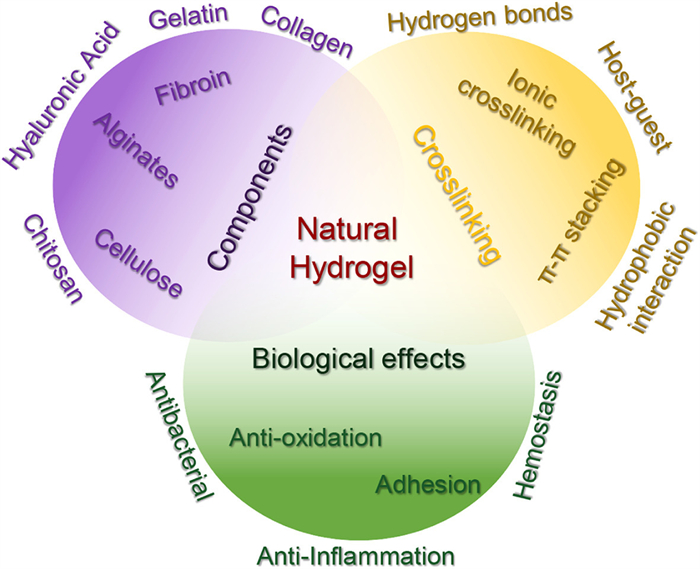
Figure 1.
Schematic representation of a natural hydrogel used as a wound dressing. It encompasses the composition, principles underlying the formation of the hydrogel network, as well as their biological processes.
Natural hydrogel dressings in wound care: Design, advances, and perspectives
Xiaoliu Liang , Chunliu Huang , Hui Liu , Hu Chen , Jiabao Shou , Hongwei Cheng , Gang Liu

Wound therapy has long been a perplexing challenge within the medical field. The process of wound healing is a complex and multifaceted biological phenomenon involving various components such as cellular activity, growth factors, and dynamic alterations in the extracellular matrix. Generally, it is divided into four main stages [1]. The initial phase, known as hemostasis, occurs when the skin sustains damage, triggering blood coagulation at the wound site to form a clot that seals the wound and prevents further blood loss. Simultaneously, the release of chemical substances attracts other cells to assist in wound repair [2]. The subsequent inflammatory stage typically begins several hours to days after the injury and primarily focuses on preventing infection and clearing away dead and damaged cells. During this phase, leukocytes, particularly neutrophils and macrophages, migrate to the wound site, engulfing pathogens and cellular debris. The proliferative stage typically ensues several days to weeks post-wounding, with the principal objective of wound filling and function restoration. During this phase, fibroblasts migrate to the wound site, producing collagen and other extracellular matrix components to fill the wound [3]. Concurrently, the formation of new blood vessels furnishes nutrients and oxygen, facilitating the growth of new tissue. Furthermore, epithelial cells commence growth from the wound margins towards the center, reinstating the skin’s barrier function. The remodeling stage, which can begin several weeks to years after the injury, focuses on optimizing and strengthening the newly formed tissue. In this phase, collagen undergoes rearrangement and cross-linking, enhancing tissue strength at the wound site. Meanwhile, excess cells and blood vessels are eliminated through programmed cell death [4]. However, the stages of wound healing do not always progress flawlessly in a sequential manner and can be influenced by various factors. Excessive inflammation, burns, infections, diabetes, and other factors can potentially disrupt normal wound repair processes, leading to delayed or impaired wound healing [5]. Further research is needed to gain a deeper understanding of the complex mechanisms involved in wound healing and to develop innovative interventions that address the challenges encountered in clinical practice.
Wound dressings play a crucial role in the intricate process of wound healing, fulfilling various essential functions such as wound protection, exudate absorption, moisture regulation, infection prevention, and targeted delivery of bioactive molecules to facilitate healing [4,6,7]. An exemplary wound dressing should possess the following characteristics: (1) Exceptional biocompatibility and biodegradability, minimizing potential toxic side effects on the human body; (2) Creation of a suitable moisture environment conducive to cellular proliferation and migration; (3) Superior mechanical properties to prevent dressing rupture and bacterial infections; (4) Capability to serve as a drug delivery system, releasing growth factors, anti-inflammatory agents, or other bioactive molecules to directly promote wound healing. Traditional wound dressings such as gauze and bandages, have inherent limitations. They can adhere to the wound surface, causing secondary damage and hindering epidermal migration. Additionally, they may lead to desiccation of granulation tissue, and their removal can be time-consuming and uncomfortable for patients. In light of the wetness theory, which emphasizes the maintenance of appropriate wound moisture levels to promote faster cellular growth and migration, and prevent scab formation, dressings have evolved [8]. Advanced formulations, including hydrocolloid dressings, foam dressings, alginate dressings, film dressings, and hydrogels, are increasingly being employed in clinical settings to address these challenges and optimize wound healing outcomes [9].
Hydrogels are characterized by their exceptional absorbency, biocompatibility, pliable and resilient texture, as well as their porous architecture [10]. They can be classified into natural and synthetic hydrogels based on the origin of their raw materials. Natural hydrogels, such as those derived from collagen, gelatin, hyaluronic acid, chitosan, and fibrin, exhibit exceptional biocompatibility and biodegradability, making them widely used in biomedical applications [11-13]. Natural hydrogels, owing to their high-water content, superior moisture retention, excellent biocompatibility, and microstructure analogous to the extracellular matrix (ECM), are regarded as biomaterials closely resembling living tissue [14]. As wound dressings, natural hydrogels overcome limitations associated with traditional dressings in clinical application [15,16]. Firstly, the high-water content of hydrogels affords a moist environment that fosters wound healing [17]. Next, they also continuously absorb wound exudates, preventing secondary infections. The transparency or semi-transparency of hydrogels allows for easy monitoring of the wound healing progress. Furthermore, various drugs can be encapsulated within the hydrogels, enabling gradual and sustained release at the wound site, facilitating the healing process [5,18]. Beyond that, natural hydrogel dressings offer wound protection, pain relief, and improve patient comfort. These unique properties render natural hydrogel dressings as one of the most competitive functional materials in medical fields like wound care and skin regeneration over the past few decades [19]. Their growing adoption and revolutionary impact in wound treatment have been evident [20].
Despite the numerous advantages of natural hydrogels, they face several challenges in practical applications. These include enhancing their mechanical properties to suit different wound types, improving their antibacterial capabilities to prevent infections, and increasing their biodegradability to facilitate natural degradation after use, thereby eliminating the need for surgical removal. These issues are currently being addressed by researchers. Furthermore, despite extensive laboratory-based studies that support the superior effectiveness of natural hydrogels in wound healing, translating these findings into clinical applications remains a demanding endeavor. This review elucidates the fundamental principles of natural hydrogels as wound dressings, and their revolutionary impact on wound treatment. We initially present the essential characteristics of natural hydrogels, including their structure, properties, and notable advantages as wound dressings. Subsequently, we focus on their application in wound healing, emphasizing their pivotal role in facilitating wound healing processes and their effectiveness in combating infections. Ultimately, we address the challenges that natural hydrogels encounter and propose future research directions to overcome these hurdles. This review provides a comprehensive summary and analysis of the composition, mechanisms of hydrogel formation, and biological functions of natural hydrogels. The findings highlight natural hydrogels as a unique biomaterial with promising potential for clinical translation and widespread application (Fig. 1). We aspire to offer valuable references and insights for researchers, clinicians, and those involved in the development and production of wound dressings, thereby catalyzing the advancement of natural hydrogel dressings in the field of wound treatment, and achieving more efficient, safer, and personalized wound care.
The rise in prominence of natural hydrogels as the favored option of next-generation wound dressings can be attributed to their exceptional properties. Derived from natural resources, these hydrogels exhibit inherent biocompatibility, effectively addressing the issue of discomfort associated with prolonged application. And the characteristic of natural hydrogels could fulfill the primary demand for sustained and localized drug release in wound dressings. Furthermore, the biocompatible nature of natural materials enables direct contact with human skin, minimizing inflammation or immune responses in the host tissues. Moreover, these naturally sourced hydrogels can be gradually absorbed or degraded within the wound environment, mitigating the risk of wound tearing necessitated by dressing removal during clinical applications. Simultaneously, the use of biodegradable and renewable natural materials in wound dressings helps mitigate environmental concerns. Compared with synthetic polymers, natural products encompass an abundance of active groups such as hydroxyl, carboxyl, and amino groups, providing ample opportunities for molecular modification and construction of three-dimensional cross-linked networks. This versatility enables the development of hydrogels with tunable functions, including mechanical properties that mimic human tissues, self-adhesion, self-healing, and 3D printability, thus meeting the manufacturing requirements for wound dressings. In this section, we present a concise overview of the common natural products utilized in the design and fabrication of hydrogels, focusing on their distinct functionalities and cross-linking networks.
Collagen, a biopolymer synthesized by animal fibroblasts, holds a prominent position as the most abundant and widely distributed functional protein in mammals. It is predominantly found in various connective tissues such as skin, cartilage, tendons, and bones, playing an indispensable role in supporting and protecting bodily structures. To date, more than 30 distinct types of collagen have been discovered and named, including Type Ⅰ, Ⅱ, Ⅲ, and so on, collagen’s classification is based on the order of their discovery [21]. Type Ⅰ collagen predominantly exists in skin, tendons, and bones, Type Ⅱ collagen is mainly found in cartilage, and Type Ⅲ collagen is abundant in skin, while Type Ⅳ and Ⅴ collagen are exclusive to certain animal organs [22].
Collagen exhibits remarkable nutritive value, hemostatic properties, biocompatibility, biodegradability, and low immunogenicity, making it widely applicable in diverse fields such as food, medicine, tissue engineering, and cosmetics. The functionalities of collagen are rooted in its unique quaternary structure. Its primary structure denotes the amino acid sequence, while the secondary structure embodies the alpha helix of the polypeptide chain. The tertiary structure is composed of three left-handed alpha chains wrapped around a common axis in a right-handed spiral, forming a distinctive triple helix structure of collagen molecules. The quaternary structure involves cross-linking of collagen molecules through covalent bonds, leading to the formation of collagen fibers [23]. The exceptional structure of collagen imparts it with superior biological capabilities, including low antigenicity, excellent biocompatibility, coagulation properties [24], and the ability to promote cell proliferation and growth [25]. Composed solely of collagen monomers, collagen hydrogels can spontaneously form collagen fibers under certain conditions via a neutralization process. In the presence of an aqueous solvent, these fibers form a hydrogel. The ultimate structure and properties of collagen hydrogels are influenced by the self-assembly behavior of collagen, the collagen concentration, pH, temperature, crosslinking methods, and ionic strength. The concentration of collagen affects the mechanical properties and porosity of the hydrogel, with higher concentrations generally yielding more elastic and stable hydrogels at the expense of reduced porosity [26]. Crosslinking between collagen molecules can be achieved chemically or physically using various methods, such as enzymes, ultraviolet light, or chemical reagents [27]. The choice and extent of crosslinking affect the stability, elasticity, and degradation rate of the hydrogel. During the preparation of collagen hydrogels, pH and temperature are pivotal factors in determining collagen solubility and the formation of triple-helix structures [28]. However, the mechanical properties and thermal stability of collagen hydrogels formed by self-assembly are relatively poor, imposing limitations on their application in biomaterials. To overcome these limitations, chemical modifications are commonly performed [29]. Conventional crosslinking agents like aldehydes, carbodiimides, and isocyanates, as well as less toxic natural crosslinking agents like genipin and transglutaminase, are employed to chemically crosslink collagen and enhance its mechanical properties [30].
Gelatin, a natural polymer derived from the hydrolysis of collagen, can be obtained through chemical processing of tendons, bones, skin, and loose connective tissues from large mammals like pigs and cows [31]. Its molecular weight ranges from tens to hundreds of thousands. While homologous to collagen, gelatin possesses a significantly reduced antigenicity [32]. In its natural state, it appears as a yellow or pale yellow, semi-transparent or transparent solid with a certain luster, available in flakes or granules. It is soluble in hot water but insoluble in solvents like ethanol [33]. Under specific conditions, gelatin can form a quasi-triple helix structure, which is key for its gel-sol behavior and provides advantageous properties such as gelation, film formation, surfactant activity, and hydrophilicity [34]. The gelatin molecular chain contains active functional groups (amines, carboxyls, hydroxyls, etc.) from various amino acid residues, making it easily soluble in water and amenable to modification [35]. Furthermore, gelatin’s network structure bears remarkable resemblance to biological tissue, making it a safe material recognized by the FDA. Additionally, gelatin exhibits superior cell adhesion and promotes cell proliferation, making it widely utilized as wound dressings [36].
However, the mechanical properties of gelatin alone are insufficient to meet practical demands. To enhance the mechanical properties of gelatin, researchers typically use chemical or physical methods to bestow new structures upon it, thereby creating gelatin-based hydrogels with varying properties and functionalities. This includes employing chemical crosslinking, utilizing dual-network, interpenetrating network, and nanocomposite network structures to reinforce the gelatin network [37]. In a study, a biocompatible dual-network hydrogel was developed by combining gelatin and bacterial cellulose (BC) as the first and second networks, respectively. This gelatin-based hydrogel exhibited mechanical properties that closely matched the fracture strength and Young’s modulus of articular cartilage, marking it a highly promising candidate for tissue engineering such as artificial cartilage [38].
Hyaluronic acid (HA), a linear polysaccharide, was first purified from bovine vitreous humor in 1934. Subsequent research revealed that HA consists of repeating units of d-glucuronic acid and N-acetylglucosamine, linked by β−1,4-glycosidic bonds, maintaining a consistent molar ratio [39]. HA is ubiquitously present in the extracellular matrix throughout the human body and plays vital physiological roles [39]. Although molecular weight may vary due to different origins and extraction methods, the fundamental structure of HA remains unchanged. Diverse molecular weight HA can elicit distinct cellular responses. High molecular weight HA (above 5000 kDa) generally exhibits anti-angiogenic and immunosuppressive properties. Medium molecular weight HA (20–1000 kDa) is associated with biological processes such as embryogenesis, wound healing, and regeneration [40]. Low molecular weight HA (6–20 kDa) is linked to various cellular activities, including pro-inflammatory responses, angiogenesis, and gene expression [41]. Short chains of HA can also act as inducers for heat shock proteins and anti-apoptotic agents [42].
As a natural polysaccharide material, HA possesses numerous advantageous properties, including biocompatibility, biodegradability, and bioresorbability [43]. Notably, it serves as a significant intracellular component within connective tissues, playing a vital role in lubrication, cell differentiation, and cell growth. Additionally, its linear chain structure contains functional groups like carboxyl and hydroxyl, which can be used to introduce functional domains, allowing for the formation of multifunctional natural hydrogels [44]. Importantly, HA is a crucial participant in the wound healing process, rendering exogenous HA beneficial for wound recovery [45]. However, natural HA is susceptible to degradation and exhibits subpar mechanical properties, necessitating chemical modifications or transformations [46]. Derivatives obtained by modifying natural HA often retain biocompatibility, although their physicochemical properties may change. Covalent crosslinking is frequently employed to modify HA, involving the hydroxyl groups through ether linkages and carboxyl groups via ester linkages, thereby enhancing its stability and mechanical properties [47].
Chitosan, a multifaceted hydrophilic natural polysaccharide, is primarily derived from the shells of crustaceans, such as shrimp and crab. It is a linear compound with a high molecular weight formed by the β−1,4-glycosidic linkage of glucosamine molecules [48]. Chitosan exhibits exceptional biological properties, including biocompatibility, biodegradability, non-toxicity, and broad-spectrum antimicrobial activity, making it highly valuable in biomedicine, antimicrobial agents, and wound care adjuncts [49]. However, its widespread application is hindered by challenges such as its high molecular weight, limited storage time, and poor solubility. As a result, recent research efforts have focused on the preparation of low molecular weight chitosan to enhance solubility and expand its range of applications. Various techniques, such as physical crosslinking, chemical crosslinking, and enzymatic crosslinking, are employed for preparing chitosan hydrogels, with the first two methods being the most commonly used [50]. However, chitosan often exhibits shortcomings during application, such as excessive brittleness, inadequate viscoelasticity, and insufficient water absorption. Therefore, improving the mechanical properties of chitosan has become a central research focus.
Several methods have been identified to alter the mechanical properties of chitosan: (1) Introducing crosslinking agents, such as aldehydes, ketones, epoxides, isocyanates, and multifunctional compounds, which can form a network structure and reinforce the mechanical properties of the hydrogel. For instance, glutaraldehyde is commonly used as a crosslinking agent [51]. (2) Developing chitosan composite hydrogels by combining chitosan with other polymeric materials or nanomaterials, significantly enhancing its mechanical properties. Examples include combinations with polyvinyl alcohol, polyacrylic acid, nanosilicates, nano-hydroxyapatite [52]. (3) Implementing chemical or physical modifications, such as acylation, carboxylation, sulfonation, or exposure to ultraviolet radiation and high-pressure treatment, to alter the molecular structure and physical properties of chitosan, thereby improving its mechanical properties [53]. (4) Optimizing the preparation process by adjusting solution pH, temperature, concentration, and gelation time, which can influence the crosslinking density and extent, leading to improved mechanical properties of chitosan hydrogel [54]. (5) Modifying the pore structure of the gel through techniques like freeze-drying or supercritical drying to control pore size and porosity, thereby enhancing the mechanical properties of the gel with oriented pores or porous structures [55]. (6) Introducing covalent crosslinking to form a more stable network structure in the chitosan hydrogel, thereby enhancing its mechanical properties [51]. For example, Tan et al. established a covalent crosslink between HA and chitosan via a Schiff base reaction. In summary, by employing the aforementioned methods to enhance the mechanical properties of chitosan hydrogel, its potential applications in biomedicine, environmental engineering, and the food industry can be significantly expanded [56].
Alginate is a natural linear high molecular weight compound isolated from brown algae, consisting of β-D-mannuronic acid (M units) and α-L-guluronic acid (G units) residues linked by 1,4-glycosidic bonds [57,58]. The arrangement of G and M units within the chain determines the physical and chemical properties of the alginate. The sequence structure of the alginate consists of contiguous G units (GGGGGG), contiguous M units (MMMMMM), and alternating M and G units (GMGMGM), with its content and length related to the extraction source [59]. The formation of alginate hydrogel mainly involves ionic cross-linking. Initially, alginate is dissolved in water to form an alginate solution. Sodium alginate is typically used as the raw material due to its excellent water solubility. This alginate solution is then mixed with a multivalent cation solution, such as calcium ions (Ca2+), aluminum ions (Al3+), or iron ions (Fe3+) [60]. These multivalent cations interact with the carboxyl groups (-COO-) in the alginate, inducing an ionic crosslinking reaction and forming a net-like structure. Following the ionic cross-linking reaction, alginate molecules within the solution form a three-dimensional cross-linked network structure that can encapsulate a significant volume of water molecules, leading to the formation of an alginate hydrogel [61]. Studies have demonstrated that ionic mediums are an effective means of modulating the polyelectrolyte effect and viscosity characteristics of alginate [62].
With its biocompatibility, hydrophilicity, and non-toxicity, alginate hydrogel finds extensive application in biomedicine. Notably, alginates hold immense potential in promoting wound healing and are used in various forms of wound dressing, including hydrogels, films, and nanofibers. Alginate dressings can absorb excess fluid from the wound, maintaining a moist wound environment and minimizing bacterial infection at the wound site [63]. Nevertheless, studies have indicated that the addition of alginate reduces the mechanical properties of the hydrogel dressing. To address this drawback, the cross-linking density of the gel can be increased by elevating the concentration of the alginate solution or the amount of cross-linker [64]. However, excessively high cross-linking density may diminish the porosity and water absorption of the gel. Therefore, combining alginate with other materials, such as natural polymers, synthetic polymers, or inorganic nanomaterials, can be beneficial to improve its mechanical properties [65]. For instance, alginates can be combined with chitosan, gelatin, collagen, or nano-hydroxyapatite, the introduction of the covalent crosslinking, such as through “click” chemistry reactions or Schiff base reactions, enables the formation of a more stable network structure within the alginate hydrogel, thereby enhancing its mechanical properties [66,67].
Cellulose, a complex carbohydrate, is an abundant organic compound found in plant cell walls, playing a vital role in their growth and structural integrity. It consists of linear chains of β-D-glucose units linked by β−1,4-glycosidic bonds. Through hydrogen bonding, these chains form microcrystalline structures that give cellulose remarkable tensile strength, supporting plant structures [68]. Owing to an abundance of hydroxyl groups present on the cellulose molecular chains, intra-molecular and inter-molecular hydrogen bonding is easily facilitated, leading to the agglomeration of cellulose molecular chains, creating a crystalline protofibril structure, characteristically exhibiting a helical alignment [69]. Cellulose and its derivatives can be chemically crosslinked to form robust three-dimensional networks, such as with succinic anhydride, citric acid, maleic anhydride, ethylene glycol diglycidyl ether, epichlorohydrin, and divinyl sulfone. These modifications facilitate the formation of covalent ester or ether bonds between the cellulose chains [70]. Furthermore, cellulose and its derivatives can be combined with synthetic polymers like polyvinyl alcohol, polyethylene glycol, and poly(N,N-dimethylacrylamide), or natural polymers like alginate, chitosan, and hyaluronic acid, to create interpenetrating polymer networks with superior mechanical properties and excellent processing capabilities [71].
However, the extensive hydrogen bonds between cellulose molecules and the tightly packed crystalline structure pose challenges in dissolving cellulose in typical solvents, limiting its transformation and utilization. Carboxymethyl cellulose, a derivative of cellulose, can address this issue by chemically grafting carboxymethyl groups onto cellulose molecules. These additional groups enhance polarity, enabling the formation of hydrogen bonds with water molecules. Consequently, carboxymethyl cellulose exhibits distinct properties and finds applications in various fields. Despite their similar main chain structures, the properties and applications of carboxymethyl cellulose significantly diverge from those of cellulose [72]. The water-solubility of carboxymethyl cellulose makes it useful in numerous applications, such as a thickener, stabilizer, and emulsifier in food and pharmaceutical industries, given its ability to form a dense gel in water [73].
Silk protein, also known as sericin, is a natural protein predominantly derived from domesticated silkworm silk. It consists of two proteinaceous components: fibroin and sericin [74]. Of these, fibroin makes up approximately 70%–80% of the mass, while sericin accounts for around 20%–30% [75]. Fibroin, a fibrous protein, is assembled from two identical heavy chains and two light chains, bound by a disulfide bond situated at the N-terminus of the heavy chain. The structural hallmark of fibroin lies in its abundant, repetitive sequences enriched with glycine (Gly) and alanine (Ala), which contribute to its excellent crystallinity and mechanical properties [76]. Conversely, sericin, a high-molecular-weight protein abundant in serine and asparagine, acts as an adhesive within the silkworm silk, binding the fibroin fibers together within the silk. Sericin is irregular in structure and is primarily found in the exterior and interior voids of the fibroin fibers [77].
Silk protein molecules interact through hydrogen bonding and van der Waals forces, forming a three-dimensional network structure and resulting in the formation of a hydrogel [78]. The hydrogel formation of silk protein occurs under mild reaction conditions without the use of chemical cross-linking agents, but the gelation is promoted by various factors, including ultrasonic effects, shear forces, deprotonation, electric fields, temperature, and the addition of organic solvents or surfactants [79]. Typically, fibroin protein is usually combined with synthetic polymers to form hydrogels with physical or chemically cross-linked networks [80]. Moreover, introducing active groups onto the main chain of fibroin protein can induce the formation of a stable covalent cross-linked network. Enzymatic cross-linking is a promising methodology for preparing silk protein hydrogels, offering advantages such as excellent biocompatibility, mild operating conditions, and controllable reactions, while avoiding the use of traditional chemical cross-linking agents, organic solvents, and toxic substances [81]. One of the most commonly used enzymes is horseradish peroxidase, which can effectively enhance the gelation kinetics and mechanical properties of silk protein hydrogels by increasing the number of phenolic groups available for enzymatic cross-linking. Due to its superior biosafety, biodegradability, natural antioxidative, anti-inflammatory, anticoagulant, antimicrobial properties, apoptosis inhibition, and cell differentiation promotion, silk hydrogels find extensive application in wound dressings [82].
The hydrogel, often described as “Base of Water, Boundary of Gel”, is a three-dimensional network established through key crosslinking mechanisms that allow it to immobilize copious water without dissolving. Therefore, the selection of crosslinking strategies plays a crucial role in hydrogel preparation. Natural hydrogels, characterized by their abundant functional groups along the molecular chains, primarily rely on aggregation processes for their assembly. These processes involve the interaction of building units through weak non-covalent bonds [83]. These non-covalent bonds include hydrogen bonds, hydrophobic interactions, metal ion-mediated crosslinking, and host-guest interactions. Despite these non-covalent interactions between hydrogel monomers are relatively weak, they often demonstrate additivity and directionality, resulting in a total binding strength comparable to that of covalently crosslinked materials. In this section, we deliberate and summarize the crosslinking strategies employed in the preparation of natural hydrogels (Fig. 2). Besides, we also summarize the crosslinking mechanisms of common natural hydrogels and their mechanical properties (Table S1 in Supporting information), which could enable the modification and enhancement of natural hydrogels, opening up a wide range of applications in fields such as tissue engineering, drug delivery, biomedical devices, and beyond. Simultaneously, it is imperative to consider the biocompatibility of natural hydrogels while investigating their potential biological applications, as various crosslinking strategies employ distinct physical and chemical interactions.
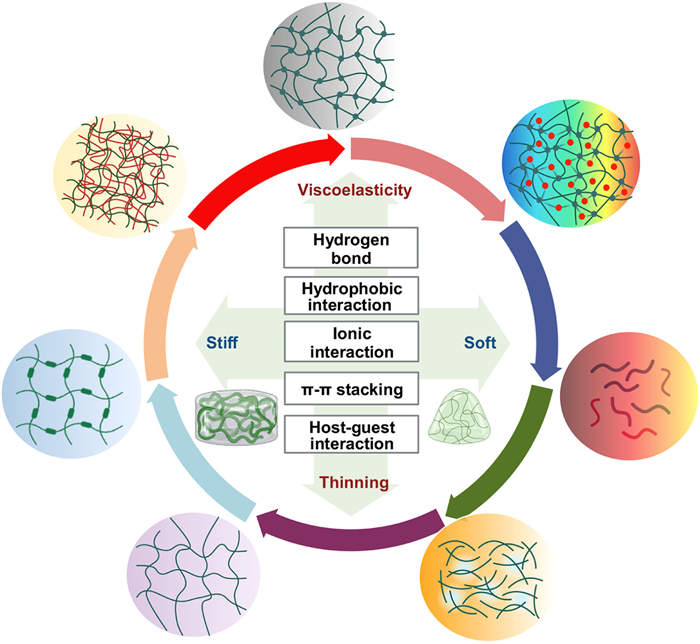
A plethora of natural polymers boast an abundance of functional groups such as hydroxyl, carboxyl, carbonyl, and amino groups, which have the potential to form hydrogen bonds intermolecularly or intramolecularly [84]. As a principal non-covalent bond, hydrogen bonding plays an indispensable role in the formation of natural hydrogels [85]. The density and strength of hydrogen bonds significantly influence the properties of the hydrogel, including its mechanical strength, swelling capacity, degradation behavior, and biological characteristics [86]. Typically, an individual hydrogen bond has relatively weak bonding energy, resulting in hydrogels with shear-thinning and injectable properties. For example, the amalgamation of natural polymers like gelatin and agarose, starch, and carboxymethyl cellulose demonstrate these shear-thinning properties whilst maintaining injectability [87]. Nonetheless, when multiple hydrogen bonds are arranged in an organized manner, the binding constant of each hydrogen bond increases, leading to the formation of highly stable self-assembled structures, as observed in cellulose and chitosan nanofibers [88].
Hydrophobic interaction is non-covalent interaction that drive the aggregation of hydrophobic structures. The interaction plays a crucial role in various biological processes and structures, such as cellular membranes, protein folding, subcellular vesicles formation, and the embedding of membrane proteins into lipid environments. In recent years, researchers have developed amphiphilic materials that possess both hydrophilic and hydrophobic portions. These substances can self-assemble in aqueous solutions, forming micelles, nanotubes, nanorods, nanosheets, and vesicles. They have found widespread applications in nanodevices, drug or gene delivery, template synthesis, and cell imaging [89]. The cellulose molecule, a natural polymer, contains an abundance of hydroxyl structures that readily form fibrous structures with a high degree of crystallinity. The ordered crystal region of cellulose exhibits hydrophobic properties and is resistant to swelling in solvents. On the other hand, chitosan, a polysaccharide, undergoes physical crosslinking primarily through hydrophobic modifications [90]. In an alkaline solvent, the amino groups on the chitosan-based hydrogel’s molecular chain form hydrogen bonds and deprotonated hydrophobic interactions between molecules, culminating in the formation of a three-dimensional network structure [91].
Ion-mediated crosslinking strategies offer a method to create hydrogels by leveraging electrostatic interactions between multivalent ions and natural polymers [92]. This approach provides several advantages, including mild reaction conditions, low toxicity, and reversibility. The most quintessential natural hydrogel based on ion crosslinking is alginate salts. Multivalent cations such as Ca2+, Fe3+, and Al3+ interact with the carboxyl groups in sodium alginate molecules, resulting in the formation of a stable hydrogel structure [93]. Importantly, this crosslinking process can be completed at room temperature and physiological pH, with the density and strength of the crosslinks influenced by ion concentration. Additionally, the introduction of multivalent cations (e.g., Ca2+, Mg2+) into a collagen and chitosan solution induces electrostatic interactions between collagen and chitosan molecules, leading to the formation of a stable hydrogel structure [94]. This type of hydrogel holds great potential in various applications such as tissue engineering and wound treatment.
Strategies based on π-π assembly serve as a potent method for facilitating molecular self-assembly through interactions between π-electron clouds [95]. The merits of this approach encompass operational simplicity, substantial control, and a multifarious array in both composition and structure, making it highly promising in materials science, nanotechnology, and biomedicine [96]. π-π interactions are van der Waals forces that occur between conjugated π-electron clouds, typically observed among organic molecules with conjugated structures. Although these interactions are relatively weak, they can realize robust assembly forces through appropriate design [97]. A plethora of natural compounds with conjugated systems can be assembled into hydrogels via π-π stacking, the combination of baicalin and sanguinarine is one example, π-π stacking allows them to form a stable fibrous nanocluster structure in addition to hydrogen bonds and electrostatic adsorption [95].
The host-guest assembly strategy constitutes a molecular self-assembly process that is fundamentally predicated in geometric coordination and hydrophobic interactions [98]. Host molecules are substantial entities characterized by the presence of discrete binding sites and a central cavity, which collectively facilitate complexation [99]. Conversely, guest molecules, which are endowed with an array of binding sites, can range from diminutive cations or anions to elaborate organic molecules. Distinct from hydrogen bonding and π-π interactions, host-guest interactions are garnering burgeoning interest owing to their unique molecular recognition capabilities and augmented complexation strength. Cyclodextrins, quintessential examples of host molecules, are naturally occurring cyclic oligosaccharides composed of multiple glucose units linked through α−1,4-glycosidic bonds, and are distinguished by internal cavities that can accommodate a diverse spectrum of compounds. Owing to these properties, cyclodextrins are harnessed as vehicles for the encapsulation of anticancer therapeutics, such as doxorubicin (DOX), paclitaxel (PTX), and camptothecin (CPT), thereby ameliorating the solubility, stability, and bioavailability of these chemotherapeutic agents [100]. Consequently, natural hydrogels synthesized through host-guest crosslinking strategies hold considerable promise for application in the delivery of hydrophobic pharmaceuticals.
Natural polymers confer an array of advantages such as superior biocompatibility, biodegradability, and wide prevalence. These polymers are eco-friendly, non-toxic, and cost-effective, thereby making them ideal candidates for application in wound dressings. Furthermore, the molecular chains of natural polymers typically abound with hydrophilic groups (including hydroxyl, amino, and carboxyl), favoring their crosslinking with water and other functional polymers. Owing to these superior properties, hydrogels derived from natural polymers are frequently employed in wound healing applications. Given the broad spectrum of skin wound types and the intricate nature of the skin wound healing process, hydrogel dressings must accommodate different requirements under diverse wound conditions. In the subsequent section, we explore several applications of natural polymer-based hydrogels in wound healing, encompassing antimicrobial hydrogels, antioxidative and anti-inflammatory hydrogels, and adhesive hemostatic hydrogel dressings. This section lays the theoretical foundation for the advancement of hydrogel wound dressings derived from natural sources.
Bacterial infections and wound healing continue to be pervasive challenges in day-to-day life. Medical dressings have the potential to partially mimic the protective function of skin, thereby cultivating a conducive microenvironment for wound repair. Within this domain, hydrogel dressings have recently emerged as a novel class of wound dressings, distinguished by their adaptability, moisture retention capabilities, and biodegradability, making them highly competitive candidates for wound management. However, a notable limitation is that the majority of extant hydrogel dressings lack intrinsic antimicrobial properties, thus restricting their clinical utility. Hence, the development of antimicrobial hydrogel dressings is of paramount research importance and holds considerable socioeconomic value. Through the synergistic amalgamation of antimicrobial substances with hydrogel dressings, the resulting dressings are effectively imbued with the ability to counteract bacterial infections. The design rationale for antimicrobial hydrogels encompasses three elements: (1) The hydrogel inherently exhibits antimicrobial properties, serving dually as a therapeutic agent and delivery carrier; (2) The hydrogel functions as a drug carrier, loaded with antimicrobial agents; (3) The hydrogel exhibits stimuli-responsive characteristics.
Intrinsically antimicrobial natural hydrogel dressings encompass a series of hydrogel dressings that are constituted by the self-assembly of a three-dimensional network structure, derived from naturally occurring polymers and their derivatives possessing antimicrobial properties [101]. Compared to hydrogels loaded with antimicrobial agents, these intrinsically antimicrobial natural hydrogel dressings potentially offer enduring and effective antimicrobial activity, concurrently mitigating cytotoxicity to tissues [102]. Generally, most natural biopolymers lack inherent antimicrobial properties. However, certain natural macromolecules, such as chitosan, antimicrobial peptides, and essential oils from plants, demonstrate commendable biocompatibility and degradability. The functional groups inherent in their structures confer antimicrobial activity. This facilitates the controlled release of medicinal compounds at the target site, ensuring protracted antimicrobial activity, which is congruent with the requisites for wound healing dressings.
Tannic acid (TA) is a polyphenolic molecule with antioxidant, antimicrobial, anti-inflammatory, and biodegradable properties [103]. As a crosslinker for natural hydrogels, its abundant phenolic groups can form hydrogen bonds, ionic bonds, coordination bonds, and hydrophobic interactions [104]. In the preparation of natural hydrogels, TA has been used as a crosslinking and anti-inflammatory additive. Quaternized chitosan (QCS) is a cationic polymer derived from natural sources, exhibiting potent tissue adhesion and antimicrobial capabilities, and is adept at forming ionic bonds with anionic groups, rendering it invaluable in the creation of multifunctional engineered wound dressings with considerable implications for healthcare. Guo et al. cleverly employed TA to prepare multifunctional QCS/TA hydrogels via a simple one-step mixing method [105]. This hydrogel, characterized by the expeditious, reversible formation of ionic and hydrogen bonds between QCS and TA, demonstrates shear-thinning and in-situ gelation properties, adeptly conforming to irregular wound surfaces. Antimicrobial activity of the QCS/TA hydrogel was found to be positively correlated with the TA content, enabling customization of antimicrobial properties through the modulation of TA content (Fig. S1a in Supporting information).
Jelleine-1 is a natural antimicrobial peptide initially isolated from royal jelly. It is a typical amphiphilic peptide that demonstrates broad-spectrum antimicrobial activity against Gram-positive bacteria, Gram-negative bacteria, and fungi through a membrane lysis mechanism. Interestingly, the antimicrobial activity of jelleine-1 remains unscathed by conventional resistance mechanisms. Zhou et al. prepared a jeleline-1 hydrogel in phosphate-buffered saline (PBS), elucidated the plausible formation mechanism, and further investigated its antimicrobial activity [106]. The hydrogel exhibited antimicrobial activity comparable to clinical drugs, effectively neutralizing Staphylococcus aureus (S. aureus), methicillin-resistant Staphylococcus aureus (MRSA), Escherichia coli (E. coli), and Candida albicans (C. albicans) (Fig. S1b in Supporting information). Glycyrrhizic acid (GA) is a naturally occurring triterpenoid saponin with diverse biological activities, its monomer molecule composed of the hydrophobic glycosyl moiety, 18β-glycyrrhetinic acid, and the hydrophilic disaccharide glucuronic acid. Zhao et al. discovered that under the influence of non-covalent bonds, GA molecules in a physiological phosphate buffer saline (PBS, 10 mmol/L, pH 7.4) solution at 37 ℃ show unique self-assembly characteristics, forming stable long nanofibers, which further assemble into supramolecular hydrogels with a three-dimensional network structure [107]. Upon evaluation of its antimicrobial properties, it was observed that GA completely inhibited the growth of Gram-positive S. aureus, but had no effect on gram-negative E. coli (Fig. S1c in Supporting information). However, the GA hydrogel lacks sufficient mechanical strength, adhesion, and self-healing capabilities, restricting its practical application in biomedicine. To enhance its mechanical properties, adhesiveness, self-healing, and other characteristics to improve its clinical translational prospects, Li et al. developed a hybrid GA hydrogel constructed from an aldehydic GA (AGA) self-assembled hydrogen bond fiber network, combined with a dynamic covalent network formed by a biopolymer carboxymethyl chitosan (CMC) through a Schiff base reaction [108]. The hybrid GA hydrogel demonstrates excellent stability and mechanical properties, as well as multifunctional features such as injectability, shape adaptability, remoldability, self-healing, and adhesion (Fig. S1d in Supporting information). Hydrogels formed through the self-assembly of natural edible and medicinal small molecules, attributable to their inherent biological activity and superlative biocompatibility, are posited as some of the most promising dressings for combating bacterial infections and facilitating skin wound healing.
Nat Wound healing is a protracted and complex process. The limitations of intrinsic antibacterial hydrogels gradually manifest, such as their competence in exhibiting antibacterial properties only upon direct contact with bacteria and their inability to eliminate bacteria from the surrounding milieu, thus posing a potential risk for escalating bacterial resistance. Consequently, the integration of antibacterial into hydrogel wound dressings emerges as an efficacious strategy for mitigating infection or achieving bacterial eradication.
Natural hydrogel dressings that carry metallic antibacterial agents have been created. Inorganic metal ions (e.g., silver, zinc, gold, copper, and magnesium) and their metallic oxide nanoparticles, which are well-acknowledged for their antibacterial properties, have been extensively deployed in wound treatment [109]. The antibacterial mechanism of these metal ions principally hinges on two aspects [110]. The interaction of the metal ions with the bacterial cell membrane causing membrane damage, and subsequent loss of bacterial protein and DNA. The generation of reactive oxygen species (ROS) based on Fenton’s reaction, thereby increasing oxidative stress and disrupting the bacterial cell membrane to achieve antibacterial purposes. Despite the need for a comprehensive resolution of issues such as cytotoxicity and long-term in vivo retention of metal-based biomaterials, metal-based composite antibacterial hydrogels remain a research hotspot in antibacterial biomaterials [111]. Moreover, the metal constituents play a critical role in regulating the physiological and mechanical properties of the hydrogel, while the three-dimensional network structure of the hydrogel can control the release rate of antibacterial metal agents. Silver, a venerable antibacterial material, exhibits excellent bioactivity and biocompatibility [112]. Tuomas P. J. Knowles employed regenerated silk fibroin (RSF), a protein characterized by its ability to self-assemble into nanofiber structures, as a fundamental ingredient. By leveraging the tyrosine residues within the protein, AgNO3 was introduced to form silver nanoparticles (AgNPs). Subsequently, through the employment of droplet microfluidic techniques, they constructed an amalgamation of organic and inorganic nanofiber microgels, replete with AgNPs, denoted as silk-silver microgels. Upon the creation of these microgels, their antimicrobial efficacy against E. coli was evaluated in vitro. The results uncovered that a concentration of 3% silk-silver microgels effectively inhibited bacterial growth entirely. Moreover, lesser concentrations of silk-silver microgels were found to suppress bacterial proliferation in a dose-dependent manner. This antimicrobial effect was further corroborated within a mouse surgical site infection model in vivo (Fig. S2a in Supporting information). Besides, zinc ions are widely recognized for their antibacterial properties, and exert their effects through diverse mechanisms including disrupting bacterial metabolic processes, inhibiting bacterial growth and reproduction, and enhancing immune system function. Furthermore, they play a promotional role in the wound healing process, which makes them a sought-after inorganic antibacterial material [113]. Hu et al. reported a multifunctional, injectable hydrogel (CMCS-brZnO) synthesized by incorporating zinc oxide nanorods (brZnO) into carboxymethyl chitosan (CMCS), where brZnO served both as a crosslinking agent and a nanofiller [114]. The CMCS matrix and brZnO exhibited a synergistic antibacterial effect. The hydrogel demonstrated a slow and continuous release of Zn2+ ions, significantly promoted wound healing, and attenuated inflammation (Fig. S2b in Supporting information). In addition, antibacterial hydrogel dressings loaded with metals like copper and gold have been sequentially developed and are being applied for wound healing.
Natural hydrogel dressings loaded with antibiotics have been developed. Antibiotics are secondary metabolites that are produced by microorganisms (including bacteria, fungi, and actinomycetes) or higher flora and fauna during their life cycle, possessing pathogen-fighting or other active properties, primarily used to treat infections caused by bacteria [115]. The operating mechanisms of antibiotics are multifarious, ranging from inhibiting the synthesis of bacterial cell walls and protein to interfering with the synthesis of nucleic acids and metabolic pathways. However, overuse of antibiotics can lead to antibiotic resistance. Hence, the three-dimensional network structure of hydrogels makes them excellent carriers for antibiotic loading for local treatment of bacterial infections, reducing adverse reactions [116]. Tobramycin (TOB) is an antibiotic primarily utilized clinically to treat severe infections caused by sensitive bacteria, especially those prompted by Gram-negative bacteria. However, since TOB is a concentration-dependent antibiotic, improper or excessive usage could lead to severe irreversible side effects. Li et al. prepared a sprayable hydrogel by employing ionic interactions between aminoglycosides like TOB and cellulose nanocrystals (CNC) [117]. The rigid nanostructure of the CNC can reinforce the crosslinked network, which in turn amplifies the stability of the hydrogel and impedes the diffusion of TOB molecules (Fig. S2c in Supporting information). This showcases an “impedance effect” and “locking effect”, prolonging the antibiotic release. The released TOB molecules maintain their antibacterial effect against S. aureus and P. aeruginosa. Furthermore, this hydrogel preparation strategy is also effective for other types of aminoglycoside drugs (such as amikacin, gentamicin, ribostamycin, neomycin). This research provides a promising option for the treatment of surgical and trauma-related infections. This kind of hydrogel-antibiotic combination can be incredibly beneficial as it allows for the controlled and sustained release of antibiotics at the wound site. This not only ensures that the bacteria are effectively dealt with but also reduces the likelihood of systemic side effects that are often associated with antibiotic use. Moreover, using antibiotics in this controlled manner may reduce the probability of bacteria developing resistance to them. It is important to note that while incorporating antibiotics in hydrogel dressings is promising, it is also crucial to use them judiciously and under proper medical supervision to prevent the emergence of antibiotic-resistant strains of bacteria.
Smart responsive hydrogels, which respond to external environmental stimuli, exhibit rapid and remarkable antibacterial properties. Notably, these hydrogels can incorporate phototherapeutic agents that are activated by light, such as those used in photothermal therapy (PTT) and photodynamic therapy (PDT). Both PTT and PDT are characterized by high selectivity and a broad spectrum of antibacterial activity, making them viable alternatives to antibiotics for wound care applications [118]. By integrating photothermal agents and photosensitizers into the hydrogel matrix, a targeted delivery system is achieved, which efficiently directs the therapeutic agents to wound sites, consequently improving the efficacy of the treatment [119].
PTT, owing to its non-invasive nature, remote control capability, excellent biocompatibility, and deep tissue penetration, has garnered significant attention in antibacterial applications [120]. Yang et al. designed and prepared a chitosan composite hydrogel endowed with antibacterial, adhesive, and self-healing properties, tailored for treating infected wounds [121]. Initially, Fe3+ and TA undergo self-assembly to form TFe-NPs nanoparticles, characterized by efficient photothermal properties. The nanoparticles were subsequently incorporated into a hydrogel network crosslinked with acrylamide (AM), 3-acrylamidephenylboronic acid (AAPBA), and chitosan (CS) to obtain the composite hydrogel (Fig. S3a in Supporting information). This hydrogel, denoted as PBAM/CS/TFe, is replete with free catechol, phenylboronic acid, amine, and hydroxyl groups, and encompasses an array of reversible and dynamic bonds, including hydrogen bonds and boronic ester bonds. Consequently, it exhibits exceptional mechanical properties, rapid self-healing capabilities, and optimal tissue-adhesive performance, and it can sustain repeated stretching (Figs. S3b–d in Supporting information). The high photothermal conversion efficiency of TFe-NPs endows the hydrogel with remarkable antibacterial activity against both gram-positive and gram-negative bacteria (Fig. S3e in Supporting information). It is noteworthy that PTT, while effective in thermally inactivating bacteria, poses a risk of damaging surrounding healthy tissues due to its operational temperatures ranging from 55 ℃ to 60 ℃. Additionally, there is a potential for the production of heat shock proteins, which may confer resistance to PTT in bacteria [122].
PDT, emerging as a promising alternative especially for combating bacteria resistant to conventional antibiotics, has been garnering escalating interest in the realm of antibacterial treatments [123]. In antibacterial PDT therapy, a photosensitizer is absorbed by bacteria, then illuminated with light of a specific wavelength, activating the photosensitizer to generate ROS capable of damaging the bacterial cell wall and internal structure, leading to bacterial death [124]. The advantages of this approach include [125]: (1) Its unique antibacterial mechanism, making it difficult for bacterial strains to develop resistance; (2) high efficiency; (3) low toxic side effects and strong bacterial targeting; and (4) the ability to kill bacteria locally, minimizing widespread damage. Natural hydrogels, being the biomaterials closest to living tissue, have attracted significant research attention in PDT antibacterial therapy, where they serve as carriers for the photosensitizer. Encapsulated within the hydrogel, the photosensitizer can be slowly released into the infected area, prolonging its presence and increasing therapeutic effectiveness, while reducing potential damage to surrounding healthy tissue [126]. During PDT, light irradiation can be conducted through the hydrogel, thereby activating the photosensitizer while safeguarding the wound from direct light-induced harm. Inspired by emerging antimicrobial hydrogels, Li et al. constructed an injectable therapeutic hydrogel based on antibacterial peptide ε-polylysine (ePL) and PDA nanoparticles, designed for real-time diagnosis of infected wounds, image-guided antibacterial PDT therapy, and as-needed bacterial debris clearance [127]. A PLU hydrogel was generated via quadruple hydrogen bonding of uracil with ePL, with Schiff base bonds introduced into the PLU@PTc hydrogel via inoculation of four (4-carboxyphenyl)porphyrin (TCPP)-loaded PDA (PTc) nanoparticles (Fig. S4a in Supporting information). The dual cross-linked network improved the hydrogel’s mechanical properties, adhesive strength, and self-healing performance (Fig. S4b in Supporting information). Injection of PLU precursors and PTc NPs induced in-situ sol-gel transformation, with acid-triggered TCPP release restoring fluorescence emission for real-time imaging of infected wounds under 410 nm light. Subsequently, TCPP released in infected wounds was precisely used for antibacterial PDT under 660 nm light irradiation (Fig. S4c in Supporting information). However, current PDT antibacterial therapies have limitations, such as the transmembrane capability of the photosensitizer and its effective accumulation within bacteria, directly affecting PDT efficacy, and the short lifespan of ROS, demanding higher ROS production rates.
Combining these phototherapeutic agents with hydrogels facilitates targeted delivery to wound sites, thus enhancing the efficiency of the treatment. The hydrogels can be engineered to release the phototherapeutic agents in a controlled manner in response to specific environmental triggers. This also allows for the possibility of repeated treatments with a single application of the hydrogel dressing, as the agents can be activated by light as needed. This integration of smart responsive hydrogels with phototherapeutic agents holds great promise as an alternative or complementary approach to antibiotics for treating bacterial infections in wounds. It offers the benefits of high precision, reduced side effects, and efficacy against antibiotic-resistant strains of bacteria. However, it is also essential to consider the potential side effects and the optimization of the light parameters to ensure that the surrounding tissues are not harmed. As with any emerging technology, further research and clinical studies are needed to refine these therapies and evaluate their long-term efficacy and safety.
ROS, a critical mediator in human metabolic processes, plays a crucial role in maintaining normal physiological activities. However, their overexpression can trigger a cascade of inflammatory responses, leading to the onset of associated diseases [128]. With the deepening understanding of ROS mechanisms in recent years, the utilization of antioxidant materials to clear ROS has gradually become a significant strategy in treating inflammatory diseases. Among these, antioxidant hydrogels, owing to their remarkable biocompatibility and multifunctionality, are considered as one of the pivotal materials for ROS clearance in the body. The antioxidant components in these hydrogels vary and can be broadly categorized into natural polyphenols, polysaccharides, amino acids, and other high molecular weight antioxidant components. These components can either be individually introduced into the hydrogel to form antioxidant hydrogels, or they can be ingeniously designed into a multifunctional antioxidant hydrogel with excellent functionality through simple combinations, modifications, and polymerization [129]. Zhao et al. devised a strategy of encapsulating rhein fibers in a HA-based polysaccharide hydrogel (Rh@OR-S gel), which self-assembles through ferrocene (Fc) and β-cyclodextrin (β-CD) [130]. This hydrogel not only enhances the stability of rhein fiber, but also accelerates the exposure of rhein fiber at the inflammation site by responding to the oxidative microenvironment of the inflamed wound through Fc. The exposed rhein fiber interacts with cells at the inflammation site, enabling effective inflammation suppression, and facilitating the transition of intractable wounds from the inflammatory phase to a normal wound healing process (Fig. S5a in Supporting information). In another study, Zhang et al. proposed a glycosaminoglycan-based hydrogel delivery system to modulate the wound microenvironment [131]. As the scaffold of the hydrogel, heparin can capture inflammatory chemokines at the wound site, while HA can mimic the function of the ECM. The hydrophobic drug curcumin is cleverly encapsulated within the hydrogel through gelation, thereby demonstrating commendable ROS clearing ability and anti-inflammatory activity. Evaluations on diabetic mice revealed that this antioxidant and anti-inflammatory hydrogel can effectively reduce the influx of immune cells at the wound site and diminish the inflammatory response. By investigating the healing of diabetic wounds, faster re-epithelialization and improved ECM remodeling were achieved (Fig. S5b in Supporting information). This hydrogel presents a multi-faceted approach to modulate the wound microenvironment, providing a potentially innovative therapeutic strategy for chronic wounds.
Uncontrolled hemorrhage is a primary contributor to fatalities in emergency scenarios, including warfare, accidents, and natural calamities. Swift hemorrhage control is paramount in preserving lives under such circumstances. Numerous natural hydrogels possess inherent adhesiveness, enabling effective wound contact [132]. Although hydrogel-based tissue adhesives and hemostatic agents have found clinical applications, the intricate relationship between the microstructure of hydrogels and their roles in hemostasis and wound healing is not fully elucidated. Moreover, engineering high-performance hydrogels that effectively address wound closure, bleeding cessation, and healing is an intricate endeavor [133]. Inspired by the microstructures of extracellular matrices and mussel-mimetic chemistry, Teng et al. [134] devised a simple method to create two types of coordination and covalent glycopeptide hydrogels (Fig. S6a in Supporting information). These hydrogels exhibit adjustable tissue adhesion strength, microporous structures, high water absorption and swelling capacities, excellent injectability, practical irregular shape filling, and low hemolysis. Experimental results demonstrate that hydrogels with larger pore sizes (16–18 µm) possess exceptional hemostatic effects, with the minimum blood loss recorded at approximately 6%, indicating that micropore size is a critical factor for controlling in vivo hemostasis (Fig. S6b in Supporting information). Moreover, the synergistic effect of biocompatibility and hemostasis can have a positive impact on the hydrogel’s wound healing efficacy. This was exemplified by the complete healing of full-thickness skin defects in rats within 14 days, characterized by robust dermal and epidermal tissue regeneration and partial hair follicle restoration (Fig. S6c in Supporting information).
Due to their remarkable properties, natural hydrogels are extensively employed in wound treatment, particularly in the therapy of chronic wounds, such as burns and ulcers, where they exhibit a significant advantage. First and foremost, natural hydrogel dressings possess exceptional biocompatibility, biodegradability, and abundant availability. Moreover, they provide a moist environment for the wound, which is crucial for its healing. A damp milieu promotes cellular migration and proliferation, thereby expediting the wound healing process. The moist environment also aids in reducing bacterial growth, minimizing the risk of infection. Furthermore, hydrogel dressings exhibit impressive cooling effects, effectively alleviating a patient’s pain. When applied to a wound, the temperature drops, generating a soothing sensation and lessening discomfort. In addition, hydrogel dressings demonstrate exceptional absorptive capability, effectively retaining excess fluids emitted by the wound, helping maintain a stable wound environment and preventing excessive moisture. Importantly, these dressings can be replaced without causing secondary damage to the wound, making them an appealing option for patients susceptible to pain or with large wound areas. Lastly, natural hydrogels can serve as carriers for therapeutic agents, enabling the delivery of antibiotics, anti-inflammatory medications, and growth factors directly to the wound site. In addition, natural hydrogels are environment friendly as medical dressings due to their biodegradable properties, which is significant in the context of medical waste management.
Despite these numerous advantages, the practical application of hydrogel dressings presents certain challenges. For instance, their absorptive capacity may be insufficient for wounds with excessive fluid exudate, potentially compromising the moist environment and impeding healing. In comparison to other dressings, hydrogel dressings may require more frequent changes, increasing patient care responsibilities. This may pose a challenge for those unable to access regular hospital visits for dressing replacement. Additionally, the cost of hydrogel dressings may be slightly higher than traditional alternatives, potentially adding to patients’ financial burdens, especially those requiring long-term use. Furthermore, in specific scenarios, hydrogels may necessitate being used in conjunction with other dressings for added protection and absorption capability. For example, a highly exuding wound may require an absorptive dressing applied over the hydrogel. This not only complicates the changing process but can also increase financial strain.
Moving forward, the future perspectives of natural hydrogel dressings are centered around addressing the challenges and capitalizing on the advantageous properties they hold for wound treatment. The development of next-generation hydrogels should focus on innovative materials and designs that can offer enhanced absorption capacities. This could involve the incorporation of superabsorbent polymers or the creation of nano-structured hydrogels, which would allow for more effective management of wounds with heavy exudate without compromising the moist environment essential for healing. Furthermore, a greater focus on customizability and personalization of hydrogel dressings could be invaluable. Developments in biofabrication and 3D printing technologies could pave the way for hydrogel dressings tailored to the specific wound topography and exudate levels of individual patients. This can ensure that the dressings provide optimal contact and adhesion, which is vital for efficient wound healing. The integration of smart technologies into hydrogel dressings is another avenue that holds great promise. The development of ‘smart’ hydrogels with embedded sensors capable of monitoring wound pH levels, temperature, or bacterial contamination could enable real-time monitoring of the wound healing process. This, in turn, would allow healthcare providers to make timely and informed decisions regarding the need for dressing changes or additional interventions. In addition, reducing the cost of hydrogel dressings should be a priority, which can be achieved through material innovations and economies of scale in manufacturing. Recent studies show that supercritical fluid technology can be used to separate and purify compounds and homogeneously manufacture pharmaceuticals [135], we believe it might improve raw material purification in natural hydrogels and reduce organic solvent residues and natural hydrogel processing costs. The development of cost-effective hydrogel dressings would make them more accessible to a broader population, especially in low-resource settings. Lastly, the incorporation of multifunctionality into a single hydrogel dressing will be crucial. This includes combining hemostatic properties, antimicrobial agents, and growth factors into the hydrogel matrix, creating an all-in-one solution that can address various facets of wound healing. Such multifunctional dressings would simplify the wound treatment process and potentially reduce the need for additional dressings or interventions. However, due to the complexity of wound healing, the physiological state of the wound varies dynamically [1], and the mechanism of dressings that boost wound healing is still unclear, further study into dynamic mechanisms is crucial. In conclusion, the future of hydrogel dressings lies in the development of advanced materials that are highly absorbent, customizable, integrated with smart technologies, cost-effective, and multifunctional. This would not only enhance the efficacy of wound treatment but also reduce the burden on healthcare systems and improve patient quality of life. Collaboration among researchers, material scientists, clinicians, and industry will be pivotal to driving these innovations from the laboratory to clinical applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 52203146, 81925019, U1705281, and U22A20333), the Fundamental Research Funds for the Central Universities (No. 20720200019), and the Program for New Century Excellent Talents in University, China (No. NCET-13-0502).
Supplementary material associated with this article can be found, in the online version, at doi:
S. Dekoninck, C. Blanpain, Nat. Cell Biol. 21 (2019) 18–24. doi: 10.1038/s41556-018-0237-6
B.K. Sun, Z. Siprashvili, P.A. Khavari, Science 346 (2014) 941–945. doi: 10.1126/science.1253836
W. Hn, H. Mj, Open Biol. 10 (2020) 200223. doi: 10.1098/rsob.200223
R. Dong, B. Guo, Nano Today. 41 (2021) 101290. doi: 10.1016/j.nantod.2021.101290
X. Qi, Y. Xiang, E. Cai, et al., Chem. Eng. J. 439 (2022) 135691. doi: 10.1016/j.cej.2022.135691
B. Guo, R. Dong, Y. Liang, M. Li, Nat. Rev. Chem. 5 (2021) 773–791. doi: 10.1038/s41570-021-00323-z
M. Farahani, A. Shafiee, Adv. Healthc. Mater. 10 (2021) e2100477. doi: 10.1002/adhm.202100477
Z. Zeng, M. Zhu, L. Chen, et al., Compos. Part B Eng. 247 (2022) 110313. doi: 10.1016/j.compositesb.2022.110313
L.I.F. Moura, A.M.A. Dias, E. Carvalho, H.C. de Sousa, Acta Biomater. 9 (2013) 7093–7114. doi: 10.1016/j.actbio.2013.03.033
S. Li, S. Dong, W. Xu, et al., Adv. Sci. 5 (2018) 1700527. doi: 10.1002/advs.201700527
Z. Wang, H. Wei, Y. Huang, Y. Wei, J. Chen, Chem. Soc. Rev. 52 (2023) 2992–3034. doi: 10.1039/d2cs00813k
D. Yang, Chem. Mater. 34 (2022) 1987–1989. doi: 10.1021/acs.chemmater.2c00188
K. Gul, R.Y. Gan, C.X. Sun, et al., Rev. Food Sci. Nutr. 62 (2022) 3817–3832. doi: 10.1080/10408398.2020.1870034
Z. Bao, C. Xian, Q. Yuan, G. Liu, J. Wu, Adv. Healthc. Mater. 8 (2019) 1900670. doi: 10.1002/adhm.201900670
Y. Xu, H. Chen, Y. Fang, J. Wu, Adv. Healthc. Mater. 11 (2022) 2200494. doi: 10.1002/adhm.202200494
V. Brumberg, T. Astrelina, T. Malivanova, A. Samoilov, Biomedicines 9 (2021) 1235. doi: 10.3390/biomedicines9091235
Y. Wang, Y. Zhang, Z. Lin, et al., Compos. Part B Eng. 222 (2021) 109047. doi: 10.1016/j.compositesb.2021.109047
P. Jia, Y. Zou, J. Jiang, ACS Appl. Mater. Interfaces 15 (2023) 22929–22943. doi: 10.1021/acsami.3c02241
L. Qi, C. Zhang, B. Wang, J. Yin, S. Yan, Macromol. Biosci. 22 (2022) e2100475. doi: 10.1002/mabi.202100475
Y. Liang, J. He, B. Guo, ACS Nano 15 (2021) 12687–12722. doi: 10.1021/acsnano.1c04206
B. Ge, H. Wang, J. Li, et al., Mar. Drugs 18 (2020) 178. doi: 10.3390/md18040178
A. Sorushanova, L.M. Delgado, Z. Wu, et al., Adv. Mater. 31 (2019) e1801651. doi: 10.1002/adma.201801651
A.M. Ferreira, P. Gentile, V. Chiono, G. Ciardelli, Acta Biomater. 8 (2012) 3191–3200. doi: 10.1016/j.actbio.2012.06.014
M. Salamone, S. Rigogliuso, A. Nicosia, et al., Biomedicines 9 (2021) 739. doi: 10.3390/biomedicines9070739
H. Kim, S.H. Han, Y.M. Kook, et al., J. Mater. Chem. B 8 (2020) 9481–9491. doi: 10.1039/d0tb01770a
J. Hu, J. Wang, X. Zhu, et al., Biomacromolecules 22 (2021) 3440–3450. doi: 10.1021/acs.biomac.1c00520
J. Pupkaite, M. Griffith, J. Hilborn, E. Suuronen, A. Samanta, J. Mol. Cell. Cardiol. 124 (2018) 104.
J. Wang, W. Liu, G. Luo, et al., Energy Environ. Sci. 11 (2018) 3375–3379. doi: 10.1039/c8ee02656d
H. Park, S.N. Nazhat, D.H. Rosenzweig, Biomaterials 286 (2022) 121606. doi: 10.1016/j.biomaterials.2022.121606
Z. Tian, W. Liu, G. Li, Polym. Degrad. Stab. 130 (2016) 264–270. doi: 10.1016/j.polymdegradstab.2016.06.015
L. Michelini, L. Probo, S. Farè, N.C. Negrini, Mater. Lett. 272 (2020) 127865. doi: 10.1016/j.matlet.2020.127865
V.H. Segtnan, T. Isaksson, Food Hydrocoll. 18 (2004) 1–11. doi: 10.1016/S0268-005X(02)00096-6
A. Duconseille, T. Astruc, N. Quintana, F. Meersman, V. Sante-Lhoutellier, Food Hydrocoll. 43 (2015) 360–376. doi: 10.1016/j.foodhyd.2014.06.006
P.L. Thi, Y. Lee, D.H. Nguyen, K.D. Park, J. Mater. Chem. B 5 (2017) 757–764. doi: 10.1039/C6TB02179D
S.T. Koshy, R.M. Desai, P. Joly, et al., Adv. Healthc. Mater. 5 (2016) 541–547. doi: 10.1002/adhm.201500757
D.S. Yoon, Y. Lee, H.A. Ryu, Y. Jang, et al., Acta Biomater. 38 (2016) 59–68. doi: 10.1016/j.actbio.2016.04.030
S. Ge, Q. Liu, M. Li, et al., Food Hydrocoll. 75 (2018) 1–12. doi: 10.1016/j.foodhyd.2017.09.023
F.A. Ngwabebhoh, R. Patwa, O. Zandraa, N. Saha, P. Saha, J. Drug Deliv. Sci. Technol. 63 (2021) 102415. doi: 10.1016/j.jddst.2021.102415
T. Kobayashi, T. Chanmee, N. Itano, Biomolecules 10 (2020) 1525. doi: 10.3390/biom10111525
R. Al-Khateeb, I. Olszewska-Czyz, Heliyon 6 (2020) e03722. doi: 10.1016/j.heliyon.2020.e03722
C.G. Boeriu, J. Springer, F.K. Kooy, L.A.M. van den Broek, G. Eggink, Int. J. Carbohydr. Chem. 2013 (2013) e624967.
A. Asari, S. Miyauchi, S. Matsuzaka, T. Itoh, Y. Uchiyama, Osteoarthritis Cartilage 4 (1996) 213–215. doi: 10.1016/S1063-4584(96)80018-5
A. Marinho, C. Nunes, S. Reis, Biomolecules 11 (2021) 1518. doi: 10.3390/biom11101518
K. Kocourková, L. Musilová, P. Smolka, et al., Carbohydr. Polym. 254 (2021) 117307. doi: 10.1016/j.carbpol.2020.117307
Y. Kawano, R. Kageyama, T. Iyoda, F. Fukai, T. Hanawa, New Biotechnol. 44 (2018) S104.
L.D. Morton, D.A. Castilla-Casadiego, A.C. Palmer, A.M. Rosales, Acta Biomater. 155 (2023) 258–270. doi: 10.1016/j.actbio.2022.11.027
M. Jurtík, B. Gřešková, Z. Prucková, et al., Carbohydr. Polym. 313 (2023) 120872. doi: 10.1016/j.carbpol.2023.120872
Z. Shariatinia, Adv. Colloid Interface Sci. 263 (2019) 131–194. doi: 10.1016/j.cis.2018.11.008
J. Cao, P. Wu, Q. Cheng, et al., ACS Appl. Mater. Interfaces 13 (2021) 24095–24105. doi: 10.1021/acsami.1c02089
I. Stefanov, D. Hinojosa-Caballero, S. Maspoch, J. Hoyo, T. Tzanov, J. Mater. Chem. B 6 (2018) 7943–7953. doi: 10.1039/c8tb02483a
B. Li, J. Elango, W. Wu, Appl. Sci. 10 (2020) 4719. doi: 10.3390/app10144719
Y. Guo, C. Zhao, C. Yan, L. Cui, Cellulose 28 (2021) 10013–10023. doi: 10.1007/s10570-021-04149-2
Y. Jiang, X. Meng, Z. Wu, X. Qi, Carbohydr. Polym. 144 (2016) 245–253. doi: 10.1016/j.carbpol.2016.02.059
D. Xu, S. Hein, L.S. Loo, K. Wang, Ind. Eng. Chem. Res. 50 (2011) 6343–6346. doi: 10.1021/ie101987w
N. Sereni, A. Enache, G. Sudre, et al., Langmuir 33 (2017) 12697–12707. doi: 10.1021/acs.langmuir.7b02997
H. Tan, C.R. Chu, K.A. Payne, K.G. Marra, Biomaterials 30 (2009) 2499–2506. doi: 10.1016/j.biomaterials.2008.12.080
K.Y. Lee, D.J. Mooney, Prog. Polym. Sci. 37 (2012) 106–126. doi: 10.1016/j.progpolymsci.2011.06.003
M. Zhang, X. Zhao, Int. J. Biol. Macromol. 162 (2020) 1414–1428. doi: 10.1016/j.ijbiomac.2020.07.311
H.H. Tønnesen, J. Karlsen, Drug Dev. Ind. Pharm. 28 (2002) 621–630. doi: 10.1081/DDC-120003853
J. Qin, M. Li, M. Yuan, X. Shi, et al., ACS Appl. Mater. Interfaces 14 (2022) 22426–22442. doi: 10.1021/acsami.2c02497
X. Xue, G. Song, C. Chang, Carbohydr. Polym. 288 (2022) 119376. doi: 10.1016/j.carbpol.2022.119376
F. Abasalizadeh, S.V. Moghaddam, E. Alizadeh, et al., J. Biol. Eng. 14 (2020) 8. doi: 10.1186/s13036-020-0227-7
F. Ahmad, B. Mushtaq, F.A. Butt, A. Rasheed, S. Ahmad, Cellulose 28 (2021) 7941–7951. doi: 10.1007/s10570-021-04043-x
J.L. Drury, R.G. Dennis, D.J. Mooney, Biomaterials 25 (2004) 3187–3199. doi: 10.1016/j.biomaterials.2003.10.002
M. Zhang, X. Qiao, W. Han, et al., Carbohydr. Polym. 266 (2021) 118100. doi: 10.1016/j.carbpol.2021.118100
L. Aguero, S. Alpdagtas, E. Ilhan, D. Zaldivar-Silva, O. Gunduz, Eur. Polym. J. 160 (2021) 110807. doi: 10.1016/j.eurpolymj.2021.110807
C. García-Astrain, L. Avérous, Carbohydr. Polym. 190 (2018) 271–280. doi: 10.1016/j.carbpol.2018.02.086
S.H. Zainal, N.H. Mohd, N. Suhaili, et al., J. Mater. Res. Technol. 10 (2021) 935–952. doi: 10.1016/j.jmrt.2020.12.012
L.H. Fu, C. Qi, M.G. Ma, P. Wan, J. Mater. Chem. B 7 (2019) 1541–1562. doi: 10.1039/c8tb02331j
H. Xu, Y. Liu, Y. Xie, et al., Cellulose 26 (2019) 8645–8654. doi: 10.1007/s10570-019-02689-2
M. He, H. Chen, X. Zhang, et al., Cellulose 25 (2018) 1987–1996. doi: 10.1007/s10570-018-1683-9
H. Namazi, R. Rakhshaei, H. Hamishehkar, H.S. Kafil, Int. J. Biol. Macromol. 85 (2016) 327–334. doi: 10.1016/j.ijbiomac.2015.12.076
A. Pettignano, A. Charlot, E. Fleury, Polym. Rev. 59 (2019) 510–560. doi: 10.1080/15583724.2019.1579226
Q. Lu, B. Zhang, M. Li, et al., Biomacromolecules 12 (2011) 1080–1086. doi: 10.1021/bm101422j
T.S. Khire, J. Kundu, S.C. Kundu, V.K. Yadavalli, Soft Matter. 6 (2010) 2066–2071. doi: 10.1039/b924530h
S. Chen, M. Liu, H. Huang, L. Cheng, H.P. Zhao, Mater. Des. 181 (2019) 108077. doi: 10.1016/j.matdes.2019.108077
A.Z.S.A. B, J.M. A, M.R. B, et al., Int. J. Biol. Macromol. 153 (2020) 317–326. doi: 10.1016/j.ijbiomac.2020.02.316
C.B. Oral, B. Yetiskin, O. Okay, Int. J. Biol. Macromol. 161 (2020) 1371–1380. doi: 10.1016/j.ijbiomac.2020.08.040
H. Zheng, B. Zuo, J. Mater. Chem. B 9 (2021) 1238–1258. doi: 10.1039/d0tb02099k
C. Niu, X. Liu, Y. Wang, X. Li, J. Shi, Eur. Polym. J. 146 (2021) 110267. doi: 10.1016/j.eurpolymj.2021.110267
O. Hasturk, K.E. Jordan, J. Choi, D.L. Kaplan, Biomaterials 232 (2020) 119720. doi: 10.1016/j.biomaterials.2019.119720
C. Yang, S. Li, X. Huang, et al., Oxid. Med. Cell. Longev. 2022 (2022) e2076680.
J. Yang, Y. Chen, L. Zhao, J. Zhang, H. Luo, Polym. Rev. 63 (2022) 574–612.
M.X. Wang, Y.M. Chen, Y. Gao, et al., ACS Appl. Mater. Interfaces 10 (2018) 26610–26617. doi: 10.1021/acsami.8b06567
S. Yao, Y. Zhao, Y. Xu, et al., Adv. Healthc. Mater. 11 (2022) 2200516. doi: 10.1002/adhm.202200516
M. Ma, J. Yang, Z. Ye, et al., Macromol. Mater. Eng. 306 (2021) 2000577. doi: 10.1002/mame.202000577
A. Khabibullin, M. Alizadehgiashi, N. Khuu, et al., Langmuir 33 (2017) 12344–12350. doi: 10.1021/acs.langmuir.7b02906
H.P.S. Abdul Khalil, Y. Davoudpour, M.N. Islam, et al., Carbohydr. Polym. 99 (2014) 649–665. doi: 10.1016/j.carbpol.2013.08.069
J. Yao, M. Yang, Y. Duan, Chem. Rev. 114 (2014) 6130–6178. doi: 10.1021/cr200359p
P. Wei, X. Yu, Y. Fang, et al., Small 19 (2023) 2301204. doi: 10.1002/smll.202301204
M.N.V.R. Kumar, R.A.A. Muzzarelli, C. Muzzarelli, H. Sashiwa, A.J. Domb, Chem. Rev. 104 (2004) 6017–6084. doi: 10.1021/cr030441b
L. Xu, L. Cui, M. Jia, et al., Carbohydr. Polym. 195 (2018) 593–600. doi: 10.1016/j.carbpol.2018.04.078
Y. Gao, X. Jin, Mar. Drugs 17 (2019) 557. doi: 10.3390/md17100557
S. Chen, D. Zhao, L. Chen, et al., Small Struct. 2 (2021) 2100082. doi: 10.1002/sstr.202100082
Z. Wang, J. Lu, Z. Yuan, et al., Small 19 (2023) 2205528. doi: 10.1002/smll.202205528
B. Yang, J. Song, Y. Jiang, et al., ACS Appl. Mater. Interfaces 12 (2020) 57782–57797. doi: 10.1021/acsami.0c18948
X. Dou, N. Mehwish, C. Zhao, et al., Acc. Chem. Res. 53 (2020) 852–862. doi: 10.1021/acs.accounts.0c00012
S. Bai, M. Zhang, X. Huang, et al., Chem. Eng. J. 413 (2021) 127512. doi: 10.1016/j.cej.2020.127512
G. Yu, K. Jie, F. Huang, Chem. Rev. 115 (2015) 7240–7303. doi: 10.1021/cr5005315
J. Zhou, G. Yu, F. Huang, Chem. Soc. Rev. 46 (2017) 7021–7053. doi: 10.1039/C6CS00898D
J. Yan, Y. Ji, M. Huang, et al., ACS Mater. Lett. 2 (2020) 1375–1380. doi: 10.1021/acsmaterialslett.0c00304
S. Cheng, H. Wang, X. Pan, et al., ACS Appl. Mater. Interfaces 14 (2022) 11144–11155. doi: 10.1021/acsami.1c25014
W. Jing, C. Xiaolan, C. Yu, Q. Feng, Y. Biomed. Pharmacother. 154 (2022) 113561. doi: 10.1016/j.biopha.2022.113561
X. Zhou, Q. Zhou, Q. Chen, et al., ACS Biomater. Sci. Eng. 9 (2023) 437–448. doi: 10.1021/acsbiomaterials.2c00997
S. Guo, Y. Ren, R. Chang, et al., ACS Appl. Mater. Interfaces 14 (2022) 34455–34469. doi: 10.1021/acsami.2c08870
J. Zhou, R. Cha, Z. Wu, et al., Nano Today 49 (2023) 101801. doi: 10.1016/j.nantod.2023.101801
X. Zhao, H. Zhang, Y. Gao, Y. Lin, J. Hu, ACS Appl. Bio Mater. 3 (2020) 648–653. doi: 10.1021/acsabm.9b01007
Q. Li, S. Zhang, R. Du, et al., ACS Appl. Mater. Interfaces 15 (2023) 17562–17576. doi: 10.1021/acsami.2c23231
Y.P. Li, I. Ben Fekih, E.C. Fru, et al., Annu. Rev. Microbiol. 75 (2021) 175–197. doi: 10.1146/annurev-micro-032921-123231
H. Huang, X. Zhang, Z. Dong, X. Zhao, B. Guo, J. Colloid Interface Sci. 625 (2022) 817–830. doi: 10.1016/j.jcis.2022.06.058
E. Zhang, X. Zhao, J. Hu, et al., Bioact. Mater. 6 (2021) 2569–2612.
L. Schnaider, Z. Toprakcioglu, A. Ezra, et al., Nano Lett. 20 (2020) 1590–1597. doi: 10.1021/acs.nanolett.9b04290
S. Shankar, J.W. Rhim, Environ. Chem. Lett. 17 (2019) 1105–1109. doi: 10.1007/s10311-018-00835-z
T. Hu, G.P. Wu, H. Bu, et al., Chem. Eng. J. 450 (2022) 138201. doi: 10.1016/j.cej.2022.138201
L.S. Munoz-Price, J.F. Frencken, S. Tarima, M. Bonten, Clin. Infect. Dis. 62 (2016) 1558–1563. doi: 10.1093/cid/ciw191
Y. Fan, M. Lüchow, Y. Zhang, et al., Adv. Funct. Mater. 31 (2021) 2006453. doi: 10.1002/adfm.202006453
L. Li, X. Cheng, Q. Huang, et al., Adv. Healthc. Mater. 11 (2022) e2201286. doi: 10.1002/adhm.202201286
Y. Wang, S. Zhang, J. Wang, Chin. Chem. Lett. 32 (2021) 1603–1614. doi: 10.1016/j.cclet.2020.11.073
S. Cheng, M. Pan, D. Hu, et al., Chin. Chem. Lett. 34 (2023) 108276. doi: 10.1016/j.cclet.2023.108276
Y. Chen, Y. Gao, Y. Chen, et al., J. Control. Release 328 (2020) 251–262. doi: 10.1016/j.jconrel.2020.08.055
K. Yang, X. Zhou, Z. Li, et al., ACS Appl. Mater. Interfaces 14 (2022) 43010–43025. doi: 10.1021/acsami.2c13283
T. Sun, X. Chen, X. Wang, et al., Mater. Chem. Front. 3 (2018) 127–136.
E. Polat, K. Kang, Biomedicines 9 (2021) 584. doi: 10.3390/biomedicines9060584
M. Piksa, C. Lian, I.C. Samuel, et al., Chem. Soc. Rev. 52 (2023) 1697–1722. doi: 10.1039/d0cs01051k
X. Hu, H. Zhang, Y. Wang, et al., Chem. Eng. J. 450 (2022) 138129. doi: 10.1016/j.cej.2022.138129
Z. Feng, S. Lin, A. McDonagh, C. Yu, Curr. Med. Chem. 27 (2020) 2681–2703. doi: 10.2174/0929867326666191016112828
P. Ran, H. Zheng, W. Cao, et al., ACS Appl. Mater. Interfaces 14 (2022) 49375–49388. doi: 10.1021/acsami.2c15561
S.J. Forrester, D.S. Kikuchi, M.S. Hernandes, Q. Xu, K.K. Griendling, Circ. Res. 122 (2018) 877–902. doi: 10.1161/circresaha.117.311401
Z. Xu, S. Han, Z. Gu, J. Wu, Adv. Healthc. Mater. 9 (2020) 1901502. doi: 10.1002/adhm.201901502
W. Zhao, X. Zhang, R. Zhang, et al., ACS Appl. Mater. Interfaces 12 (2020) 56898–56907. doi: 10.1021/acsami.0c19492
X. Zhang, J. Feng, W. Feng, et al., ACS Appl. Mater. Interfaces 14 (2022) 31737–31750. doi: 10.1021/acsami.2c08593
J. Yang, H. Yu, L. Wang, et al., Eur. Polym. J. 172 (2022) 111241. doi: 10.1016/j.eurpolymj.2022.111241
G. Chen, Y. Yu, X. Wu, et al., Adv. Funct. Mater. 28 (2018) 1801386. doi: 10.1002/adfm.201801386
L. Teng, Z. Shao, Q. Bai, et al., Adv. Funct. Mater. 31 (2021) 2105628. doi: 10.1002/adfm.202105628
Y. Zheng, Y. Huang, J. Luo, et al., Chin. Chem. Lett. 35 (2024) 109169. doi: 10.1016/j.cclet.2023.109169

Figure 1 Schematic representation of a natural hydrogel used as a wound dressing. It encompasses the composition, principles underlying the formation of the hydrogel network, as well as their biological processes.

Figure 2 Crosslinking mechanisms in the formation of natural hydrogels. Mechanical strength, viscoelasticity, and thinning qualities are susceptible to variation across natural hydrogels due to their crosslinking mechanism and distinctive structural features. Created with BioRender.com.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: