds had consequently prevented the [2 + 2] dimerization between adjacent MSTs in the nano-porous column. Moreover, the columns were orderly packed in line with intercalating dodecyl chains (Fig. 2c), which further hampered the possible [2 + 2] photo-dimerization between adjacent columns.
Figure 2
Figure 2.
(a) Crystal structure of monocyclic MST. (b, c) Packing patterns of MST in the crystal with the distance between adjacent C = C bond in each column labeled.
In order to elucidate the cyclic topology of these conjugated macrocycles, their geometries were also studied with density functional theory (DFT) calculations using functional B3LYP with the 6–31G(d) basis set. The optimized structures clearly revealed their cyclic shaped topology and nano-porous backbone with highly twisted phenyl rings (Fig. S3 in Supporting information). The diameter of the void within the cyclic backbone for MST and DMTPE was approximately 0.4 nm. The homodesmotic calculations of MST and DMTPE were also carried out to evaluate their strain energy within the rigid cyclic backbones (Fig. S4 in Supporting information) [42]. With their corresponding acyclic strain-released counterparts as comparison, bicyclic DMTPE exhibited obviously raised strain relative to its monocyclic analogue MST. The strain energies of DMTPE and MST were calculated to be 3.83 kcal/mol and 6.06 kcal/mol.
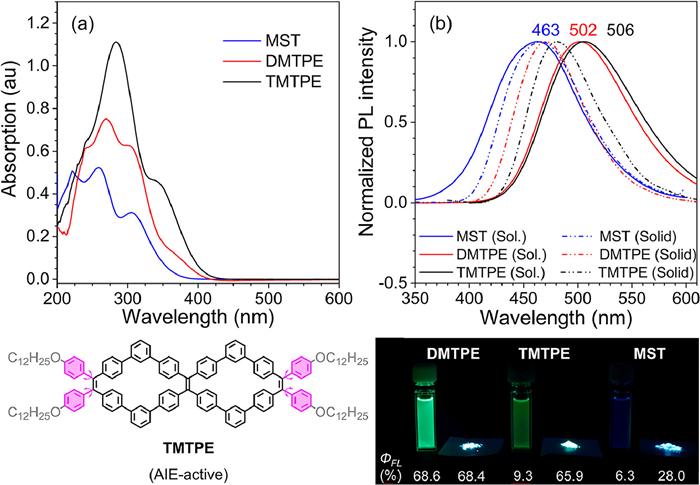
The UV–vis absorption and photoluminescence (PL) spectra of these PVMs were further recorded to investigate the impacts of cyclic topology on their photo-physical properties (Fig. 2 and Table S2 in Supporting information). To better understand topological impacts, compound TMTPE as a derivative of DMTPE with four peripheral free-rotating phenyl rings, was synthesized as a comparison (Scheme S3 in Supporting information). As recorded, the absorption spectra had gradually red-shifted following the order of MST, DMTPE and TMTPE owing to the π-conjugation extension from monocyclic to bicyclic backbone and attachment of phenyl rings (Fig. 3a). This result agreed well with their simulated UV–vis absorption spectra based on the time-dependent DFT calculation using functional B3LYP with the 6-31G(d) basis set (Figs. S5-S7 in Supporting information). The emission spectra of these PVMs in THF also exhibited a remarkable bathochromic shift from 462 nm for MST, to 502 nm for DMTPE and 506 nm for TMTPE (Fig. 3b). The red-shifted absorption and emission was further supported via theoretical simulation of the molecular orbital and band gap for PVMs (Fig. S8 in Supporting information). The band gaps had decreased from 4.06 eV for MST, to 3.63 eV for DMTPE, and finally to 3.43 eV for TMTPE. Additionally, the absolute emission quantum yield (ФFL) of these PVMs was recorded to evaluate their photo-physical performance. The ФFL of monocycles MST, bicyclic DMTPE and TMTPE were determined to be 6.3%, 68.6% and 9.3% in their THF solution, and 28.0%, 68.4%, 65.9% in the solid state, respectively (Fig. 3c). This indicated that the monocyclic MST and bicyclic TMTPE were evidently AIEgens, whereas bicyclic DMTPE without peripheral phenyl rings was proved to be a bright dual-phase emitter.
Figure 3
Figure 3.
(a) UV–vis absorption spectra of π-conjugated macrocyclic fluorophores DMTPE, MST and TMTPE in the THF solution. (b) PL spectra of π-conjugated macrocyclic fluorophores in the THF solution and solid. Inset: chemical structure and images (with ФFL) of macrocyclic luminogens and TMTPE in the solution and solid under UV lamp. Concentration = 10 µmol/L. Excitation wavelength: 310 nm for MST and DMTPE, and 340 nm for TMTPE.
Regarding their different emission capability in solution and solid, the PL spectra of PVMs in THF/H2O mixture was further studied. The emission of MST was slightly enhanced with high water content (f) due to aggregates formation, whereas the ФFL at f = 90% reached 14.1% (Fig. S9 in Supporting information). The diameter of the nano-aggregates was recorded to be 459 nm (Figs. S10 and S11 in Supporting information) with increased emission lifetime of 1.46 ns relative to 0.40 ns in pure THF (Fig. S12 in Supporting information). By contrast, bicyclic DMTPE was unveiled to be highly emissive in both the THF solution and as nano-aggregates (Fig. S13 in Supporting information). With raising water content in the THF-H2O mixture, the PL spectra of DMTPE remained nearly identical when f < 20%. Then, the emission was slightly enhanced and obviously blue-shifted from 502 nm to 476 nm parallel with raising f. The ФFL of DMTPE as nano-aggregates (f = 90%) was determined to be 86.7%, whereas the emission lifetime had also slightly increased from 2.63 ns in THF to 3.29 ns as nano-aggregates (Fig. S12). The blue-shifted emission after aggregates formation was probably owing to the packing of fluorophroes in aggregates with diameter of approximate 295 nm (Figs. S10 and S11), which driven the fluorophore to adopt a more twisted conformation in the solid [43]. As a bright dual-phase emitter, bicyclic DMTPE had combined the advantages of both AIE-active and ACQ-active fluorophores [44–46]. By contrast, bicyclic analogue TMTPE with peripheral decoration had exhibited classical AIE behavior with obviously enhanced and blue-shifted emission along with the addition of water in THF solution (Fig. S14 in Supporting information), whereas its ФFL when fw = 90% reached as high as 83.3%.
This divergence in the photo-physical properties for PVMs was probably attributed to the cyclic topology. Although the cis-trans isomerization of stilbene moiety within these PVMs was prohibited owing to the topological constrain, intramolecular motions (like rotation and vibration) and photochemical reactions were still possible for monocyclic MST with less strain. This finally resulted in the non-radiative decay of MST from its excited state and low emission efficiency in THF solution, whereas these motions and reactions would be frozen in nano-aggregate or solid leading to emission enhancement observed. For bicyclic DMTPE, the constrained bicyclic skeleton had significantly restricted the intramolecular motions (like flipping of phenyl rings) in solution. Non-radiative decay pathway from the excited states of DMTPEvia molecular motions was consequently hampered and resulted in intense emission in THF [47]. In solid state, the highly twisted structure of DMTPE had diminished the intramolecular π-π interaction and detrimental stacking like classical ACQ fluorophores, and thus bright emission was retained for DMTPE in the solid state. By contrast, the free rotation of peripheral phenyl rings had converted TMTPE back to AIEgens, although it shared identical cyclic backbone with DMTPE. On the other hand, the structure flexibility of luminogens could also be reflected via their mechanochromic luminescence (MCL) behavior, whereas conformational flexible fluorophores could be enforced to adapt more twisted conformations with densely packing in crystal and ultimately led to blue-shifted emission in crystalline phase [43]. After grinding and solvent annealing, the emission of DMTPE remained unchanged, whereas its analogue TMTPE exhibited obvious red-shifted emission after grinding and blue-shifted emission with solvent fuming (Fig. S15 in Supporting information). This distinction in the MCL behavior of DMTPE and its analogue TMTPE had further confirmed the shape-persistence and structural rigidity for bicyclic DMTPE.
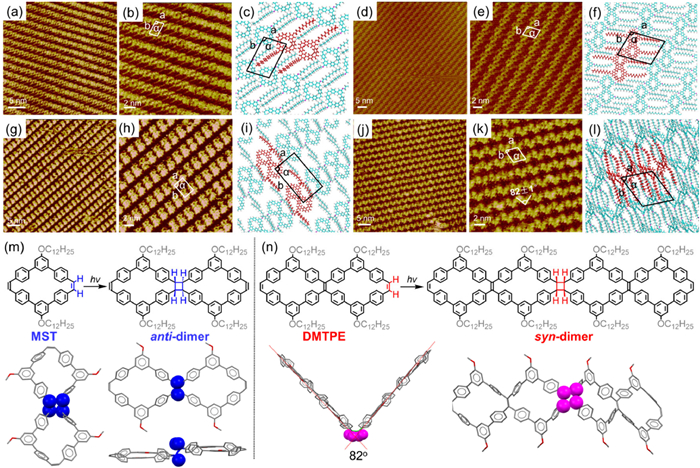
To further study the structures of PVMs, monolayers of these macrocycles on highly oriented pyrolytic graphite (HOPG) were investigated by STM at the liquid/solid interface (Fig. 4). As shown in Figs. 4a and b, the backbone of mono-cyclic MST appeared as a ring which was arranged head-to-tail closely and lined in arrays, whereas the void within each MST is darker in the STM image owing to low electron density. By contrast, the backbone of bicyclic DMTPE appeared as an 8-shaped dual-ring as shown in Figs. 4d and e, and each dual-ring was arranged head-to-tail closely and lined in arrays. The middle portion of each DMTPE backbone is brighter in the STM image, which corresponds to the central TPE moiety with higher electron density. Referring to the unit cell parameters (Table S6 in Supporting information) and corresponding molecular models (Figs. 4c and f), it can be seen that the ditches between the adjacent arrays were filled with the interlaced long alkyl side chains. The supramolecular self-assembling was stabilized by the π-π interaction between the PVMs and the HOPG substrate, as well as the van der Waals force between the dodecyl side chains supported by the energy calculation (Table S7 in Supporting information).
Figure 4
Figure 4.
STM image, high-resolution STM image, and proposed molecular model for the assembly of MST (a-c) and DMTPE (d, f), and UV-light irradiated assembly of MST (g-i) and DMTPE (j-l) at the 1-phenyloctane/HOPG interface. Imaging parameters: Iset = 366.2 pA, Vbis = 742.8 mV for MST; Iset = 381.5 pA, Vbis = 556.6 mV for DMTPE; Iset = 338.7 pA, Vbis = 661.3 mV for irradiated MST; Iset = 213.6 pA, Vbis = 856.3 mV for irradiated DMTPE. Photo-chemical [2 + 2] cycloaddition reaction and optimized geometry for the anti or syn-dimer of MST (m) and DMTPE (n) observed in STM investigation, whereas the dodecyl chain was replaced with methyl group for clarity.
Considering the possibility of [2 + 2] photo-dimerization for stilbene, the self-assembled monolayer of PVMs was further irradiated with UV light to explore the on-surface photochemistry of these PVMs. Surprisingly, the STM images of self-assembled structures for MST had obviously transformed from the head-to-tail arrayed ring topology to shoulder-by-shoulder lined 8-shaped dual-ring array, whereas the experimental unit cell parameter had expanded from a = 1.4 ± 0.1 nm; b = 2.3 ± 0.1 nm, α = 100° ± 1° to a = 2.0 ± 0.1 nm, b = 3.4 ± 0.1 nm, α = 90° ± 1° after photo-irradiation (Figs. 4g-i). Assisted by the structure modeling, the newly observed species was attributed to the anti-dimer of MST (Fig. 4m) which originated from the [2 + 2] photo-dimerization of alkene-terminated mono-cyclic MST. By contrast, the dual-ring structure of DMTPE had evolved to lined "V"-shaped bright spots after UV light irradiation (Figs. 4j-l). The experimental unit cell parameter had transformed from a = 2.9 ± 0.1 nm, b = 2.1 ± 0.1 nm, α = 104° ± 1° to a = 2.3 ± 0.1 nm, b = 2.7 ± 0.1 nm, α = 65° ± 1° According to the stereochemistry of [2 + 2] dimerization, this observed species was consistent with the syn-dimer of DMTPE from [2 + 2] cyclo-addition of terminal double bond. The angle of "V"-shaped spot was determined to be 82° in the STM image, which was close to the value determined from optimized geometry of the syn-dimer (Fig. 4n). Furthermore, the experimental unit cell parameters for the UV-irradiated samples of the macrocycles MST and DMTPE agreed well with their theoretical models as shown in Figs. 4i and l. Therefore, these two PVMs could undergo stereo-selective [2 + 2] dimerization on the HOPG interface depending on the topology of the macrocycles. To reveal the underlying reason for this selectivity, the energy of the anti- and syn-dimers for these two PVMs were calculated based on theoretical simulation, and the anti-dimers for MST and DMTPE were both thermodynamically more stable than their syn-dimers with an energy difference of approximate 10.5 kcal/mol (Fig. S16 in Supporting information). Hence, the stereo-selectivity observed in on-surface photochemistry of PVMs was probably attributed to the reaction mechanism rather than thermodynamic factors.
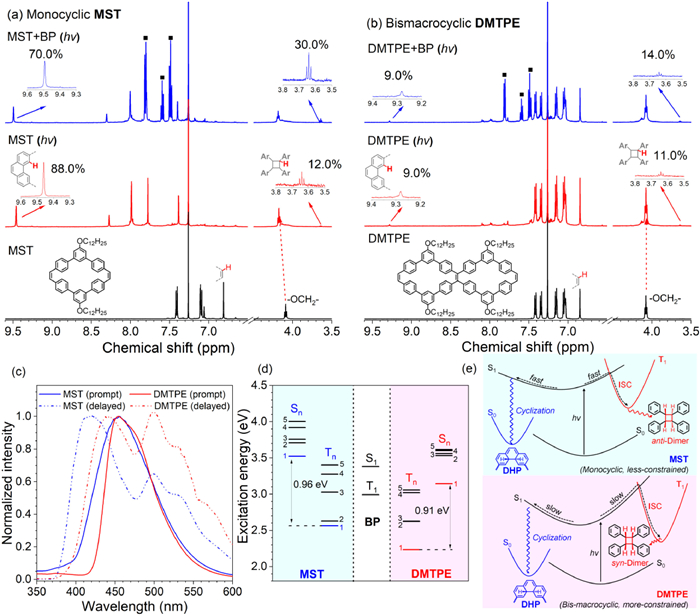
To reveal the photochemical process for PVMs, the 1H NMR of MST and DMTPE was investigated after UV light irradiation, whereas benzophenone (BP) was introduced as the triplet sensitizer (Figs. 5a and b). For mono-cyclic MST, the PVM was extremely ready to photochemical reaction while the signal at 6.81 ppm corresponding to the proton on terminal alkene bond had completely disappeared after UV light irradiation (Fig. 5a). To evaluate the photochemical product, two characteristic peaks at 9.46 and 3.46 ppm were studied in detail, which were attributed to the phenanthrene ring from photocyclization and the cyclobutane unit from [2 + 2] photo-dimerization, respectively. This was also confirmed via MALDI-TOF mass spectra of the irradiated samples (Fig. S17 in Supporting information). The mass spectra suggested the dominating specie with mass charge ratio (m/z) of 873.03 after light irradiation, which agreed well with the molar mass of photo-cyclized macrocycles MST-4H, which emitted at 385 nm in THF and 450 nm as solid (Fig. S18 in Supporting information). Meanwhile, a trace peak had also been observed around m/z = 1760, which confirmed the formation of dimers after photo-irradiation.
Figure 5
Figure 5.1H NMR spectra of macrocycles MST (a) and DMTPE (b) after UV light irradiation (hv) for 30 min in absence or presence of triplet sensitizer benzophenone (BP, ■) in CDCl3. (c) PL spectra of macrocycles MST and DMTPE in toluene at 77 K. (d) Energy diagrams of singlet (Sn) and triplet (Tn) excited states for BP, MST and DMTPE. (e) Illustrative scheme for the photochemical reactions of macrocycles MST and DMTPE. S0 and S1 stand for the singlet ground state and the first singlet excited state, whereas T1 is the triplet state.
According to the integral of these two characteristic peaks, the conversion of the PVMs along two photochemical approaches was estimated. Obviously, photo-cyclization was the dominating pathway for MST under UV light, whereas the high yield of photo-cyclized product was probably owing to the inherent cis-configuration of the stilbene moiety. By contrast, only approximate 12% of the alkene bond in MST had engaged in the competitive [2 + 2] photo-dimerization, and conversion toward the photo-dimer was obviously increased to 30% after addition of BP. This trend clearly demonstrated that triplet excited state was probably involved in the [2 + 2] photo-dimerization of MST. In addition, the casted film of MST was exposed to UV irradiation for 2 h, however, no cyclized product could be observed in the 1H NMR spectrum whereas only small amount (approximate 7%) of stilbene moiety was involved in [2 + 2] photo-dimerization (Fig. S19 in Supporting information). The absence of oxidant (like O2) in the solid state had prohibited the photo-cyclized process, whereas low molecular mobility and unfavorable packing patterns (Fig. 2) of MST in solid had accordingly disfavored the photo-dimer formation.
By contrast, bicyclic DMTPE was almost inert to photo-chemical reactions, whereas only trace amount of the PVMs had underwent the photo-cyclization and dimerization reactions (Fig. 5b). According to the integral at 9.28, 6.84 and 3.64 ppm, it was found that only about 9% of the terminal cis-stilbene unit in DMTPE had been involved in photo-cyclization regardless of the existence of triplet sensitizer BP in solution. The yield of cyclobutane from [2 + 2] dimerization was slightly promoted from 11% to 14% after the addition of BP. The lower photo-chemical reactivity of DMTPE was probably attributed to its rigid bicyclic skeleton, which hampered the structural reorganization required in photochemical reaction. The small impact of BP on cyclobutane formation also indicated that the role of triplet excited state in the photo-dimerization reactions for DMTPE was trivial. Solid sample of DMTPE was also irradiated while no cyclized product was observed in the 1H NMR spectra identical to the case for MST (Fig. S19). However, obviously nearly 22% of stilbene moiety was involved in [2 + 2] photo-dimerization. This remarkably raised yield compared with the investigation in solution was probably owing to the favorable packing orientations and close distance between adjacent DMTPEs.
For cyclic alkenes (such as coumarin [48], chromone [49], and acenaphthylene [50]), direct UV irradiation which only involved singlet excimer would yield the syn-dimer. By contrast, the triplet state which usually generate from the triplet sensitized reaction would give both syn and anti-dimers depending on the solvent or other factors. Hence, the nature of the excited intermediate was probably responsible for the stereo-regularity observed in on-surface photo-dimerization of MST and DMTPE. The singlet and triplet excited state for these PVMs were firstly studied via their prompt and delayed PL spectra measured in toluene at 77 K (Fig. 5c), as well as time-dependent density functional theory (TD-DFT) calculation using functional B3LYP with the 6–31G(d) basis set (Fig. 5d). Both PVMs displayed fluorescence around 450 nm and well-resolved delayed luminescence. Unexpectedly, the first delayed emission peak for both PVMs was blue shifted relative to their prompt emission peak. This was probably attributed to that this delayed emission signal was originated from the m-terphenyl segment instead of the whole macrocycle [51]. And thus, the second delayed emission at 500 nm was attributed to the delayed emission from the PVMs. The similar prompt and delayed emission for both PVMs indicated that these two PVMs had identical energy splitting (ΔEST11) between the lowest singlet (S1) and triplet (T1) excited states. This was also confirmed by the TD-DFT results, ΔEST11 for MST and DMTPE was calculated to be 0.96 and 0.91 eV, respectively. Therefore, ΔEST of MST and DMTPE was not the driving force for their different tendency toward triplet state. Usually, the intersystem crossing (ISC) from the excited singlet state to the triplet state depend on their energy splitting and structural reorganization. Therefore, relatively more flexible conjugated monocyclic MST with lower reorganization energy was more ready to approach T1via ISC or sensitization reaction with BP, and subsequently the triplet state involved photo-dimerization of MST yielded the anti-dimer (Fig. 5e). By contrast, more rigid bicyclic DMTPE with highly constrained geometry and higher reorganization energy was unable to approach the triplet state which required structure reorganization in excited state, and thus could only experience the [2 + 2] photo-dimerization process via singlet excimer to form only syn-dimer on the surface.
Similar topological impacts on photochemical reactivity of PVMs were also observed in the photocyclizaton process. For stilbene and related alkenes, their excited cis-S1 states could also undergo an electrocyclic rearrangement to a DHP S1 state, which would then relax to the DHP ground states and be converted to phenanthrene by oxidation. To approach the DHP state, structural rearrangement of cis-stilbene moiety was inevitably required. After UV light irradiation of MST solution, a new absorption peak at 457 nm, which was attributed to the DHP intermediate, had appeared in its UV–vis absorption spectra (Fig. S20 in Supporting information). By contrast, the absorption spectra of DMTPE remained unchanged after irradiation. Therefore, the skeleton rigidity of the PVMs would be the dominating factor controlling their reactivity toward photo-cyclization (Fig. 5e). Mono-cyclic MST with more relaxed structure and inherent cis-stilbene unit could smoothly rearrange to the DHP intermediate and then be oxidized to phenanthrene. By contrast, highly constrained bicyclic DMTPE was quite difficult to approach the DHP intermediate via structural reorganization, and thus only 8% stilbene moiety was involved in photocyclization. This was also consistent with their photo-physical properties, whereas bicyclic DMTPE exhibited intense emission in solution due to restricted intramolecular motions and the emission efficiency for mono-cyclic MST was quite low owing to non-radiative intramolecular motions.
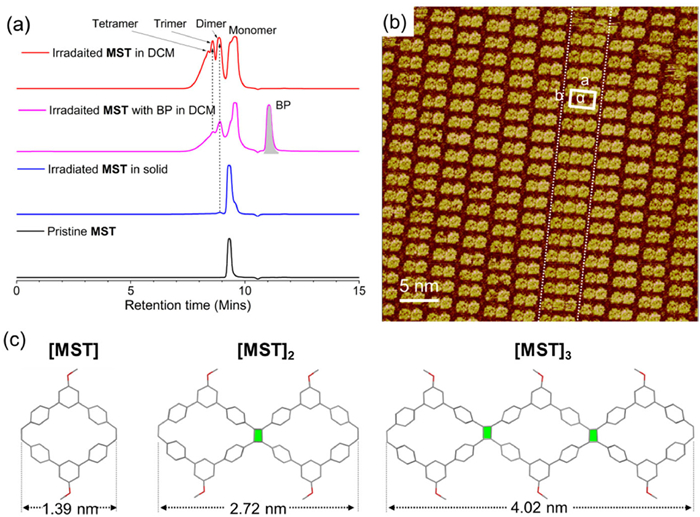
Concerning the relative lower yield for the photo-dimerization of DMTPE in CDCl3, the photo-polymerization of MST was conducted in anhydrous DCM solution under argon atmosphere to build up the multicyclic carbon-rich ribbons connected via four-membered ring. The inert atmosphere would prohibit the oxidation of DHP intermediate, and thus [2 + 2] photo-dimerization of MST would be the dominating process under UV irradiation instead of the photo-cyclization reaction. Gel permeation chromatography (GPC) was subsequently applied to evaluate the degree of polymerization (Fig. 6a). After irradiating the solution with UV light for 1 h under inert atmosphere, the species of dimer, trimer, and tetramer were obviously observed in the GPC curve. The relative lower polymerization degree and existence of monomer species was probably attributed to the slow reaction rate for the [2 + 2] dimerzation. Additionally, the triplet sensitizer BP showed neglectable impacts on the photo-polymerization process under inert gas probably because the competitive photo-cyclized by-reaction had been already prohibited. By comparison, only a trace of dimer had been observed for MST sample with UV irradiation in solid state, and this result was also consistent with the NMR study in Fig. S19.
Figure 6
Figure 6.
(a) GPC curve of macrocyclic MST after UV light irradiation for 1 h in absence or presence of BP in anhydrous DCM under Argon atmosphere, or in solid state. [MST] = 10 mg/mL. (b) STM images of MST after irradiating in anhydrous DCM without BP. Imaging parameters: Iset = 198.4 pA, Vbis = 779.7 mV. (c) Optimized geometry and chain length for anti-dimer and anti-trimer of MST.
To confirm the multicyclic topology of oligomers, the MST derivatives were further studied via STM imaging on HOPG interface. As shown in Fig. 6b, the tricyclic moiety was arrayed in line and dispersed in the network of bicyclic species, where the experimental unit cell parameter for tricyclic units was estimated to be a = 3.2 ± 0.1 nm; b = 2.2 ± 0.1 nm, α = 90° ± 1° Based on STM images, the length of the monocyclic MST and its dimer, trimer was recorded to be 1.4 ± 0.1, 2.8 ± 0.1, and 4.4 ± 0.1 nm, respectively. As indicated by the optimized geometry of MST, and its dimer and trimer in which the rings were connected via anti-[2 + 2] dimerization, the length of the monomer, anti-dimer and anti-trimer were theoretically calculated to be 1.39, 2.72, 4.02 nm (Fig. 6c). These results agreed well with the observed parameter in STM images, and confirmed that the observed tricyclic carbon-rich ribbon was originated from the anti-trimer of MST. However, owing to the complicated conformation of polymer chains and complex stereochemistry of [2 + 2] photo-dimerization, the structure corresponding to multicyclic tetramer and polymers had not been observed in STM images.
In summary, a series of π-conjugated macrocycles with phenylene vinylene backbone and different topology were successfully synthesized by intramolecular McMurry coupling. Their chemical identities and cyclic topology were further confirmed via NMR, MALDI-TOF MS, single crystal analysis and STM technique. Owing to different cyclic topology of the PVMs, relatively more flexible monocyclic MST showed low emission efficiency in the solution and AIE behavior in the solid state, whereas more constrained bicyclic DMTPE showed dual-phase emission behavior with high fluorescence quantum yield of 68% both in the solution and solid state. Additionally, these two PVMs had shown distinctive photochemical behaviors on the HOPG surface and in solution owing to the topological impacts. Monocyclic MST with less strained geometry was more ready for structural reorganization, and thus easier to approach the DHP intermediate and triplet state which yielded photo-cyclized product and anti-dimer. By contrast, highly constrained bicyclic DMTPE showed extremely low photochemical reactivity and produced the syn-dimer via singlet excited complex. Based on solution-mediated photo-polymerization of MST, multicyclic porous carbon-rich ribbons connected by four-membered ring had been constructed and directly visualized in STM imaging. These results had illustrated the significance of topology on the photo-physical and photo-chemical properties of conjugated macrocycles, and opened up a new avenue for fabricating novel porous carbon-rich materials based on conjugated macrocycles. Synthesis of nano-porous graphene nano-ribbon with four-membered ring connections was also in progress in our lab.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is partially supported by the National Natural Science Foundation of China (Nos. 21704065, 22272039), National Key Basic Research Program of China (No. 2016YFA0200700), Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515030228). We thank the Instrumental Analysis Centre of Shenzhen University (Xili Campus) for NMR measurement. The authors wish to acknowledge Professor Zikai He for his helpful discussions regarding the photochemical mechanism.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109571.
Figure 2
(a) Crystal structure of monocyclic MST. (b, c) Packing patterns of MST in the crystal with the distance between adjacent C = C bond in each column labeled.
Figure 3
(a) UV–vis absorption spectra of π-conjugated macrocyclic fluorophores DMTPE, MST and TMTPE in the THF solution. (b) PL spectra of π-conjugated macrocyclic fluorophores in the THF solution and solid. Inset: chemical structure and images (with ФFL) of macrocyclic luminogens and TMTPE in the solution and solid under UV lamp. Concentration = 10 µmol/L. Excitation wavelength: 310 nm for MST and DMTPE, and 340 nm for TMTPE.
Figure 4
STM image, high-resolution STM image, and proposed molecular model for the assembly of MST (a-c) and DMTPE (d, f), and UV-light irradiated assembly of MST (g-i) and DMTPE (j-l) at the 1-phenyloctane/HOPG interface. Imaging parameters: Iset = 366.2 pA, Vbis = 742.8 mV for MST; Iset = 381.5 pA, Vbis = 556.6 mV for DMTPE; Iset = 338.7 pA, Vbis = 661.3 mV for irradiated MST; Iset = 213.6 pA, Vbis = 856.3 mV for irradiated DMTPE. Photo-chemical [2 + 2] cycloaddition reaction and optimized geometry for the anti or syn-dimer of MST (m) and DMTPE (n) observed in STM investigation, whereas the dodecyl chain was replaced with methyl group for clarity.
Figure 51H NMR spectra of macrocycles MST (a) and DMTPE (b) after UV light irradiation (hv) for 30 min in absence or presence of triplet sensitizer benzophenone (BP, ■) in CDCl3. (c) PL spectra of macrocycles MST and DMTPE in toluene at 77 K. (d) Energy diagrams of singlet (Sn) and triplet (Tn) excited states for BP, MST and DMTPE. (e) Illustrative scheme for the photochemical reactions of macrocycles MST and DMTPE. S0 and S1 stand for the singlet ground state and the first singlet excited state, whereas T1 is the triplet state.
Figure 6
(a) GPC curve of macrocyclic MST after UV light irradiation for 1 h in absence or presence of BP in anhydrous DCM under Argon atmosphere, or in solid state. [MST] = 10 mg/mL. (b) STM images of MST after irradiating in anhydrous DCM without BP. Imaging parameters: Iset = 198.4 pA, Vbis = 779.7 mV. (c) Optimized geometry and chain length for anti-dimer and anti-trimer of MST.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
