Figure 1.
CPL-triggered enantioselective synthesis, including chiral organic molecules and inorganic nanostructures.

Chirality, the basic characteristics in biological system, is the main driving force for the evolution and exists in various biomolecules, such as proteins, nucleic acids, and polysaccharides [1]. However, these enantiomers of bioactive molecule usually exhibit different pharmacological activities, metabolic pathways and cytotoxicity, which deserve careful study. Generally, there are three approaches for synthesizing chiral compounds, including chiral resolution, biosynthesis and stereoselective synthesis.
Stereoselective synthesis is a key process in modern chemistry and is particularly significant in the field of pharmaceuticals, since different enantiomers usually take different biological activity. Generally, in the absence of external chiral influence, chiral product is a racemic mixture with equal amount of constituent enantiomers. However, chiral environments driven either by chemical reactions or physical forces can potentially affect the equilibrium of enantiomers resulting in enantiomer enrichment. In organic chemistry, chiral product with high enantiomeric excess (ee) could be obtained with the assistance of chiral catalysts, which are typically chiral coordination compounds. However, these chiral catalysts are invariably endowed with chirality by using extra chiral ligands, which requires additional asymmetric catalytic synthesis. Meanwhile, these chiral catalysts, usually belong to metal complex, would remain in products, resulting in additional post-processing. Therefore, it is urgent and necessary to development a green synthesis method using circularly polarized light (CPL) as the sole chiral source.
As we all know, there are various physical forces such as linearly polarized light, CPL, magnetic and gravitational fields in the world have been tested to induce chirality in molecular system. However, only CPL functioned as a truly chiral physical force successfully produces product with detectable optical activity, and left-handed CPL (l-CPL) and right-handed CPL (r-CPL) generate products with opposite handedness due to photo-resolution or asymmetric absorption of enantiomers. For a chiral molecule capable of absorbing ultraviolet (UV) or visible light, in principle, it is reasonable to enrich one enantiomer from a racemic mixture by CPL irradiation; since circular dichroism (CD) arises from a difference in the absorption of l-CPL and r-CPL by an optically active molecule, l-CPL and r-CPL would preferentially interact with one enantiomer of substances that exhibit circular dichroism.
CPL-triggered asymmetric photochemical reaction takes several advantages. For example, continuous exposure of amino acids or sugars under CPL irradiation could generate biased chirality in small enantiomeric excess, which then undergoes chiral amplification to enrich single-handed chiral structures [2,3]. Therefore, CPL is considered to be a true chiral entity and has been suggested as a possible explanation for the origin of homochirality in nature. Meanwhile, photochromic system and its reversible switching of molecular chirality by CPL has the potential application in chiroptical memory devices for data storage [4]. Thus, CPL cannot only provide the required energy to a photochemical reaction, but also introduce some chiral bias without extra impurities, which can be regarded as a green chiral initiator. It should be note that, we easily obtain CPL source through a physical method by using a linear polarizer and a quarter-wave plates. CPL may also be generated from chiral luminescent materials, called circularly polarized luminescence [5-7]. Herein, we are talking about asymmetric photochemical reactions induced by CPL generated by the physical method.
In this review, we summarize the recent status of absolute enantioselective synthesis to generate chiral organic molecules and inorganic nanostructures under CPL irradiation (Fig. 1). We begin with three mechanisms of CPL-triggered asymmetric photochemical reactions. Subsequently, three detecting techniques that widely used in determining and quantifying chiral matters, including chiral high-performance liquid chromatography (HPLC), optical rotation and CD spectroscopy are introduced. Next, organic molecules induced by CPL through the mechanism of photo-resolution or asymmetric absorption are discussed in detail, and further extended to helical polymer and self-assembly systems. Meanwhile, CPL-induced chiral inorganic nanostructures are highlighted and exhibit different mechanism comparing with organic systems. We end this review with perspectives that enhance the enantioselectivity by using the recently emerged superchiral light, including superchiral near-field and far-field. We hope this review could guide the further development of CPL-triggered absolute enantioselective synthesis in organic and inorganic systems.
Generally, CPL-induced absolute asymmetrically photochemical reactions can be divided into three modes [8,9]:
(1) Photochemical asymmetric degradation: the pair of enantiomers in a racemic substrate (SS and SR) take different decomposition rates during the photochemical process, and the enantiomer with a lower decomposition rate would be selectively retained. In theory, however, a high ee values of substrate in this pathway can only be obtained when the photolysis reaction of one enantiomer is almost complete. Therefore, the recovery of the substrate is usually very low (Fig. 2A).

(2) Photochemical deracemization: one starting enantiomer (SR) of a racemic substrate is selectively excited by CPL (e.g., r-CPL in Fig. 2B), and the excited state of enantiomer (SR*) can be converted into the opposite enantiomer (SS*). In this case, both excited states could transform into the ground states (SR & SS), and finally form a photo-stationary state. Compared with asymmetric degradation, the main difference is the total concentration of substrate does not change during photo-deracemization and reaches the same photo-stationary state after irradiation, regardless of the initial enantiomeric composition ratio.
(3) Photochemical asymmetric fixation: one enantiomer of the racemic substrate is selectively excited by CPL and generates a chiral product through a photochemical process (Fig. 2C). Different from the above two approaches, this method can directly generate chiral products (PR).
HPLC is a versatile separation technique with the characteristics of sensitivity, specificity and precision. Generally, a mixture dissolved in solvent (mobile phase) passes through the packing material (stationary phase), which is hold on the column in HPLC. The mixture would be separated into individual components by the successively flow out from the stationary phase, due to the different interactions (adsorption, distribution, ion attraction, exclusion, affinity) of each component with the stationary phase. The concentration of each component could be responded by the detector and recorded as a peak on the spectrum (Fig. 3A). More importantly, the peak's area represents the quantity of component, and the content of each component can be further analyzed by integrating the corresponding peak area. Specifically, using a chiral stationary phase in HPLC, the enantiomers can be selectively separated. Therefore, the enantiomeric excess could be determined: where [R] stands for the peak area of R-enantiomer, and [S] stands for that of S-enantiomer. If it is a S-dominated product, switching the formula. The negative ee value indicates that this reaction generates the product with the opposite configuration. We must note that chiral HPLC cannot determine the absolute configuration for an unknown chiral compound, which usually determined by single crystal X-ray diffraction analysis (XRD). Except for the direct XRD method, circular dichroism spectra (CD), nuclear magnetic resonance (NMR), enzymatic method and other techniques are also developed for determining the absolute configuration [10].
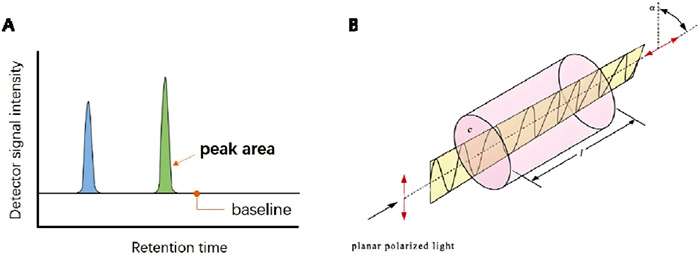
Chiral compounds show optical rotation, which is the rotation of the orientation of the plane of polarization about the optical axis of linearly polarized light as it travels through certain chiral materials (Fig. 3B). Specific rotation is defined as the change in orientation of monochromatic plane-polarized light, per unit distance–concentration product, as the light passes through a chiral solution. Compounds rotate the plane of polarization beam clockwise are said to be dextrorotary, and correspond with positive specific rotation values (+[α]), while compounds rotate the plane of polarization of plane polarized light counterclockwise are said to be levorotary, and correspond with negative values (−[α]). The rotation angle (α) of the plane polarized light measured at a path length (l) and concentration (c). This is a typical characteristic of a chiral substance. The formula of specific rotation [α] is:
|
|
where, T is the detection temperature and λ is the wavelength of the incident light.
In some case, the ee value of a sample could be calculated by using the optical rotation. If we do not know the quantities of the enantiomers in the mixture, we can calculate them by first determining the observed rotation of the sample. This number then is used in the following formula linking the ee, the observed specific rotation, and the specific rotation of the pure enantiomer that is in excess:
|
|
CD spectroscopy is the differential absorption (∆ε) plotted as a function of wavelength: ∆ε = εL - εR, where ε and ε refer to the extinction coefficients for l-CPL and r-CPL, respectively. Different from HPLC and optical rotation technique, which only detect solution sample, both sample in solution and solid state can be sensitively detected by CD spectroscopy. Therefore, CD signal at different wavelengths take different applications: in UV region is used to investigate the secondary structure of proteins; in UV-vis region is used to investigate charge-transfer transitions; in near-infrared region is used to investigate geometric and electronic structure by probing metal d→d transitions.
The magnitude of CD signal can be quantified by the absorptive dissymmetry factor: gabs, which is the ratio of the CD to the molar extinction coefficient: gabs = ∆ε/ε
Experimentally, the value of gabs is defined as:
|
|
where the ellipticity (unit: mdeg) and the absorbance can be directly obtained from CD spectroscopy. Therefore, gabs eliminates the effect of concentration of sample and shows the absolute magnitude of chirality.
CPL-induced asymmetrically organic synthesis was pioneered from the early 1970s. Kagan [11] and Calvin [12] groups reported the CPL-triggered annulation to synthesize optically active helicenes (Eq. 1). This breakthrough reaction was carried out in an aromatics solution under CPL irradiation in the presence of I2 (~0.21% ee for [6]helicene 2). After altering the handedness of CPL, the product showed opposite optical activity. This result demonstrated firstly that the configuration of products could be controlled by a chiral physical field. Followed this pioneering report, some better enantioselective reactions (~0.28% ee for [8]helicene and 0.37% ee for [9]helicene) was successfully achieved [13].
|
|
(1) |
As we all know, the photochemical electrocyclization is a reversible reaction. Fuchter successfully promoted an asymmetric cyclization of dihydrohelicene 4 and its reverse ring-opening reaction by using two wavelengths of CPL beam, in which r-CPL at 355 nm and l-CPL at 532 nm induced the asymmetric cyclization and ring-opening reaction respectively [14]. Meanwhile, an "addictive" effect, that stepwise accumulation of enantiomers through the formed photo-stationary state for each individual reaction, could ultimately increase the enantioselectivity of the overall reaction. This strategy can double the ee value of [6]helicene obtained by Kagan (Eq. 2). By analyzing the mechanism, we observe that the enantiomers could be selectively excited by CPL in a rapid equilibrium due to the rotation of the single bond, resulting in low enantioselectivity in synthesis of helicene. In contrast, the configuration of the double bond is relatively fixed, and the interconversion of the cis/trans configurations is usually achieved via a planar excited state.
|
|
(2) |
Feringa and co-workers reported a CPL-induced photo-resolution of overcrowded alkenes. Compared with a low enantiomer excess in hexane (0.07% ee), 0.14% ee could be obtained after CPL irradiation for a mixture of alkene 6 assisted with cholesteric liquid crystal (Eq. 3). In this case, although low ee value of 6 was obtained, it acted as a chiral dopant in liquid crystal. As a result, the achiral liquid crystal turned to a chiral mesoscopic phase (cholesteric liquid crystal). The long-range ordered cholesteric liquid crystal amplified the enantioselectivity of CPL-induced photo-resolution of alkene in reverse [4]. Therefore, the alkene and liquid crystal complement each other in this enantioselective photoreaction.
|
|
(3) |
Generally, carbonyl compounds showed higher absorptive dissymmetry factor (gabs) comparing with other organic molecules due to the partially forbidden n-π* transition [9]. In 1995, Schuster and co-workers provided that CPL-excited ketone group to afford a chiral ketone 7 after 47 h with 1.6% ee (Eq. 4) [15]. On the basis of this resolution, Soai developed a CPL-induced, alkene-catalyzed autocatalytic reaction (Eq. 5) [16,17]. This autocatalysis can give an excellent enantioselectivity (> 90% ee) due to the characteristic of nonlinear amplification of chirality [18,19]. However, nonlinear chiral amplification is quite rare in asymmetric organic synthesis. Except for the asymmetric nucleophilic addition of alkyl zinc to pyrimidine aldehyde (Soai reaction), another example is a similar asymmetric nucleophilic addition of alkyl zinc to benzaldehyde, which was reported by Bellemin-Laponnaz et al. in 2020 [20].
|
|
(4) |
|
|
(5) |
It is well known that azobenzene can undergo a reversible cis-trans isomerization by irradiation at different wavelengths [21]. Here, CPL-induced asymmetric cis-trans isomerization of a prochiral diazobenzne to generate chiral compound [22]. Tamaoki provided a new idea for the construction of a chiral azobenzene. Macrocyclic azobenzene 10 bearing with a dimethylbenzene unit is a planar chiral molecule, when the azobenzene unit is in the trans-conformation, which is due to the restricted free rotation of the dimethylbenzene rotor [23]. In contrast, cis-azoisomer allowed the rotation and lack of chirality, but showed unstable comparing with trans-isomer. As the photochemical cis-trans isomerization, Tamaoki utilized CPL at 488 nm to promote the reversible construction of chiral trans-azomacrocycle with 0.3% ee through the rapidly racemization in the cis-isomer (Eq. 6) [24].
|
|
(6) |
CPL-triggered enantioselective thiol–ene reactions, which combine absolute asymmetric synthesis with click reactions. In 2017, Zou group firstly reported this asymmetric photoreaction due to the asymmetric absorption of CPL by racemic thiol enantiomers [25]. Then, in 2021, Zou further investigated its mechanism in detail by testing this reaction in serials of small organic molecules [26]. They found that the enantioselectivity is extremely low (ee < 2%) and ascribed it to an undesirable side-effect in its unique reaction kinetics. The key intermediate in thiol–ene photoreaction is thiyl radicals. Based on this understanding, they designed a new strategy to improve enantioselectivity of CPL-triggered asymmetric thiol–ene reaction, by mitigating the generation of racemic thiyl radical upon adding extra hydrogen atom donor, which should quench the carbon-centered radical directly and suppress the chain transfer step, breaking the conventional cycle of thiol–ene reaction. As a result, a two-fold improvement in enantioselectivity of this reaction was achieved (Eq. 7).
|
|
(7) |
Based on the above discussion, the enantioselectivity in CPL-triggered photochemical reaction to synthesis chiral small organic molecule is extremely low (ee < 2%), indicating that the chiral photon-matter interaction is very faint. This is owing to the arrangement of molecular bonds giving rise to chiral features, is over a smaller distance than the helical pitch of the CPL. Therefore, it is necessary to develop a new strategy to increase the ee of product. Some scientists turned to CPL-triggered asymmetrical photopolymerization or self-assembly, which shown better light-matter interaction and higher optical activity. Following are some examples about CPL-induced chiral polymer and self-assembly.
In 2000, Selinger and co-workers introduced this axial styrene to polyisocyanates, which exist equally dynamical interconverting right- and left-handed helices. These helices rotated in opposite direction were separated by infrequent helical reversals, whose relative populations were extraordinarily sensitive to chiral perturbation. Notably, Green found that these chiral side chains in low ee, near-racemic polyisocyanates often cluster together randomly to form a short chiral helix, that is "majority rules" [27]. Taking advantage of this properties, a slight enantiomeric excess of axial styrene was induced by CPL, and amplified through the chiral response of the polymer (Fig. 4) [28]. On the CD spectrum, the chiral polyisocyanate initially exhibited a CD maximum at 255 nm. However, after CPL irradiation, the CD signal of para-13 disappeared, indicating that partial resolution of the para-linked ketone group in the polymer-13 didnot affect the change of the helical structure of the backbone. In contrast, meta-14 or ortho-15 polymer maintained its CD signals after r-CPL irradiation, and showed opposite features. Furthermore, both CD signals of meta-14 and ortho-15 could be reversibly switched upon irradiated with opposite-handedness CPL, and disappeared when irradiated by unpolarized light. This result indicated that the optical activities of polymer could be fine-tuned by chiral external fields.

In 1997, Nikolova and co-workers reported the first CPL-induced chirality in films of side-chain azobenzene polyester, and the rotation direction of the polyester could be maintained even the excitation was switched off after a few minutes [29]. In addition, the incident light with small ellipticity might induce changes in the orientations of the macroscopic optical axis, but no optical axis can be induced when light ellipticity is ~1 [30]. Followed this breakthrough, CPL-induced ordered chiral azo-polymer was developed rapidly [31,32].
Royes et al. carried out CPL-induced chirality in azobenzene-contained polymer (azo-polymer in Figs. 5A and B) [33]. The r- and l-CPL irradiated azo-polymers gave well-defined CD features. However, r- and l-CPL irradiation generated silent CD results for non-azo-polymer (19). It implied that azobenzene units are required for the chirality transfer from photons to molecules. By examining the CD spectroscopy of the monofunctional-unit-containing polymer 16 formed after CPL irradiation, a strong exciton pairs associated with π-π* absorption was shown. It indicated the existence of a chiral arrangement between achiral azobenzene units. The non-absorptive signal at long wavelength (500 nm) suggested that there may be selective reflection caused by long-range helical organization. Moreover, there were some slightly differences in CD signals between monofunctional- (16) and difunctional-unit-containing polymers (17, 18). For the di-azo-polymer 17, the most intense CD absorption was the exciton pairs associated with the π-π* transition of the azobenzene groups, whereas the mono-azo-polymer 18, CD signals came from the overlap of exciton couplet of 4-cyanoazobene moieties and π-π* transition of 4-cyanobiphenyl moieties. The chiral arrangement of non-photosensitive biphenyl units due to the CPL-induced asymmetric alignment of azobenzene units. This difference between 16 and 17 (or 18) indicate that CPL-induced chiral arrangement is influenced by the density of the azobenzene unit.

Fujiki and Zhang group verified that the chiral center of a photosensitive molecule was not only determined by the handedness of CPL, but also the wavelength of CPL (Fig. 5C) [34,35]. It was found that selectively exciting the first CD band (365 nm or 436 nm) or the second CD band (313 nm) of polymer through CPL irradiation, gained an opposite supramolecular chirality. Specifically, CPL at 365 and 436 nm resulted similar bisignate CD signal of resultant azo-polymer, but CPL at 313 nm afforded definitively opposite CD signal of azo-polymer (Fig. 5D). This difference originates from the polymeric aggregates (~200 nm), in which the stacks of trans-azobenzene have restricted conformational freedom. As a result, the chiroptical activity, restoration, inversion, and switching processes can be controlled by simply choosing two CPL beams with appropriate irradiation wavelength, time, and handedness.
Kulkarni and co-workers reported a photo-switchable azobenzene polymer (20, Fig. 5E) that exhibit novel properties at mesoscopic scale [36]. The helicity of cholesteric azobenzene can be reversed via CPL irradiation. For example, irradiating (P)-helical cholesteric supramolecule with l-CPL gave the (M)-helical one, and r-CPL could further make the (M)-helicity to the (P)-helicity. This reversible inversion could be achieved by alternatingly r/l-CPL over multiple cycles (Fig. 5F). Moreover, the supramolecular helicity interconversion involved a consecutive change in the twist angle between neighboring layers of polymer chains.
As mentioned above, azo-polymers formed chiral helix through asymmetric non-covalent bond stacking of side chains under CPL irradiation. Similarly, some polyaromatic compounds can also form chiral supramolecular by asymmetric non-covalent bond stacking. Although polyfluorene derivative was absent of chiral center, Nakano and co-workers achieved axial chirality around the single bonds connecting fluorene monomeric units via a CPL-induced self-assembly process (Fig. 6A) [37,38]. Racemization would be occurred when the chiral polymer solid 21 was dissolved in THF or heated at 160 ℃. With the increase of CPL irradiation time, the CD intensity of the polymer first increased and then decreased. Besides, the corresponding UV absorption decreased and blue-shifted continuously (Fig. 6B, pictures a and b). The absorptive dissymmetry factor (gabs) gradually increased and leveled off after 60 min, reaching a plateau after 90 min (0.0013 in Fig. 6B, picture c) [37]. The maximum gabs (up to 0.012 [38]) observed for the polyfluorene was comparable to that of small molecular biphenyl derivatives (in the order of 10−3–10−4 for 100% ee), suggesting that CPL-induced helices for polyfluorene was almost one-handedness due to a chiral amplification effect.
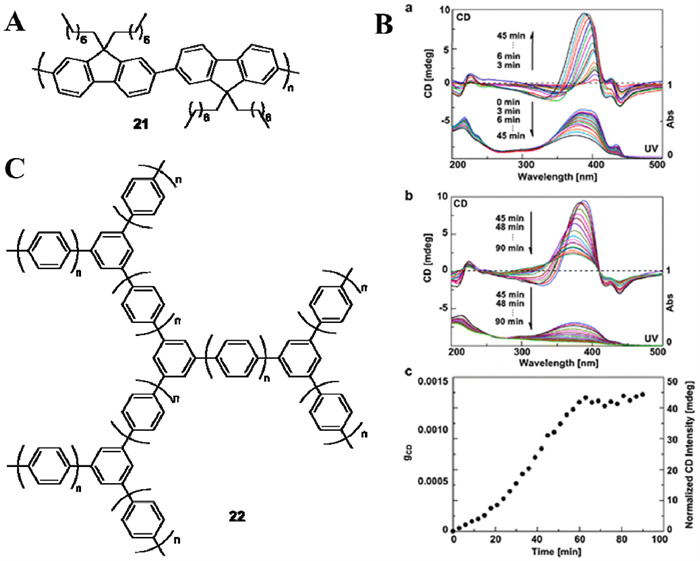
Unlike fluorene, whose steric hindered disubstituted benzylic carbon provides opportunities to obstruct the free rotation of single bonds, the biphenyl has a twisted aromatic-aromatic single bond, which can be transformed to a coplanar conformation in excited states. Nakano employed CPL to excite COFs composed of benzene-benzene junctions (Fig. 6C) [39]. One of the enantioisomeric twist is preferentially excited by CPL and converted to a racemic ground state. Correspondingly, the un-excited twists accumulate, resulting in chiral compounds.
Chirality expressed at the supramolecular level can be referred to supramolecular chirality, which can be constructed by the nonsymmetric arrangement of building blocks through weak noncovalent interactions, such as hydrogen bonding, π-π stacking, electrostatic interaction and host-guest interaction [40]. Interesting, these building blocks are not normally chiral components; supramolecular chirality can be achieved by achiral molecules or a combination of chiral and achiral molecules. Therefore, achiral components assembly with helical sense has attracted increasing attention. More interesting, the CPL-induced asymmetric assembly can easily switch the helicity of the superstructure through tuning the handedness of CPL.
Triarylamines are C3 symmetric molecules, and the three aromatic rings exhibit a twisted conformation due to steric hindrance, similar to the blades of a propeller. Although this twisted conformation could be interconverted by rapid rotation of the single bond in solution, it would be suppressed in the solid state. Kim et al. provided CPL-induced symmetry breaking to form the triaryl ammonium radicals, which adopt a more planar configuration to facilitate stacking, and stabilized by electric interaction of the radical cation-anion pairs. During the self-assembly process, the chiral photons within CPL could be transmitted into the stacking, and the supramolecular chirality was reversible by switching the handedness of CPL. The triaryl amine (23) finally stacked into helices aided by the nonlinear amplification of chiral information (Fig. 7A) [41]. The chirality amplification originates from triphenylamine moieties with the same helical arrangement (i.e., the same enantiomer) and further stabilized by the enthalpy gain. The generated supramolecular chirality was reversible (Fig. 7B) and would disappear under exposure to unpolarized light. Moreover, the authors utilized photopolymerization of diacetylene moieties bearing on the triphenylamines of the self-assembly to lock the chirality.
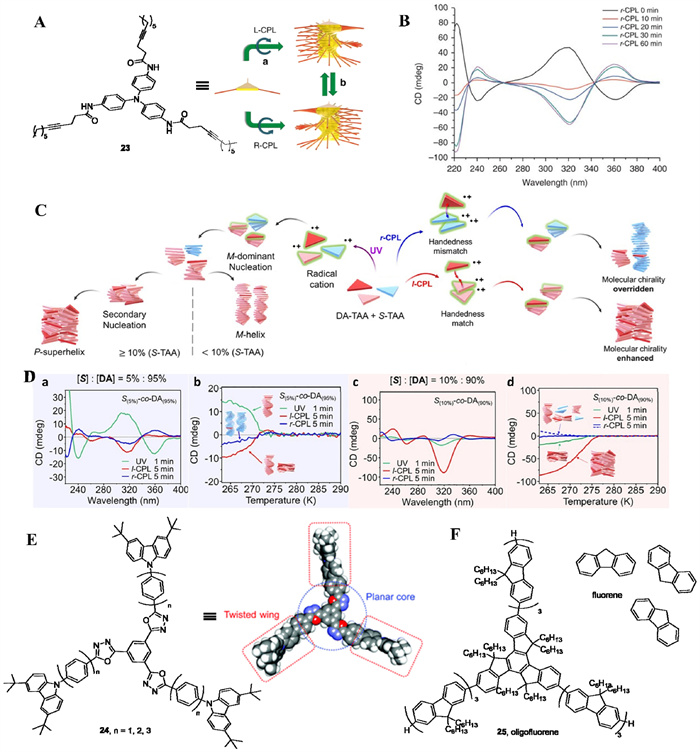
Kang et al. controlled the self-assembly of triarylamine followed by "sergeants and soldiers" process (Fig. 7C) [42]. The achiral triarylamine (DA-TAA) can be enantioselective polymerized to form left-handed (M-type) and right-handed (P-type) helices via l- and r-CPL irradiation, respectively. The CD intensity at 317 nm increased rapidly with CPL irradiation. It leveled after 5 min irradiation, but decreased beyond 20 min. Interestingly, when DA-TAA was copolymerized with S-TAA (< 10 mol% loading) as a molecular sergeant under UV irradiation, M-helix was mainly produced, and P-superhelix populated above, that is formed by opposite coiling of the M-helix and can be detected by atomic force microscopy. This demonstrated that the chirality of copolymerization was controlled by S-TAA under UV irradiation. In contrast to UV irradiation, both l- and r-CPL irradiation produced opposite CD sign in the presence of 5 mol% of S-TAA (Fig. 7D, a). Based on the spectral features of CD spectrum, P-helix and P-superhelix was identified to generate by the r- and l-CPL, respectively. The formed P-helix of DA-TAA irradiated with CPL indicated that CPL overrode the chiral information from S-TAA. Nevertheless, increasing the loading of S-TAA (10 mol%) led more M-helix populate to P-superhelix under UV irradiation, and overrode the chiral information from r-CPL, which gave a similar CD sign with lower intensity. As expected, chirality-matched l-CPL boosted the CD response (Fig. 7D, c).
Nakano group reported a CPL-induced enantioselective self-assembly of C3 symmetric 1, 3, 5-trioxadiazolylbenzene, bearing with N-phenylcarbazole (24). In contrast to triarylamine, which generated axial chirality from the core structure, trioxadizole benzenes 24 generated an axial chirality from the N-phenylcarbazole moiety on the periphery (Fig. 7E) [43]. Nevertheless, CPL-induced asymmetric self-assembly of C3 symmetric oligofluorene (25) was failed due to the difficulty in close packing of chains [44]. Nakano group selected fluorene (or phenanthrene) as an aid molecule to promote the ordered arrangement (Fig. 7F). By adjusting the ratio of fluorene and oligofluorene ([fluorene]/[oligofluorene]) to 3.2, CPL induced the best performance in asymmetric self-assembly. The crystal surface of fluorene may play a scaffold role in the chirality induction of oligofluorene. The CD intensity increased with irradiation time and reached a maximum gabs of 0.00033 after 210 min. Moreover, this CPL-induced chirality is reversible where l-CPL irradiation for 60 min gave opposite CD signals (380 nm), but changed to negative after an extended r-CPL irradiation for 60 min.
CPL-induced restructuring of inorganic nanostructures with optical activity can provide a new, powerful, versatile tool for nanoscience [45]. Chiral photons-driven synthesis of chiral plasmonic nanostructures has been more challenging due to their short lifetime of the plasmonic states [46]. Therefore, construction of chiral nanostructures using CPL as the sole chiral source is quite rarely prepared due to it is extremely difficult to convert the circular polarization into geometrically twisted nanostructures [47].
In 2014, Yeom et al. firstly demonstrated that twisted CdTe nanoribbons could be formed with an ee exceeding 30% by tuning the chirality of nanoparticles participating in the self-assembly process upon irradiation of racemic nanoparticles with CPL (Figs. 8A and B) [48]. It should be noted that, this ee value of nanostructures were not obtained by chiral HPLC technique, the author calculated this result after analyzing 100 SEM images of ~1000 nanoribbons. This study offered a new strategy for asymmetric photoresolution of racemic substrates to synthesis chiral nanostructures using electromagnetic field as the primary chiral bias. Compared with photo-resolution of racemic material, it is more challenging and meaningful to asymmetric photosynthesis of chiral nanostructures from a prochiral or achiral staring material [49]. In 2019, Kim et al. reported that CPL illumination of Au salt solution transferred the chirality of photons to the nanoparticle assemblies (Figs. 8C–F) [50]. Despite the CD signals of products were extremely small (< 2 mdeg), they exhibited opposite chiroptical activities upon switching the handedness of CPL. The author considered that transient interparticle forces dependent on the polarization of CPL generated the chiral bias in the intermediate dynamic assemblies, which was subsequently locked in irregular shapes of the nanoscale cores. In addition, recently Xu et al. synthesized Au nanoparticles (AuNPs) with controllable nanometre-scale chirality and gabs up to 0.4 using 594 nm CPL irradiation assisted with cysteine–phenylalanine dipeptide [51]. Although the handedness of the nanoparticles was determined by their surface dipeptide ligands, the maximum curvature of the blade was determined by CPL (Fig. 8G).
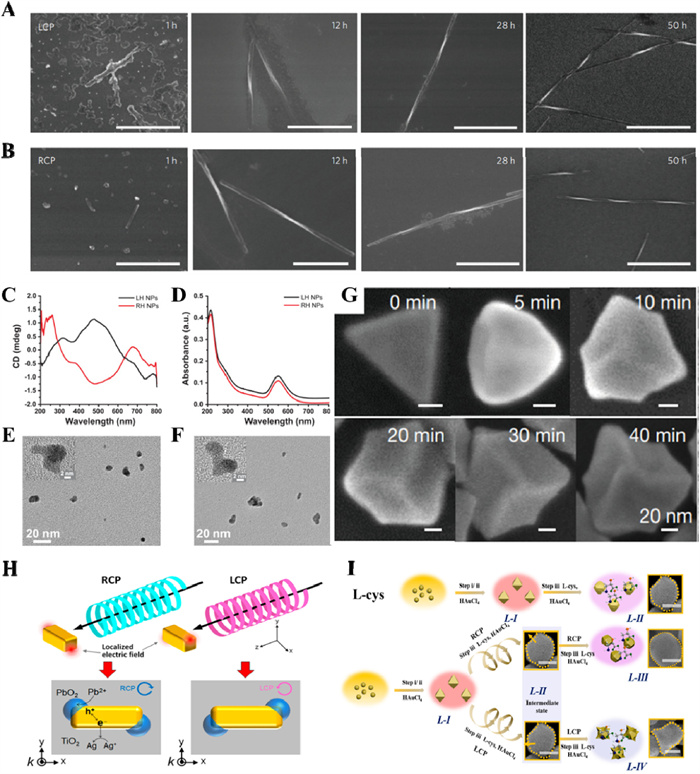
Unlike organic molecules, plasmonic nanostructures take many unique advantages, such as plasmon-induced charge separation (PICS) [52]. PICS occurs at a nanoparticle in contact with a semiconductor, allowing photoinduced site-selective reactions beyond the diffraction limit [53]. In 2018, Saito and Tatsuma exploited the twisted electric field distributions around achiral Au nanocuboids under CPL illustration, and achieved bottom-up fabrication of chiral plasmonic nanostructures by means of site-selective deposition of PbO2 assisted with PICS [54]. The chiral nanostructures eventually achieved a higher ee (43%) and ellipticity (20 mdeg) (Fig. 8H). More recently, Wang et al. using such site-selective chirality regulation on the morphology of AuNPs with assistance of cysteine under CPL irradiation (Fig. 8I) [55]. The author discovered that l-CPL enhanced the site-selective AuNPs growth in the convex region, whereas r-CPL forces chiral nanoparticles to grow on the concave position of the cysteine-assisted AuNPs.
There are two factors make chiral transfer from photon-to-matter a promising research direction for nanoscience. Firstly, the plasmonic states of nanostructures are highly polarizable, therefore, are strongly affected by CPL. Secondly, CPL could potentially affect both the growth and assembly process of plasmonic nanoparticles. Both spin and site-selective features involved in hot-electron process may lead to asymmetric particle growth of nanostructures. Therefore, CPL-induced absolute asymmetric synthesis provides a new, attractive strategy for the construction of anisotropic nanomaterials.
The absolute asymmetric synthesis based on CPL displays several advantages including the purity of products without extra chiral dopants or catalysts, and the facile adjustment of CPL parameters such as intensity, wavelength, polarization and interference. Chirality transfer from photons to matter has both fundamental and practical significance, as a process that could be involved in emergence of biomolecular homochirality, employed as a preparative method for chiral organics, and lead to new photonic devices. The enantiomeric excess of organic molecules, helical polymers, supramolecular assemblies, noble metal nanostructures, after CPL irradiation was observed. However, light-matter interaction in chirality is very faint because the arrangement of molecular bonds giving rise to chiral features, is over a smaller distance than the helical pitch of the CPL [47]. For example, a sugar molecule could exhibit strong optical rotation, but at the expense of extremely high molar concentrations. Likewise, the selective discrimination between enantiomers turns out to be a great challenge in enantioselective photochemistry. As a result, enantiomer molecules show extremely small dipole moments, only weakly coupling to an external chiral field, leading extremely small ee (< 2%) and g value (< 10−3) for products. Thus, there is an urgent need for strategy to enhance the light-matter interplays and increase the ee of photoreaction product. Listed below are three strategies that may hold great potential for enhancing the enantioselective of CPL-driven absolute asymmetric synthesis.
In naturally occurring chiral materials, such as amino acids, the scale of small organic molecules with respect to the wavelength of incident CPL is extremely small, producing faint CD response due to the CPL experience an imperceptible perturbation of the excitation rate. It may be reasonable to achieve large optical dissymmetry by squeezing the CPL period, reorienting the field lines on molecular distances. The chiral field compression gives rise to far-field superchiral light (SCL), inducing boosted chiro-optical coupling with chiral molecules, which was put forward by Tang and Cohen around 2010 (Fig. 9A) [56].

As we all know, In CPL, the field vectors rotate at a constant rate along the propagation direction, undergoing a complete revolution once per wavelength. However, in SCL, generated by two counter-propagating lasers of CPL with same frequency, opposite handedness and slightly different intensity, the field vectors rotate through nearly 180° in a distance much shorter than half the free-space wavelength, inducing boosted chiroptical coupling with small molecules [57]. To quantify this enhancement, a time-even, parity-odd and scalar quantity, called local optical chirality (C) was introduced by Lipkin, and defined, for a monochromatic electromagnetic wave, as
|
|
where ε0 and μ0 are the permittivity and permeability of free space, respectively. E and B denote the complex electric and magnetic fields, respectively. Two counter-propagating CPL beams with same frequency, different handedness and slightly different electric field amplitude E1 > E2 interfere, giving rise to a standing wave with the absorptive dissymmetry factor of SCL (gSCL) becomes:
|
|
where gCPL is the absorptive dissymmetry factor under conventional CPL, c is the speed of light, Ue is the local electric energy density and ω is the angular frequency, respectively [58].
Next, we just need to focus on C and Ue in the numerator and denominator of cC/2Ueω. Near the nodes of SCL field, C = ωε0(E12 - E22/c, independent of position, whereas U achieve its minimum value at the nodes: Ue = ωε0(E12 - E22)/c. As E2 approaches E1, Ue and C approach zero together near the nodes. But Ue approaches zero much faster than C, so the ratio C/Ue becomes extremely large, resulting gSCL/gCPL > 1 (Fig. 9A) [59]. Therefore, it is anticipated that the enhanced optical dissymmetry in SCL may be utilized to promote enantioselectivity of asymmetric synthesis. We must note that superchiral nodes with enhanced optical dissymmetry were restricted in very narrow region, limiting further application.
In 2018, He et al. firstly demonstrated experimentally that enantioselective photopolymerization of achiral diacetylene monomers within Langmuir–Blodgett films could be triggered and highly enhanced by SCL to generate optically active polydiacetylene (PDA) (Fig. 9B) [60]. A maximum about 6-times enhancement in gabs values of PDA over conventional CPL technology could be achieved. This dissymmetry enhancement in asymmetric photopolymerization was not only rely on the greater chiral bias induced by SCL, but also with the help of chiral transfer and amplification during the polymerization process. Thus, the enhanced optical dissymmetry in superchiral nodes could be transferred to closely and orderly packed monomers within Langmuir–Blodgett films, resulting in enhanced dissymmetry for macroscopic films.
Based on above discussion, it is difficult to achieve absolute asymmetric synthesis with high enantioselectivity in large scale only based on superchiral far-field because the superchiral nodes are restricted to narrow region. Thus, we need another tool to transfer the enhanced optical dissymmetry, such as chiral transfer, long-rang ordered liquid crystals as templates or plasmonic nanostructures [61-63].
Unlike superchiral far-field, another common way to generate superchiral fields lies in chiral plasmonic nanostructures to locally concentrate and tailor the incident light, enhancing the superchiral near-field [64]. Plasmonic nanoparticles always show highly efficient photo-matter interaction because of the large dipole moments of the plasmonic resonance, which would strengthen the interaction with an external light exceeds the one for molecules by magnitude orders. Such resonance modes include long-range radiating mechanism, i.e., antenna effect, and near-field electromagnetic dipole coupling. Unfortunately, the polarization of incident light is almost lost in the light-matter interaction, then the near-field polarization is determined by the excited plasmonic modes [47]. Therefore, we need to fabricate plasmonic nanostructures with chiral features, which helps to generate superchiral near-field.
Typically, there are mainly two superchiral near-fields associated with plasmonic nanostructures: one of them is chiral nanostructure with plasmonic CD at visible and near-infrared regions. As shown in Figs. 10A and B, helical nanostructures were predicted to experience incident polarization dependent local-handedness strong superchiral near-fields, as well as chiral eigenmodes generation due to the geometrical chirality in intertwined multi helical architectures, inducing enhanced optical dissymmetry [65]. Meanwhile, even achiral structures with twisted-stacking structure can also lead to near-fields with high optical chirality, which could induce absolute asymmetric synthesis [66]. For example, Wei et al. reported successful chirality-selective manipulation of dimerization products from prochiral monomers under unpolarized 365 nm UV irradiation. The chirality of products was controlled by the handedness of helical metal nanostructures, and attributed the result to a chiro-plasmonic effect (Fig. 10C) [67].
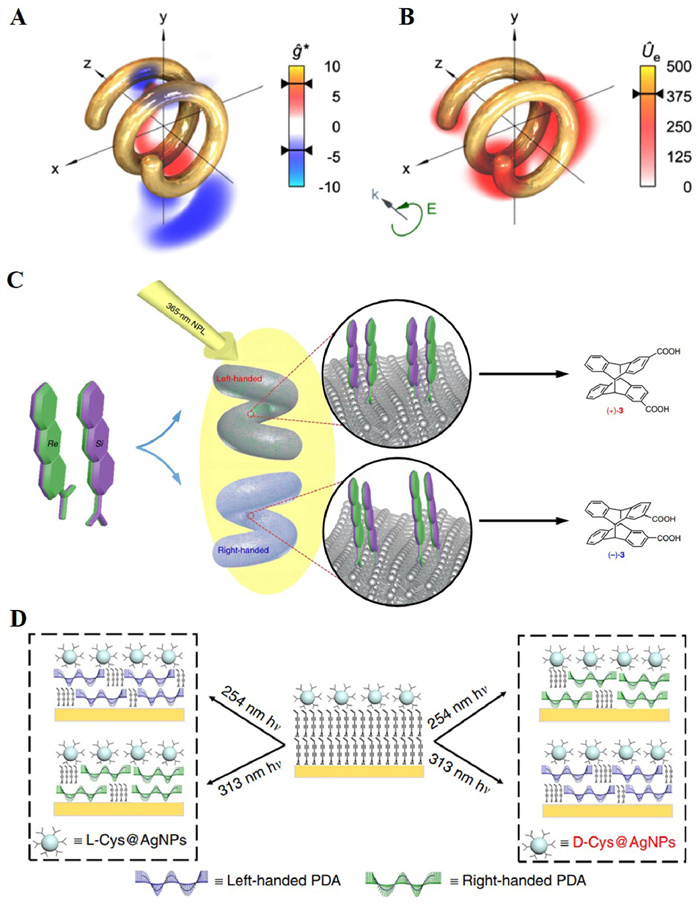
Besides the plasmonic absorption, noble metal nanoparticles also have strong intrinsic absorption due to photoexcited interband transitions in the UV range (for example 220–320 nm for Ag) [68]. This new CD resonance is further amplified through strong Coulomb coupling with the interband transition of nanoparticles and enhanced by the local electric field [69]. He et al. reported firstly that superchiral near-field generated by l/d-cysteine-modified Ag nanoparticles (L/D-Cys@AgNPs) could be utilized to induce absolute asymmetric photopolymerization of achiral monomers with high enantioselectivity and reactivity under unpolarized UV irradiation [70]. Interestingly, this superchiral near-field match the interband transition of Ag nanoparticles rather their chiroplasmonic field, showing opposite chirality when switching the excitation wavelength (Fig. 10D). Therefore, by simply varying the irradiation wavelength, one can manipulate the direction of the chiral symmetry breaking in asymmetric photochemical reactions.
Base on above discussion, both enantioselectivity and reactivity of asymmetric synthesis would be amplified assisted with chiral plasmonic nanostructures, which benefits from the resonance between the incident light and the electromagnetic field near chiral plasmonic nanostructures. Meanwhile, chiral plasmonic nanomaterials are versatile and could be further extended to other material combinations for various chiral light-matter interactions.
A typical feature in CPL-triggered absolute asymmetric synthesis is a difference coefficient in the absorption of r-CPL and l-CPL by enantiomers; r-CPL and l-CPL should preferentially interact with one enantiomer of a racemate. However, this difference in absorption coefficient is extremely small (small dipole moments of molecule couldnot strongly couple to a circularly polarized field), resulting in very low enantiomeric excess of product. This is verified by conventional CPL-triggered asymmetric thiol-ene photochemical reaction (Fig. 11A) [25]. In this reaction, the racemic thiol can be selectively excited by CPL to generate chiral thiol radical via photoresolution, which was captured by styrene to obtain a benzyl radical and subsequent produce chiral thioether via hydrogen atom abstraction. However, the carbon-centered radical seizes a hydrogen atom from the racemic thiol and regenerates racemic thiyl radical, resulting in a low enantioselectivity (2.2% ee at 50% conversion). After analyzing the mechanism, Zou et al. believe that the low enantioselectivity is an undesirable side-effect in its unique chemical kinetics. It would make a lot of sense to enhance the enantioselectivity just by regulating the chemical kinetics. Based on the understanding, a new strategy that introduction of an achiral and "green" additional, hydrogen atom donor (HAD), into the reaction should quench the carbon-centered radical directly (step 4) and suppress the chain transfer step (step 3), breaking the conventional cycle of thiol-ene reaction (Fig. 11B) [26]. Moreover, the HAD could also donate a hydrogen atom to quench the racemic thiyl radical (step 5) and the regenerated thiol molecule could be retriggered by CPL with chiral preference. As a result, the ee of the final product increased both in absolute asymmetric small molecule reactions and photopolymerization reactions, almost doubling the ee in case of no HAD. Unlike enhance both reactivity and enantioselectivity through chiral plasmonic nanostructures, the HAD would suppress the reactivity and enhance enantioselectivity, which is a unique feature for regulating asymmetric chemical kinetics.

In this review, we retrospect the historical research of CPL-induced absolute asymmetric synthesis of chiral organic molecules, helical polymers, supramolecular assemblies, noble metal nanostructures. However, the enantioselectivity is extremely low, which should be ascribed to the arrangement of molecular bonds is over a smaller distance than the helical pitch of the CPL, leading weak chiral photon-matter interaction. Therefore, conventional CPL irradiation should combine with other new emerged chiral technique, such as superchiral field, chiral plasma, as well as the regulation of chemical kinetics to promote the enantioselectivity in asymmetric synthesis. We hope in the near future, the enantioselectivity of CPL-driven absolute asymmetric synthesis could be improved and more chiral synthesis techniques would be developed.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge the support of Academic promotion program of Shandong First Medical University (No. 2019LJ003).
A. Lazcano, S.L. Miller, Cell 85 (1996) 793–798. doi: 10.1016/S0092-8674(00)81263-5
J.R. Cronin, S. Pizzarello, Science 275 (1997) 951–955. doi: 10.1126/science.275.5302.951
G. Balavoine, A. Moradpour, H.B. Kagan, J. Am. Chem. Soc. 96 (1974) 5152–5158. doi: 10.1021/ja00823a023
N.P.M. Huck, W.F. Jager, B. de Lange, B.L. Feringa, Science 273 (1996) 1686–1688. doi: 10.1126/science.273.5282.1686
C. Wang, T. Jiang, X. Ma, Chin. Chem. Lett. 31 (2020) 2921–2924. doi: 10.1016/j.cclet.2020.03.021
L. Liu, J. Chen, T. Yu, et al., Chin. Chem. Lett.34 (2023) 107649. doi: 10.1016/j.cclet.2022.06.072
C. Tu, W. Wu, W. Liang, et al., Angew. Chem. Int. Ed. 61 (2022) e202203541. doi: 10.1002/anie.202203541
H. Rau, Chem. Rev. 83 (1983) 535–547. doi: 10.1021/cr00057a003
Y. Inoue, Chem. Rev. 92 (1992) 741–770. doi: 10.1021/cr00013a001
S. Allenmark, J. Gawronski, Chirality 20 (2008) 606–608. doi: 10.1002/chir.20524
A. Moradpour, J.F. Nicoud, G. Balavoine, H. Kagan, G. Tsoucaris, J. Am. Chem. Soc. 93 (1971) 2353–2354. doi: 10.1021/ja00738a061
W.J. Bernstein, M. Calvin, O. Buchardt, J. Am. Chem. Soc. 94 (1972) 494–498. doi: 10.1021/ja00757a029
H. Kagan, A. Moradpour, J.F. Nicoud, et al., Tetrahedron Lett. 27 (1971) 2479–2482.
R.D. Richardson, M.G.J. Baud, C.E. Weston, et al., Chem. Sci. 6 (2015) 3853–3862. doi: 10.1039/C4SC03897E
Y. Zhang, G.B. Schuster, J. Org. Chem. 60 (1995) 7192–7197. doi: 10.1021/jo00127a026
I. Sato, R. Sugie, Y. Matsueda, Y. Furumura, K. Soai, Angew. Chem. Int. Ed. 43 (2004) 4490–4492. doi: 10.1002/anie.200454162
T. Kawasaki, M. Sato, S. Ishiguro, et al., J. Am. Chem. Soc. 127 (2005) 3274–3275. doi: 10.1021/ja0422108
K. Soai, T. Kawasaki, A. Matsumoto, Acc. Chem. Res. 47 (2014) 3643–3654. doi: 10.1021/ar5003208
K.P. Bryliakov, ACS Catal. 9 (2019) 5418–5438. doi: 10.1021/acscatal.9b00697
Y. Geiger, T. Achard, A. Maisse-François, S. Bellemin-Laponnaz, Nat. Catal. 3 (2020) 422–426. doi: 10.1038/s41929-020-0441-1
H.M. Dhammika Bandara, S.C. Burdette, Chem. Soc. Rev. 41 (2012) 1809–1825. doi: 10.1039/C1CS15179G
P.K. Hashim, N. Tamaoki, ChemPhotoChem 3 (2019) 347–355. doi: 10.1002/cptc.201900068
M. Mathews, N. Tamaoki, J. Am. Chem. Soc. 130 (2008) 11409–11416. doi: 10.1021/ja802472t
P.K. Hashim, R. Thomas, N. Tamaoki, Chem. Eur. J. 17 (2011) 7304–7312. doi: 10.1002/chem.201003526
G. Yang, Y.Y. Xu, Z.D. Zhang, et al., Chem. Commun. 53 (2017) 1735–1738. doi: 10.1039/C6CC09256J
C.L. He, Z. Feng, Y. Li, et al., Polym. Chem. 12 (2021) 2433–2438. doi: 10.1039/d1py00082a
M.M. Green, B.A. Garetz, B. Munoz, et al., J. Am. Chem. Soc. 117 (1995) 4181–4182. doi: 10.1021/ja00119a039
J. Li, G.B. Schuster, K.S. Cheon, M.M. Green, J.V. Selinger, J. Am. Chem. Soc. 122 (2000) 2603–2612. doi: 10.1021/ja993290w
L. Nikolova, T. Todorov, M. Ivanov, et al., Opt. Mater. 8 (1997) 255–258. doi: 10.1016/S0925-3467(97)00046-3
L. Nikolova, L. Nedelchev, T. Todorov, et al., Appl. Phys. Lett. 77 (2000) 657–659. doi: 10.1063/1.127076
S.W. Choi, S. Kawauchi, N.Y. Ha, H. Takezoe, Phys. Chem. Chem. Phys. 9 (2007) 3671–3682. doi: 10.1039/b702835k
R.M. Tejedor, L. Oriol, J.L. Serrano, T. Sierra, J. Mater. Chem. 18 (2008) 2899–2908. doi: 10.1039/b803561j
J. Royes, J. Rebolé, L. Custardoy, et al., J. Polym. Sci. Part A Polym. Chem. 50 (2012) 1579–1590. doi: 10.1002/pola.25929
M. Fujiki, Y. Donguri, Y. Zhao, et al., Polym. Chem. 6 (2015) 1627–1638. doi: 10.1039/C4PY01337A
L. Wang, L. Yin, W. Zhang, X. Zhu, M. Fujiki, J. Am. Chem. Soc. 139 (2017) 13218–13226. doi: 10.1021/jacs.7b07626
C. Kulkarni, R.H.N. Curvers, G. Vantomme, et al., Adv. Mater. 33 (2021) 2005720. doi: 10.1002/adma.202005720
Y. Wang, T. Sakamoto, T. Nakano, Chem. Commun. 48 (2012) 1871–1873. doi: 10.1039/c2cc17027b
Y. Wang, T. Harada, L.Q. Phuong, Y. Kanemitsu, T. Nakano, Macromolecules 51 (2018) 6865–6877. doi: 10.1021/acs.macromol.8b01453
Y. Wang, K. Yazawa, Q. Wang, et al., Chem. Commun. 57 (2021) 7681–7684. doi: 10.1039/d1cc02671b
M. Liu, L. Zhang, T. Wang, Chem. Rev. 115 (2015) 7304–7397. doi: 10.1021/cr500671p
J. Kim, J. Lee, W.Y. Kim, et al., Nat. Commun. 6 (2015) 6959. doi: 10.1038/ncomms7959
J.S. Kang, S. Kang, J.M. Suh, et al., J. Am. Chem. Soc. 144 (2022) 2657–2666. doi: 10.1021/jacs.1c11306
Z. Zhang, T. Harada, A. Pietropaolo, et al., Chem. Commun. 57 (2021) 1794–1797. doi: 10.1039/d0cc07898k
Y. Wang, A.L. Kanibolotsky, P.J. Skabara, T. Nakano, Chem. Commun. 52 (2016) 1919–1922. doi: 10.1039/C5CC08464D
W. Ma, L. Xu, A.F. de Moura, et al., Chem. Rev. 117 (2017) 8041–8093. doi: 10.1021/acs.chemrev.6b00755
J. Liu, L. Yang, P. Qin, et al., Adv. Mater. 33 (2021) 2005506. doi: 10.1002/adma.202005506
A. Passaseo, M. Esposito, M. Cuscunà, V. Tasco, Adv. Opt. Mater. 5 (2017) 1601079. doi: 10.1002/adom.201601079
J. Yeom, B. Yeom, H. Chan, et al., Nat. Mater. 14 (2015) 66–72. doi: 10.1038/nmat4125
H. Behar-Levy, O. Neumann, R. Naaman, D. Avnir, Adv. Mater. 19 (2007) 1207–1211. doi: 10.1002/adma.200601702
J.Y. Kim, J. Yeom, G. Zhao, et al., J. Am. Chem. Soc. 141 (2019) 11739–11744. doi: 10.1021/jacs.9b00700
L. Xu, X. Wang, W. Wang, et al., Nature 601 (2022) 366–373. doi: 10.1038/s41586-021-04243-2
K. Wu, J. Chen, J.R. MeBride, T. Lian, Science 349 (2015) 632–635. doi: 10.1126/science.aac5443
H. Tang, C.J. Chen, Z. Huang, et al., J. Chem. Phys. 152 (2020) 220901. doi: 10.1063/5.0005334
K. Saito, T. Tatsuma, Nano Lett. 18 (2018) 3209–3212. doi: 10.1021/acs.nanolett.8b00929
H. Wang, Y. Liu, J. Yu, et al., ACS Appl. Mater. Interfaces 14 (2022) 3559–3567. doi: 10.1021/acsami.1c22191
Y. Tang, A.E. Cohen, Phys. Rev. Lett. 104 (2010) 163901. doi: 10.1103/PhysRevLett.104.163901
Y. Tang, A.E. Cohen, Science 332 (2011) 333–336. doi: 10.1126/science.1202817
N. Yang, Y. Tang, A.E. Cohen, Nano Today 4 (2009) 269–279. doi: 10.1016/j.nantod.2009.05.001
N. Yang, A.E. Cohen, J. Phys. Chem. B 115 (2001) 5304–5311.
C. He, G. Yang, Y. Kuai, et al., Nat. Commun. 9 (2018) 5117. doi: 10.1038/s41467-018-07533-y
K.S. Burnham, G.B. Schuster, J. Am. Chem. Soc. 121 (1999) 10245–10246. doi: 10.1021/ja9923033
J. Yan, F. Ota, B.A. San Jose, K. Akagi, Adv. Funct. Mater. 27 (2017) 1604529. doi: 10.1002/adfm.201604529
M. Zhang, Y. Wang, Y. Zhou, et al., Nanoscale 14 (2022) 592–601. doi: 10.1039/d1nr06036h
N.A. Abdulrahman, Z. Fan, T. Tonooka, et al., Nano Lett. 12 (2012) 977–983. doi: 10.1021/nl204055r
M. Schäferling, D. Dregely, M. Hentschel, H. Giessen, Phys. Rev. X 2 (2012) 031010. doi: 10.1103/PhysRevX.2.031010
Y. Chen, W. Du, Q. Zhang, et al., Nat. Rev. Phys. 4 (2022) 113–124.
Wei X., Liu J., Xia G.J., et al. Nat. Chem. 12 (2020) 551–559. doi: 10.1038/s41557-020-0453-0
Z.Y. Bao, W. Zhang, Y.L. Zhang, et al., Angew. Chem. Int. Ed. 56 (2017) 1283–1288. doi: 10.1002/anie.201607563
K. Kolwas, A. Derkachova, Nanomaterials 10 (2020) 1411. doi: 10.3390/nano10071411
C. He, Z. Feng, S. Shan, et al., Nat. Commun. 11 (2020) 1188. doi: 10.1038/s41467-020-15082-6

Figure 1 CPL-triggered enantioselective synthesis, including chiral organic molecules and inorganic nanostructures.

Figure 2 Three modes of CPL-induced asymmetrically photochemical reactions: (A) photochemical asymmetric degradation, (B) photochemical deracemization; (C) photochemical asymmetric fixation. The uppercased S and P represent the starting materials and products, the subscript S and R represent the absolute configuration of enantiomers, and SS* and SR* represent the excited state of enantiomers of substrates.

Figure 4 CPL-induced stereoselective self-assembly of styryl-bicylics polymer. Copied with permission [28]. Copyright 2000, American Chemical Society.

Figure 5 CPL-induced asymmetric self-assembly of azo-polymer. (A) Molecular structures. (B) UV-vis and CD spectra of MFP, DAP and MAP. Copied with permission [33]. Copyright 2012, John Wiley and Sons. (C) The wavelength- controlled stereoselective self-assembly and (D) corresponding CD spectra of polymer. (E) The cholesteric azobenzene polymer and (F) reversibly conversional supramolecular helicity. (C, D) Copied with permission [35]. Copyright 2017, American Chemical Society.

Figure 6 CPL-induced asymmetric self-assembly of aromatic polymer. (A) Structure of polyfluorene. (B) CD and UV spectra of aromatic polymer measured every 3 min during l-CPL irradiation, and gabs vs. irradiation time plot. Copied with permission [37]. Copyright 2012, Royal Society of Chemistry. (C) Structure of biphenyl COFs. Copyright 2012, Royal Society of Chemistry.

Figure 7 Self-assembly of (A) triaryl amine and (B) its reversible supramolecular chirality; (C) "sergeants and soldiers" processed self-assembly of triarylamine, and (D) the CD spectral characteristic with different loadings of "sergeant". (C, D) Copied and Reproduced with permission [42]. Copyright 2022, American Chemical Society. Structures of (E) triaryl benzene and (F) oligo fluorene. Copied with permission [43]. Copyright 2021, Royal Society of Chemistry.

Figure 8 SEM images of CdTe ribbons assembled with (A) l-CPL and (B) r-CPL as a function of time exposure for 1 h, 12 h, 28 h and 50 h. All scale bars are 1 µm. (A, B) Copied with permission [48]. Copyright 2014, Springer Nature. (C) CD and (D) UV-vis spectra of dispersions formed under l-CPL and r-CPL, respectively. High resolution TEM images of NPs obtained with (E) l-CPL and (F) r-CPL irradiation, respectively. (C-F) Copied with permission [50]. Copyright 2019, American Chemical Society. (G) SEM images of L-dipeptide modified nanoparticles after 0, 5, 10, 20, 30 and 40 min of CPL illumination. Copied with permission [51]. Copyright 2020, Springer Nature. (H) Schematic illustrations of chiral nanostructure fabrication by PICS using CPL. Copied with permission [54]. Copyright 2018, American Chemical Society. (I) Synthesis schematic for L-cysteine-chiral AuNPs (L-Ⅱ) and site-selective chiral AuNPs (L-Ⅲ was irradiated by r-CPL). Copied with permission [55]. Copyright 2022, American Chemical Society. Scale bar is 50 nm.

Figure 9 (A) Enhanced chiral asymmetry in SCL. The ratio C/Ue, which determines the enantioselectivity, becomes large near a node in the superchiral far-field. Copied with permission [56]. Copyright 2010, American Physical Society. (B) Experimental set-up for far-field SCL generated by two counter-propagating CPL waves with same frequency and opposite handedness.

Figure 10 Enhancement of (A) dissymmetry factor gabs and (B) electric energy density Ue near a plasmonic helix structure with respect to CPL. Superchiral near-fields with up to 7 times higher enantioselectivity could be obtained. (C) Metallic nanohelices mediated enantioselective photo-cyclodimerization of 2-anthracenecarboxylic acid under irradiation of 365 nm unpolarized light. Copied with permission [67]. Copyright 2020, Springer Nature. (D) Schematic illustration of left-handed PDA is obtained using 254 nm irradiation assisted with L-Cys@NPs or 313 nm irradiation assisted with D-Cys@NPs. Right-handed PDA is obtained using 254 nm irradiation assisted with D-Cys@NPs or 313 nm irradiation assisted with L-Cys@NPs.

Figure 11 (A) Conventional CPL-triggered asymmetric thiol–ene click reaction and (B) "new strategy" with extra HAD. Step 1: Photo initiation; step 2: propagation; step 3: chain transfer; step 4: generation of product; step 5: recycling of thiyl radicals. Step 6: hydrogen atom exchange.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: