Shanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, Shanghai University of Electric Power, Shanghai 200090, China
b.
CAS Key Laboratory of Energy Regulation Materials, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Received Date:
22 October 2021 Accepted Date:
21 February 2022 Revised Date:
17 February 2022 Available Online:
15 November 2022
Abstract:
A new method for the preparation of fluoroalkylthioethers including trifluoromethylthioether and difluoromethylthioether by iridium(Ⅰ)-catalyzed deoxgenation of fluoroalkylsulfoxides with dimethyl diazomalonate was developed. In the reaction system, dimethyl diazomalonate was used as reducing reagent and the corresponding fluoroalkylthioethers were produced through oxygen atom transfer from fluoroalkylsulfoxides to diazomalonate. The protocol featuring effective oxygen atom transfer, mild reaction conditions and good functional groups tolerance offers an alternative strategy for the synthesis of fluoroalkylthioethers.
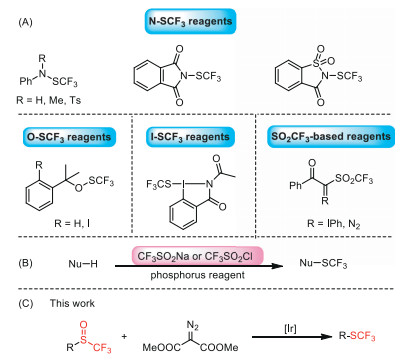
Due to its special physicochemical and biological properties, organic fluorine compounds have been widely utilized in the fields of pharmaceutical, pesticide and material science [1-10]. Among many fluoroalkyl groups, trifluoromethylthio group (CF3S-) is more prominent owing to its electron-withdrawing effect, high lipophilicity (Hansch's parameter πp = 1.44) and metabolic stability [11-14]. Over the past few decades, much effort has been made to develop efficient methods and reagents for selectively incorporating SCF3 motifs into organic molecules [15-22]. In particular, the invention of easy-to-handle electrophilic trifluoromethylthiolation reagents greatly contributed to the development of this field [23-26]. According to the skeleton, these commonly used electrophilic SCF3-transfer reagents were classified as N-SCF3 reagents [27-30], O-SCF3 reagents [31], I-SCF3 reagents [32] and SO2CF3-based reagents (Scheme 1A) [33, 34]. Although these novel reagents facilitated the introduction of trifluoromethylthiol group into organic molecules, they need to be prepared in advance with several steps.
Scheme 1
Scheme 1.
(A) Several types of electrophilic trifluoromethylthiolation reagents. (B) Trifluoromethylthiolation using CF3SO2Na or CF3SO2Cl as the SCF3 source. (C) Iridium(Ⅰ)-catalyzed deoxgenation of trifluoromethyl sulfoxides with dimethyl diazomalonate to access trifluoromethylthioether.
Very recently, commercially available CF3SO2Na or CF3SO2Cl was used as trifluoromethylthiolation reagent in the presence of phosphorus reductant for the trifluoromethylthiolation of electron-rich aromatic compounds (Scheme 1B) [35-43]. The process might involve the trifluoromethylsulfinylation of electron-rich aromatic compounds to generate trifluoromethyl sulfoxides, which were further reduced by phosphorus reagent to give the corresponding trifluoromethylthioethers [39]. However, the use of moisture or air sensitive phosphorus reductant limited their wide applications. On the other hand, trifluoromethyl sulfoxides could be easily prepared by diverse methods [44], including the direct trifluoromethylsulfinylation of aromatic compounds using triflinate salts in triflic acid [45] or CF3SO2Na/POCl3 system [42, 46], the nucleophilic trifluoromethylation of sulfinyl halides or sulfinic esters with TMSCF3 [47-49], the rearrangement of aryl triflinates in the presence of AlCl3 [50] and the oxidation of the corresponding trifluoromethylthioethers [51-55]. This led us to develop the direct reduction of trifluoromethyl sulfoxides under mild conditions to prepare trifluoromethylthioethers.
α-Diazoesters [56-58] are used as versatile reagents in organic synthesis, especially in the transition metal-catalyzed carbene transfer reactions [59, 60], such as ylide reaction [61], cyclopropanation [62, 63], insertion reaction [64, 65] and 1, 2-migration reaction [66]. Recently, oxidative dedinitrogenation of α-diazoesters has been used as a useful method to prepare α-ketoesters, where molecular oxygen [67], dimethyl sulfoxide (DMSO) solvent [68], pyridine-N-oxides [69], diphenyl sulfoxide [70] could be employed as the oxidants. Inspired by these works, we envisaged that if trifluoromethyl sulfoxides could be reduced by α-diazoesters through oxygen atom transfer to prepare trifluoromethylthioethers (Scheme 1C).
Initially, we chose phenyl trifluoromethyl sulfoxide and dimethyl diazomalonate as model substrates, with Rh2(OAc)4 (0.5 mol%) as the catalyst in dichloromethane in sealed Schlenk tube at 80 ℃ (Table 1, entry 1). Interestingly, the trifluoromethyl phenyl sulfide 2a was obtained as the major product with 54% yield, accompanied by the sulfonium ylide [71] as the minor product with 17% yield. Thus, we screened the metal catalyst to optimize this reaction. Cu(OTf)2 gave the trifluoromethyl phenyl sulfide in 60% yield and sulfonium ylide in 14% yield (Table 1, entry 2). CuI almost did not promote this reaction (Table 1, entry 3). The reaction using Cu(MeCN)4BF4 as the catalyst afforded trifluoromethyl phenyl sulfide in 57% yield, and the formation of sulfonium ylide was not observed (Table 1, entry 4). However, increasing the loading of Cu(MeCN)4BF4 failed to further improve the yield, due to the incomplete conversion of phenyl trifluoromethyl sulfoxide. To our delight, switching the catalyst to Ir(COD)2BF4, the reaction occurred to full conversion, and trifluoromethyl phenyl sulfide was obtained in 71% yield and no sulfonium ylide by product was formed (Table 1, entry 5). Further solvents screening showed the reaction in 1, 2-dichloroethane gave the 2a in the highest yield 82% (Table 1, entry 9), while the reaction in polar coordinating solvents, such as ethyl acetate (EA), Tetrahydrofuran (THF), and CH3CN, proceeded in less than 29% yield (Table 1, entries 6–8). In addition, lowing Ir(COD)2BF4 loading resulted in the decreased yield of 2a (Table 1, entries 10 and 11). Finally, no product was observed in the absence of catalyst (Table 1, entry 12).
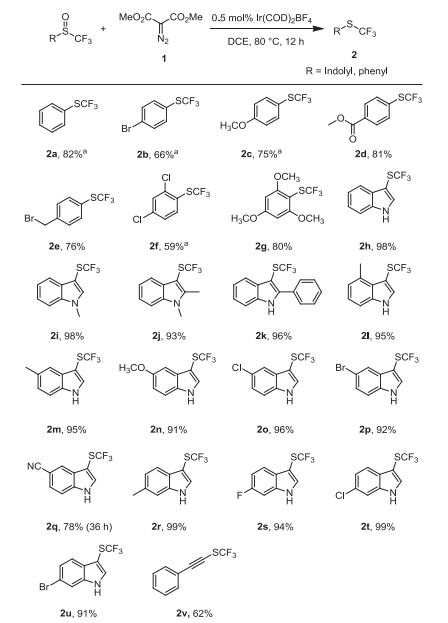
With the optimized reaction conditions in hands (Table 1, entry 9), we next investigated the substrate scope of the reaction. As summarized in Scheme 2, both aryl-trifluoromethyl and heteroaryl-trifluoromethyl sulfoxides reacted with dimethyl diazomalonate smoothly to give the corresponding trifluoromethyl sulfides in moderate to good yields. Trifluoromethyl phenyl sulfoxides with electron-donating group on the phenyl ring gave higher yield than trifluoromethyl phenyl sulfoxides with electron-withdrawing group. For example, 2, 4, 6-trimethyoxyphenyl trifluoromethyl sulfoxide gave the product 2g in 80% yield, while 2, 4-diclorophenyl trifluoromethyl sulfoxides gave the product 2f in 59% yield. The trifluoromethyl indolyl sulfoxides with substituents at 2-, 4-, 5- and 6-position reacted to give the products in good yields. Similarly, N-methylated indolyl trifluoromethyl sulfoxides could also be fully converted into 2i and 2j, with 98% and 93% yields, respectively. Moreover, sterically hindered 2-phenyl trifluoromethyl 3-indolyl sulfoxides could also convert into the desired sulfide 2k in 96% yield. Besides, phenylethynyl trifluoromethyl sulfoxide gave the product 2v in 62% yield. Furthermore, some common functional groups such as halides including fluoride (2s), chloride (2f, 2o, 2t) and bromide (2b, 2p, 2u), benzylic bromide (2e), ester (2d), cyano (2q), alkynes (2v) showed good tolerance under the conditions.
Scheme 2
Scheme 2.
Substrate scope for Ir(COD)2BF4-catalyzed deoxgenation of trifluoromethyl sulfoxides with dimethyl diazomalonate. Reaction conditions: Trifluoromethyl sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12–36 h; isolated yields. a 19F NMR Yields (due to high volatility).
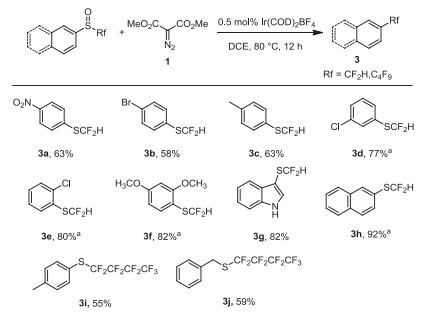
Encouraged by the success for the preparation of trifluoromethylthioethers, we next extended the approach to prepare difluoromethylthioethers and long-chain perfluoroalkylthioethers. As shown in Scheme 3, a variety of difluoromethyl phenyl sulfoxides reacted smoothly and afforded the corresponding difluoromethylthioethers in moderate to good yields (3a-3h). Notably, nitro group was not affected in this deoxgenation reaction (3a). 2-Naphthalenyl difluoromethylthioether was obtained in 92% yield (3h). To our delight, the (p-tolyl) perfluorobutylthioether 3i and benzyl perfluorobutylthioether 3j were successfully obtained with 55% and 59% yield, respectively.
Scheme 3
Scheme 3.
Substrate scope for Ir(COD)2BF4-catalyzed deoxgenation of difluoromethyl sulfoxides with dimethyl diazomalonate. Reaction conditions: Difluoromethyl sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12 h; isolated yields. a 19F NMR Yields (due to high volatility).
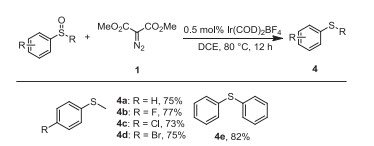
Furthermore, the protocol was also applicable for the deoxgenation of normal sulfoxides [72]. For example, reactions of substituted phenyl methyl sulfoxides or diphenyl sulfoxides, afforded the corresponding sulfides in good yields (Scheme 4, 4a-4e).
Scheme 4
Scheme 4.
Scope of deoxgenation of common aryl sulfoxides with dimethyl diazomalonate. Reaction conditions: Sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12 h; isolated yields.
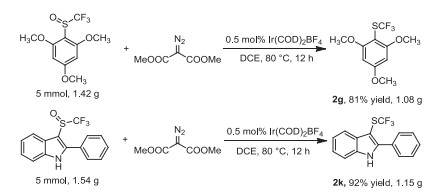
Besides, the gram scale reactions were conducted to evaluate the practical application (Scheme 5). Under the reaction conditions, the 5 mmol of 2, 4, 6-trimethyoxyphenyl trifluoromethyl sulfoxide delivered the corresponding trifluoromethylthioether product 2g in 81% yield (1.08 g). Similarly, (2-pheny)-3-indolyl trifluoromethyl sulfoxide gave the desired product 2k in high yield (1.15 g, 92%).
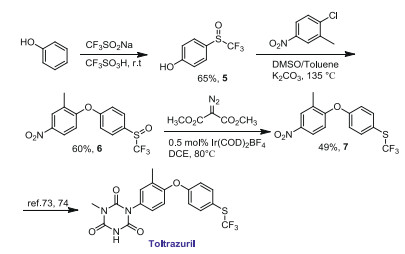
To further evaluate the utility of this method, we prepared a precursor of Toltrazuril [73, 74], an anticoccidial drug containing SCF3 moiety. In Scheme 6, CF3SO-substituted diaryl ether 6 could be easily prepared through the trifluoromethylsulfinylation of phenol and subsequent etherification reaction. Under standard conditions, compound 6 was reduced to give trifluoromethylthioether intermediate 7 in 49% yield.
Scheme 6
Scheme 6.
Synthesis of a precursor of Toltrazuril.
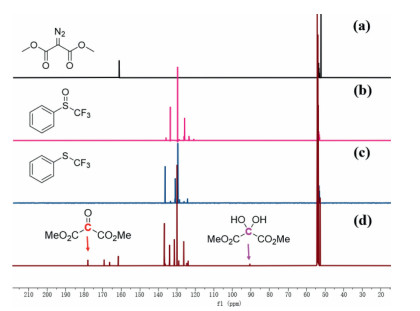
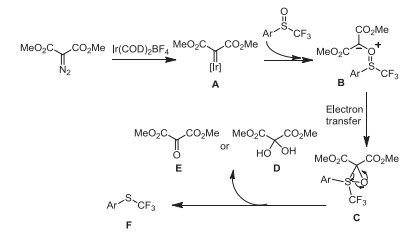
To understand the mechanism, the Ir(COD)2BF4-catalyzed deoxgenation of phenyl trifluoromethyl sulfoxide with dimethyl diazomalonate was carried out in CD2Cl2 under the standard conditions and monitored by 1H NMR (Fig. S1 in Supporting information), 19F NMR (Fig. S2 in Supporting information) and 13C NMR (Fig. 1). After 12 h, a new signal at −43.29 ppm was detected in the 19F NMR (Fig. S2c), indicating that the reaction occurred smoothly in CD2Cl2 and trifluoromethylthioether was formed. 1H NMR (Fig. S1d) experiments further showed a new peak at 3.98 ppm was found, which was corresponding to the MeO peak of dimethyl 2-oxomalonate [75]. The 13C NMR of this reaction mixture showed that two new signals at 177.6 ppm and 90.6 ppm were observed, which were ascribed to the characteristic peaks of vicinal tricarbonyl compound and its hydrate, respectively (Fig. 1d). The result indicated that phenyl trifluoromethyl sulfoxide was reduced through the oxygen atom transfer from S=O bond to dimethyl diazomalonate. Based on previous report [34, 69, 76] and our results, we proposed a possible mechanism for iridium(Ⅰ)-catalyzed deoxgenation reduction of trifluoromethyl sulfoxides by dimethyl diazomalonate. Firstly, dimethyl diazomalonate reacted with Ir(COD)2BF4 to generate a Iridium carbene complex A by losing nitrogen. Subsequently, Complex A reacted with the trifluoromethyl sulfoxide to form intermediate B. The electron transfer of intermediate B formed a three-membered ring transition state C, which further released D or E and provided trifluoromethylthioether F (Scheme 7).
Figure 1
Figure 1.13C NMR of (a) dimethyl diazomalonate, (b) trifluoromethyl phenyl sulfoxide; (c) trifluoromethyl phenyl sulfide, (d) Ir(COD)2BF4-catalyzed deoxgenation reduction of phenyl trifluoromethyl sulfoxide with dimethyl diazomalonate in CD2Cl2.
In summary, we have developed a new method for the preparation of fluoroalkylthioethers including trifluoromethylthioether and difluoromethylthioether by iridium(Ⅰ)-catalyzed deoxgenation of fluoroalkylsulfoxides with dimethyl diazomalonate. In the reaction system, dimethyl diazomalonate was used as reducing reagent and the corresponding fluoroalkylthioethers were produced through oxygen atom transfer from fluoroalkylsulfoxides to diazomalonate. The protocol featuring mild reaction conditions and good functional groups tolerance offers an alternative strategy for the synthesis of fluoroalkylthioethers.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Natural Science Foundation of Shanghai (No. 20ZR1471600), the Science and Technology Commission of Shanghai Municipality (No. 19DZ2271100) and the Open Research Fund Program of CAS Key Laboratory of Energy Regulation Materials (No. ORFP2020–06).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.061.
[1]
T. Liang, C.N. Neumann, T. Ritter, Angew. Chem. Int. Ed. 52 (2013) 8214–8264. doi: 10.1002/anie.201206566
D. Guenther Andreas, D. Mohrmann Karl-Heinrich, D. Stubbe Mathias, D. Ziemann Heinz, Patent (BAYER AG) DE3516630, 1986.
[75]
A.B. Smith 3rd, Z. Liu, Org. Lett. 10 (2008) 4363–4365. doi: 10.1021/ol801794f
[76]
T. Rajasekaran, G. Karthik, B. Sridhar, B.V. Reddy, Org. Lett. 15 (2013) 1512–1515. doi: 10.1021/ol400287q
Scheme 1
(A) Several types of electrophilic trifluoromethylthiolation reagents. (B) Trifluoromethylthiolation using CF3SO2Na or CF3SO2Cl as the SCF3 source. (C) Iridium(Ⅰ)-catalyzed deoxgenation of trifluoromethyl sulfoxides with dimethyl diazomalonate to access trifluoromethylthioether.
Scheme 2
Substrate scope for Ir(COD)2BF4-catalyzed deoxgenation of trifluoromethyl sulfoxides with dimethyl diazomalonate. Reaction conditions: Trifluoromethyl sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12–36 h; isolated yields. a 19F NMR Yields (due to high volatility).
Scheme 3
Substrate scope for Ir(COD)2BF4-catalyzed deoxgenation of difluoromethyl sulfoxides with dimethyl diazomalonate. Reaction conditions: Difluoromethyl sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12 h; isolated yields. a 19F NMR Yields (due to high volatility).
Scheme 4
Scope of deoxgenation of common aryl sulfoxides with dimethyl diazomalonate. Reaction conditions: Sulfoxide (0.5 mmol), dimethyl diazomalonate (0.75 mmol), Ir(COD)2BF4 (0.5 mol%), in solvent (5 mL) were stirred at 80 ℃ under N2 atmosphere for 12 h; isolated yields.

 DownLoad:
DownLoad:


 下载:
下载:









 下载:
下载:
