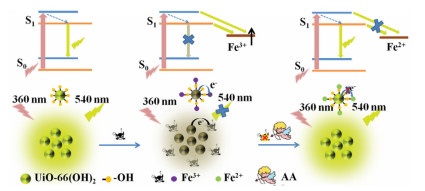
Scheme 1.
Schematic illustration the "on-off-on" fluorescent detection of Fe3+, AA using UiO-66-(OH)2 as sensor.
Metal-organic framework as a multi-component sensor for detection of Fe3+, ascorbic acid and acid phosphatase
Hao Wang , Xiuli Wang , Rong-Mei Kong , Lian Xia , Fengli Qu

Fe3+, an essential trace element in the growth and development of plants and animals, plays a vital role in life activities such as myoglobin synthesis in hemoglobin [1]. Abnormal level of Fe3+ has been reported to be related to various diseases such as diabetes, anemia, and even cancers [2]. Therefore, in order to monitor and prevent some diseases in advance, sensitive and reliable methods for Fe3+ detection are urgently required. Some traditional methods for detecting Fe3+ have been reported in the literatures including plasma-optical emission spectrometry [3], chromatography [4], inductively coupled plasma mass spectrometry (ICP-MS) [5]. However, these methods required large equipments, tedious operations, trained professionals, and higher cost. Ascorbic acid (AA) plays critical roles in many human physiological process, low level of AA in human body can cause cardiovascular disease, scurvy, and so on [6]. As an reductant, AA can reduce free radicals to non-free radicals, which is widely used in cosmetics, food and even medicine [7]. So far, various analytical methods for detecting AA have been reported, including electrochemistry [8], capillary electrophoresis [9] and ion chromatography [10]. However, as with these methods for the detection of AA, they showed disadvantages of tedious operation, heavy equipment and high cost. Fe3+ and AA are two biologically relevant substances [11]. Therefore, developing a fast, simple, and low-cost method to sensitively detect Fe3+ and AA is essential. Although carbon quantum dots have been reported to be used to detect these two substances at the same time, there are still some disadvantages such as the tedious preparation process and high detection limit [12]. Metal-organic frameworks (MOFs) composed of metal ions and organic ligands are a class of crystalline porous materials with many excellent properties, including high surface area, multifunctional composition and rich active sites [13, 14]. MOFs are involved and applied in many fields because of their unique advantages, such as gas storage and separation [15, 16], chemical biosensor [17], catalysis [18] and biomedicine [19]. Especially, MOFs showed great potential to be designed as fluorescent sensors for various targets [20, 21]. Compared with other fluorescent probes, MOF-based probes have attracted more attention due to their inherent characteristics (for example, adjustable pore size, large surface area, and different fluorescent emission mechanisms). In addition, MOFs can be functionalized with different functional groups, such as —NH2, —OH, to achieve the unique recognition of different targets [22-24].
As materials with excellent performance, MOFs have also been designed as sensors for detection of Fe3+. In our previous report, we have constructed a nanocomposite of RhB@MOF as a sensor to detect Fe3+ and AA with high sensitivity and selectivity based on inner filter effect (IFE). This kind of sensors based on IFE must meet the requirement of that the absorption spectrum of the analytes or the substrates overlaps with the excitation or emission spectrum of the fluorophores [25]. Recently, lanthanide-based MOFs (Ln-MOFs) were reported as excellent sensors for Fe3+ detection on the basis of dynamic or static quenching effect owing to its inhibition to antenna effect of ligand to lanthanide ions [26, 27]. Whereas, these MOF-based fluorescent sensors were usually constructed by coordination of lanthanide ions with complicated and expensive ligands, which involved in tedious synthesis process, limited their wide application. In addition, many MOF shapes for detecting metal ions were irregular, resulting in a relatively small interaction area between MOF and metal ions, which affected sensitivity and reaction rate [28]. However, there were few reports on the fluorescent detection of Fe3+ and AA using reactive groups functionalized MOF with direct mode. Herein, based on the high binding affinity of phenol hydroxyl group to Fe3+, we synthesized a hydroxyl group functionalized fluorescent MOF (UiO-66-(OH)2) using Zr4+ as the metal node and 2, 5-dihydroxyterephthalic acid (DHT) as the ligand through one-pot process. The abundant phenol hydroxyl groups on the surface of UiO-66-(OH)2 had efficient chelation ability to Fe3+ ions. Therefore, with addition of Fe3+, electrons/charges in the excited UiO-66-(OH)2 were transferred to the half-filled 3d orbit of Fe3+, the radiation electron transition from energy level of S0 to S1 was destroyed for UiO-66-(OH)2, leading to fluorescence quenching of UiO-66-(OH)2 due to facilitating non-radiative electron/hole recombination annihilation through an effective electron transfer process [29-31]. The quenched fluorescence by Fe3+ was then recovered by AA due to the redox reaction between them. In addition, acid phosphatase (ACP) can catalyze the hydrolysis of 2-phospho-L-ascorbic acid (AAP) to produce AA [32], which can also recover the fluorescence quenched by Fe3+. All in all, in this study, we have developed a simple, fast response and effective fluorescent probe for the detection of Fe3+, AA and ACP. So far, no literature has reported for multi-component detection of these three substances using active group functionalized MOFs through direct mode (Scheme 1).
We used the following reagents throughout the experiment, Zirconium chloride (ZrCl4), N, N-dimethylformamide (DMF), 2, 5-dihydroxyterephthalic acid (DHT), terephthalic acid, ferric chloride (FeCl3) were obtained from Sigma-Aldrich in Beijing, China. AA, ACP, AAP, L-alanine (L-Ala), DL-phenylalanine (DL-Phe phospho), L-tyrosine (L-Tyr), citric acid (CA), L-glycine (L-Gly), L-valine (L-Val), L-lysine (L-Lys), L-glutathione (GSH), glucose (Glu), L-cysteine (L-Cys), L-serine (L-Ser), dopamine (DA), bovine serum albumin (BSA), human serum albumin (HAS), glucose oxidase (GOx), horseradish peroxidase (HRP) were obtained from Shanghai Macklin Biochemical Co., Ltd. In addition, the water used in our laboratory was distilled water, which was obtained from Watson (Beijing, China). All solvents were commercially available and used as received.
The instruments used in the whole experiment are as follows, scanning electron microscopy (SEM) (JEOL, Model JSM7500 F) and transmission electron microscopy (TEM) (Philips EM 420, 120 kV) were used to measure the morphology of the materials. Fourier transform infrared (FT-IR) spectra were recorded in the 4000–500 cm−1 region using KBr pellets for tableting on a PerkinElmer spectrometer. A LabX XRD-6000 was used to record X-ray diffraction (XRD) patterns of the MOFs. The UV detection was carried out on a Lambda 750 s UV–vis spectrophotometer. Fluorescence spectroscopy was performed on a Hitachi F-7000 fluorescence spectrophotometer. The fluorescence lifetimes were measured by a SCINCO fluorescence FS-2 fluorescence spectrofluorometers. Zetasizer Nano ZS90 (Malvern, UK) was used to determine the zeta potential for this experiment.
UiO-66-(OH)2 was prepared using the reported solvothermal method and some modifications were made on the basis [33]. In short, added 0.54 mmol ZrCl4, 1 mL HCl and 15 mL DMF to a glass bottle (20 mL) with a screw cap and sonicated until all the contents were completely dissolved. Then, added 0.74 mmol of ligand DHT and sonicated 10 min for mixing well. The mixture was stirred and heated at 120 ℃ for 12 h (Scheme S1 in Supporting information). Cooled the obtained solution naturally, and the resulting yellow powder was collected by centrifugation. Then, the product was washed 3 times with DMF and EtOH, respectively. It was dried in a vacuum oven at 50 ℃ overnight.
UiO-66 was synthesized in the same way as UiO-66-(OH)2, except that the ligand was replaced with terephthalic acid. In short, the UiO-66-(OH)2 aqueous solution was first prepared by dispersion of UiO-66-(OH)2 into water (0.025 g/L). For Fe3+ detection, 100 μL of UiO-66-(OH)2 solution, 100 μL freshly prepared Fe3+ solution with different concentrations and 800 μL NaAc-HAc buffer (10 mmol/L, pH 5.5) were mixed well. To investigate whether the prepared UiO-66-(OH)2 had good selectivity, the aqueous solutions containing various interference metal ions (Ca2+, Hg2+, Mn2+, Ni2+, Pb2+, Zn2+, Cd2+, Mg2+, Zn2+, Al3+, K+, Fe2+, Co2+, Na+, Ag+, and Cu2+) were prepared in a concentration of 50 μmol/L, respectively, other operations were the same as above. Finally, fluorescence intensity measurements were performed at 360 nm excitation.
The experiment of AA sensing was performed as below: 100 μL of Fe3+ (180 μmol/L) and 100 μL of UiO-66-(OH)2 (0.025 g/L) were introduced into 700 μL of NaAc-HAc buffer (10 mmol/L, pH 5.5) to form the sensing platform denoted as UiO-66-(OH)2/Fe3+. Then added 100 μL of diverse concentrations of AA and mixed thoroughly. Incubated at room temperature for 50 min, then recorded the fluorescence recovery spectrum. To investigate whether UiO-66-(OH)2/Fe3+ system had good selectivity for AA, all potential interferences like L-Ala, DL-Phe, L-Tyr, L-Trp, CA, L-Gly, L-Val, GSH, Glu, L-Cys, L-Ser were added to the mixed solution with a final concentration of 300 μmol/L. Shook well, and performed fluorescence detection after 50 min of reaction.
To further detect ACP, the sensing platform of UiO-66-(OH)2/Fe3+ was employed. Different concentrations of ACP solution were firstly mixed with AAP (100 μmol/L), and then added to the previous sensing platform of UiO-66-(OH)2/Fe3+. After thoroughly shaking, the mixture was performed by incubation at 37 ℃ for 70 min. At last, fluorescence recovery spectra were recorded.
A serum sample analysis was performed accordingly. Human serum samples were taken from Jining People's Hospital, and the experiments we performed complied with relevant laws and regulations. Before performing the experiment, the obtained serum samples were diluted 10 times and different concentrations of Fe3+, AA and ACP were added by the standard addition method, respectively.
And then we first characterize UIO-66-(OH)2. The structural information on the prepared UiO-66-(OH)2 was firstly studied by SEM and TEM. As observed in Fig. 1, UiO-66-(OH)2 exhibited a spherical structure with average diameters of 900 nm and with uniform distribution. The as-synthesized UiO-66-(OH)2 was also well dispersed in water, which helped to quickly respond to targets.

The FT-IR spectra were showed in Fig. S2a (Supporting information). The adsorption peak at 3460 cm−1, which is attributed to the phenol hydroxyl group, was observed both in ligand (DHT) and UiO-66-(OH)2, suggesting that there was no influence on phenol hydroxyl group after the coordination. The absorption peak at 3126 cm−1, derived from the stretching vibration of O—H (νO-H) in the carboxyl group, was only observed in DHT, suggesting that the carboxyl group was coordinated to form MOF. The peaks at 1630 cm−1 and 1580 cm−1 derived from the C=O tensile vibration of the carboxyl groups in DHT and MOF, respectively [34]. After Fe3+ was added, a peak appeared at 650 cm−1, which was attributed to the coordination of Fe3+ with the phenolic hydroxyl group, forming an Fe-O bond [35]. The FT-IR comparison between UiO-66-(OH)2 and UiO-66 was also conducted. As shown in Fig. S2a, the adsorption peak for stretching vibration of hydroxyl group was observed both in UiO-66-(OH)2 and UiO-66, but UiO-66-(OH)2 showed weaker intensity. This might be due to the presence of intra-framework hydrogen bonds between the phenol hydroxyl group of ligand and Zr6O4(OH)4 unit of metal central [36, 37].
As shown in Fig. S2b (Supporting information), UiO-66-(OH)2 exhibited similar UV absorption profile to DHT, only a slight blue-shift was observed after DHT being coordinated with Zr4+ to form UiO-66-(OH)2.
The synthesized UiO-66-(OH)2 was characterized by XRD (Fig. S2c in Supporting information). The XRD patterns of the prepared UiO-66-(OH)2 had good consistency with that of simulated UiO-66, revealing that the crystalline UiO-66-(OH)2 had been successfully synthesized. The high crystallinity of UiO-66-(OH)2 help contribute to its high quality and excellent stability. Brunner–Emmet–Teller (BET) surface area of UiO-66-(OH)2 was obtained through the measurement of N2 adsorption-desorption isotherms (Fig. S2d in Supporting information). The results showed that the measurements surface area of UiO-66-(OH)2 was 57.6185 m2/g and the average pore size was 7.09 nm. Moreover, the curves belonged to type V isotherms, demonstrating that the interaction between UiO-66-(OH)2 and N2 was weak.
The element mapping results, shown in Fig. S3 (Supporting information), further indicated the successful synthesis of UiO-66-(OH)2. We measured the excitation and emission spectra of UiO-66-(OH)2, from Fig. S4 (Supporting information) we can observe that the excitation and emission wavelength of the material were 360 nm and 540 nm, respectively.
Then, we verified the stability of UIO-66-(OH)2. The stability of UiO-66-(OH)2 in aqueous solution was examined (Fig. S5 in Supporting information). We can observe from Fig. S5a, the emission spectra of UiO-66-(OH)2 suspension at 540 nm showed negligible change as the time progressed, which indicated that UiO-66-(OH)2 had good time stability in aqueous solution. Moreover, in the range of pH 5–8, the fluorescence intensity of UiO-66-(OH)2 at 540 nm is also relatively stable (Fig. S5b). In addition, as shown in Fig. S2a, after Fe3+ was added, a peak at 650 cm−1 appeared, which was attributed to the Fe-O bond formed by coordination of Fe3+ with the phenolic hydroxyl group, this indicated that the chelating band between Fe3+ and UiO-66-(OH)2 was stable during the detection. Moreover, as shown in Fig. S2b and Fig. S6 (Supporting information), after addition of Fe3+, the UV absorption and the fluorescence emission spectra had no shift, but only showed intensity change, which further demonstrated that the structure of UiO-66-(OH)2 was stable after chelating coordination with Fe3+. Therefore, excellent stability indicated the feasibility of UiO-66-(OH)2 being constructed as a fluorescent probe used in complex environments.
The fluorescence properties of UiO-66-(OH)2 and the sensing experiment of Fe3+ ions were conducted then. The fluorescence properties of UiO-66-(OH)2 were attributed to the properties of the coordinated ligand DHT in the framework. DHT has two electron-donating phenol hydroxyl groups at the para-position, which facilitates electron delocalization. Therefore, the free UiO-66-(OH)2 showed strong fluorescence emission at 540 nm in aqueous solution. Owing to high binding affinity of the phenol hydroxyl groups toward Fe3+, after the Fe3+ being added into the system, the Fe3+ was easily coordinated with the phenol hydroxyl group, electrons/charges in the excited UiO-66-(OH)2 were transferred to the half-filled 3d orbit of Fe3+, so the radiation electron transition from energy level of S0 to S1 was destroyed for UiO-66-(OH)2, resulting in quenching of UiO-66-(OH)2 fluorescence.
To evaluate the response kinetics of the probe towards Fe3+, the time response characteristics was performed. At the Fe3+ concentration of 50 μmol/L, emission spectra of UiO-66-(OH)2 excited at 360 nm was measured with different response time. Fig. S6a shows the time response fluorescence spectra and the kinetics curve within 60 s. As the response time increased from 0 to 30 s, the fluorescence intensity at 540 nm of UiO-66-(OH)2 was gradually decreased, and got a fixed value when the time reached to 30 s (Fig. S6b). After that, as the response time increased, the fluorescence intensity did not change significantly. Therefore, the time of response was selected as 30 s in the following experiments. The rapid kinetic response of the probe to the target may be attributed to the high affinity of phenol hydroxyl group to Fe3+ and the enrichment effect of the UiO-66-(OH)2 to Fe3+, as well as abundant response sites supplied by the probe owing to the high surface area. The fast response kinetics advantage of the UiO-66-(OH)2 compared with other probes was shown in Table S1 (Supporting information).
The pH value is an important factor that has complex impacts on sensors, so the effect of different pH on the fluorescence emission of UiO-66-(OH)2 solution was studied. The experiments of UiO-66-(OH)2 responding to Fe3+ (40 μmol/L) in buffer solution with various pH values were carried out and the results were shown in Fig. S7 (Supporting information). We compared the fluorescence intensity of UiO-66-(OH)2 before and after adding Fe3+ to obtain a sensitive output signal for detecting Fe3+ (Fig. S7). From the Fig. S7b, the pH 5.5 was the optimum value for sensitive signal change. Therefore, the pH value of 5.5 was selected for analysis in the following experiments.
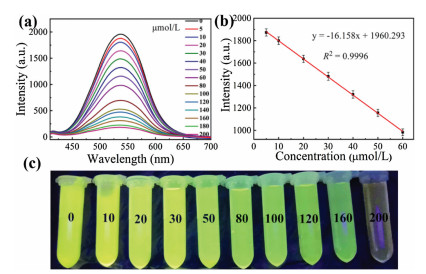
The sensitivity of the sensor was investigated under optimum assay conditions. The fluorescence intensity at 540 nm of the UiO-66-(OH)2 (0.025 g/L) was measured after 30 s response time with pH 5.5 at different concentration of Fe3+. As can be seen in Fig. 2a, as the concentration of Fe3+ increased from 0 to 200 μmol/L, the fluorescence intensity at 540 nm of the UiO-66-(OH)2 was gradually decreased. From the Fig. 2b, the fluorescence signal exhibited a good linear relationship with Fe3+ concentration in the range of 5−60 μmol/L, and the linear correlation coefficient (R2) for this relationship was 0.9996. Fig. 2c shows the photo of the detection system for detection of Fe3+ at different concentrations under UV at 365 nm. The limit of detection (LOD) for Fe3+ was 1.26 μmol/L according to the signal-to-noise (S/N) of 3.
Selectivity is one of the most important influencing factors for sensitive detection of metal ions. Therefore, experiments were performed to evaluate the selectivity of UiO-66-(OH)2. As observed in Fig. S8 (Supporting information), under the same test conditions, only Fe3+ led to significantly fluorescence quenching, while other metal ions (Cu2+, Na+, Fe2+, Ca2+, Ni2+, Co2+, Zn2+, Mn2+, K+, Mg2+, Pb2+, Hg2+ and Cd2+) showed insignificant effect on fluorescence intensity of the UiO-66-(OH)2. This revealed that UiO-66-(OH)2 exhibited high specific recognition to Fe3+. Specially, no such distinct selectivity was observed when UiO-66-(OH)2 was replaced by DHT (Fig. S9 in Supporting information). The probable reason was that DHT not only reacted with iron ions through phenol hydroxyl group, but also reacted with other metal ions, such as copper ion through carboxyl groups. In UiO-66-(OH)2 structure, the carboxyl group of the ligand had been coordinated with zirconium ion so that had no react activity to other metal ions.
Then we moved on the determination of AA based on UiO-66-(OH)2/Fe3+ probe. Besides, in the above experiment, we found that Fe2+ had no quenching effect on the fluorescence intensity of UiO-66-(OH)2. It is widely known that AA can reduce Fe3+ to Fe2+ under the physiological conditions, hence it is possible to design a "turn-on" sensor to detect AA, of which the quenched fluorescence of UiO-66-(OH)2/Fe3+ can be restored with the addition of AA.
First of all, we measured the fluorescence intensity of UiO-66-(OH)2 at 540 nm before and after reacting with AA, and found that there was no significant change after adding AA, indicating that AA had no influence on free UiO-66-(OH)2 (Fig. S10 in Supporting information). Whereas, after adding Fe3+ to the UiO-66-(OH)2 solution, the quenched fluorescence could be recovered by AA successfully. Then, the effect of incubation time for the UiO-66-(OH)2/Fe3+ detecting AA was studied. As we observed in Fig. S12 (Supporting information), when 120 μmol/L AA was added, the fluorescence intensity gradually recovered and remained stable after 50 min. Moreover, a significant linearity relationship of fluorescence intensity of the UiO-66-(OH)2/Fe3+ against the AA concentration (1−20 μmol/L) was observed with the correlation coefficient of R2 = 0.9986. According to S/N = 3, the LOD of the method to AA was calculated to be 0.37 μmol/L (Fig. S14 in Supporting information). In addition, to evaluate the advantages of the probe constructed in this study, a performance comparison between this probe and other reported MOF sensors in terms of measurement techniques, linear range, LOD and response kinetics (Table S1). As exhibited in Table S1, UiO-66-(OH)2, the newly constructed fluorescent sensor, showed much advantages including simultaneous detection of Fe3+ and AA, fast response kinetics, comparable or lower LOD. Meanwhile, the selectivity tests for UiO-66-(OH)2/Fe3+ sensing platform for AA were performed in the presence of representative interferents including L-Ala, DL-Phe, L-Tyr, L-Trp, CA, L-Gly, L-Val, GSH, Glu, L-Cys and L-Ser (Fig. S15 in Supporting information) under the same conditions. As expected, these interfering substances had negligible effect on the fluorescence intensity of UiO-66-(OH)2/Fe3+, indicating the good selectivity of this sensing platform for AA detection based on "turn-on" fluorescent principle.
In addition, we studied the performance of ACP activity assay. With the presence of ACP, AAP can hydrolyze to AA (Fig. S16 in Supporting information), so this system can also be used to detect ACP content. To achieve the highest response signals, the conditions potential to affect the sensitivity of ACP detection, including substrate AAP concentration, temperature and reaction time were optimized in detail. The results shown in Fig. S17 (Supporting information) demonstrated that AAP concentration of 100 μmol/L, temperature of 37 ℃, incubation time of 70 min were the optimum detection conditions. As the Fig. S18a (Supporting information) shows, with the gradual increase of ACP concentration, the fluorescence intensity of UiO-66-(OH)2/Fe3+/AAP was gradually enhanced. A good linear relationship (y = 26.993x + 438.770, R2 = 0.9971) between the fluorescence intensity and ACP concentration in the range of 0.2–14 U/L was achieved (Fig. S18b in Supporting information). The LOD for ACP detection was calculated to be as low as 0.05 U/L based on S/N = 3. In addition, in order to investigate the specific detection of ACP by the UiO-66-(OH)2/Fe3+/AAP system, we performed interference experiments (Fig. S19 in Supporting information). The results showed that the UiO-66-(OH)2/Fe3+/AAP system exhibited high selectivity for detection of ACP.
In order to study the feasibility of this developed method in practical sample detection, UiO-66-(OH)2 was used to determine Fe3+, AA and ACP in human serum samples. Briefly, 10-fold diluted human serum samples were respectively spiked with different standard solutions, and then measured with the developed method under the optimum conditions. As we can see from Table S2 (Supporting information), the recovery of the established method ranged from 98.30%–99.80% with the relative standard deviation (RSD, n = 3) less than 3.27% for Fe3+, from 99.50%–100.9% with RSD less than 3.02 for AA, from 92.67%–98.33% with RSD less than 4.09 for ACP, indicating the excellent ability to detect Fe3+, AA and ACP in biological samples. Above results demonstrate that the UiO-66-(OH)2 displayed the reliability and feasibility for Fe3+, AA and ACP determination in the human samples.
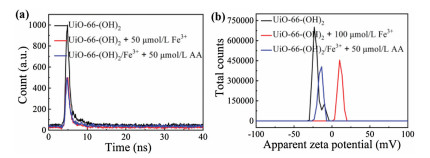
We further verified the experimental mechanism. Previous reports have confirmed that oxygen-containing groups (e.g., hydroxyl groups) showed excellent binding affinity to Fe3+ [34]. In addition, the experimental results from this study demonstrated that UiO-66 did not react with Fe3+ (Fig. S20 in Supporting information), therefore we can conclude that the phenol hydroxyl group in UiO-66-(OH)2 is the reactive site for Fe3+. In order to further confirm the response mechanism between the UiO-66-(OH)2 and Fe3+, the fluorescence lifetime experiments were carried out. According to Fig. 3a, we can see that the fluorescence lifetime of UiO-66-(OH)2 reduced from 2.94 ns to 2.33 ns after adding Fe3+, however, after adding AA, the fluorescence lifetime of UiO-66-(OH)2/Fe3+ increased from 2.33 ns to 2.51 ns, which indicated that the fluorescence quenching effect belongs to kinetic quenching. In addition, zeta potential of the UiO-66-(OH)2 before and after adding Fe3+ were measured, the results showed that the zeta potential of UiO-66-(OH)2 increased from −20.9 mV to 10.8 mV with the addition of Fe3+, whereas after adding AA, the zeta potential was decreased from 10.8 mV to −15.1 mV, further confirmed the coordination effect between the phenol hydroxyl group of UiO-66-(OH)2 and Fe3+ (Fig. 3b). Therefore, after Fe3+ chelating with the phenol hydroxyl group on the surface of UiO-66-(OH)2, electrons/charges in the excited UiO-66-(OH)2 were transferred to the half-filled 3d orbit of Fe3+, so the radiation electron transition from energy level of S0 to S1 was destroyed for UiO-66-(OH)2, resulting in fluorescence quenching of UiO-66-(OH)2 [29, 31]. The AA detection system of UiO-66-(OH)2/Fe3+ was based on redox reactions. After AA adding to the system, Fe3+ was reduce to Fe2+, non-radiation electron transition from UiO-66-(OH)2 to Fe3+ could not take place, resulted in the fluorescence recovery of UiO-66-(OH)2.
In summary, we used Zr4+ as the metal node and DHT as the ligand successfully synthesized a hydroxyl group functionalized fluorescent MOF (UiO-66-(OH)2) through one pot process as an "on-off-on" fluorescent probe for detection of Fe3+, AA and ACP. Fluorescence of UiO-66-(OH)2 can be quenched by Fe3+ with high specificity due to electron transfer from UiO-66-(OH)2 to Fe3+, therefore the electron radiation transition of the probe was destroyed. Moreover, the quenched fluorescence can be recovered as Fe3+ was reduced to Fe2+ by AA. Therefore, the probe can also be used to detect ACP based on the enzymolysis of AAP to produce AA. Benefitting from the hydroxyl group and the characteristics of UiO-66, including the high porosity and large surface area, the developed UiO-66-(OH)2 showed extensive advantages as a fluorescent probe for detection of multi-component, such as high sensitivity and selectivity, colorimetric detection, fast response kinetics and easy to operate, economical and secure. This is the first time to use active group functionalized MOFs as a multi-component sensor for these three substances detection.
There are no conflicts of interest to declare.
This work is supported by grants awarded by the National Natural Science Foundation of China (Nos. 21505084, 21775089), Natural Science Foundation Projects of Shandong Province (No. ZR2014BM029), Key Research and Development Program of Shandong Province (No. 2017GSF19109), Innovation Project of Shandong Graduate Education (No. SDYY16091), Outstanding Youth Foundation of Shandong Province (No. ZR2017JL010) and Taishan Scholar of Shandong Province.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.017.
H. Qi, M. Teng, M. Liu, et al., Colloid Interface Sci. 539 (2019) 332-341. doi: 10.1016/j.jcis.2018.12.047
W. Xuan, L. Ruiyi, F. Saiying, et al., Sens. Actuators B: Chem. 243 (2017) 211-220. doi: 10.1016/j.snb.2016.11.150
P. Liu, P.F. Borrell, M. Božic, et al., J. Hazard. Mater. 294 (2015) 1779-185.
E. Bakkaus, R.N. Collins, J. -L. Morel, B. Gouget, J. Chromatogr. A 1129 (2006) 208-215. doi: 10.1016/j.chroma.2006.07.004
G.L. Arnold, S. Weyer, A. Anbar, Anal. Chem. 76 (2004) 322-327. doi: 10.1021/ac034601v
X. Guo, G. Yue, J. Huang, et al., ACS. Appl. Mater. Interfaces 10 (2018) 26118-26127. doi: 10.1021/acsami.8b10529
K. Ghanbari, S. Bonyadi, New J. Chem. 42 (2018) 8512-8523. doi: 10.1039/C8NJ00857D
A. Abellán-Llobregat, C. González-Gaitán, L. Vidal, A. Canals, E. Morallón, Biosens. Bioelectron. 109 (2018) 123-131. doi: 10.1016/j.bios.2018.02.047
Y. Tang, M. Wu, Talanta 65 (2005) 794-798. doi: 10.1016/j.talanta.2004.08.013
M. Şahin, L. Özcan, B. Usta, Y. Şahin, Biosens. Bioelectron. 24 (2009) 3492-3497. doi: 10.1016/j.bios.2009.05.005
K.Y. Wu, L. Qin, C. Fan, et al., Dalton Trans. 48 (2019) 8911-8919. doi: 10.1039/C9DT00871C
X. Gao, X. Zhou, Y. Ma, et al., Appl. Surf. Sci. 469 (2019) 911-916. doi: 10.1016/j.apsusc.2018.11.095
S.L. Yao, S.J. Liu, X.M. Tian, et al., Inorg. Chem. 58 (2019) 3578-3581. doi: 10.1021/acs.inorgchem.8b03316
Y.P. Li, X.H. Zhu, S.N. Li, et al., ACS Appl. Mater. Interfaces 11 (2019) 11338-11348. doi: 10.1021/acsami.8b20410
D.X. Xue, Q. Wang, J. Bai, Coord. Chem. Rev. 378 (2019) 2-16. doi: 10.1016/j.ccr.2017.10.026
X.Y. Xu, B. Yan, ACS Appl. Mater. Interfaces 7 (2015) 721-729. doi: 10.1021/am5070409
J.N. Hao, B. Yan, Chem. Commun. 51 (2015) 7737-7740. doi: 10.1039/C5CC01430A
A.W. Stubbs, L. Braglia, E. Borfecchia, et al., ACS Catal. 8 (2018) 596-601. doi: 10.1021/acscatal.7b02946
R. Riccò, W. Liang, S. Li, et al., ACS Nano 12 (2018) 13-23. doi: 10.1021/acsnano.7b08056
B.H. Liu, D. -X. Liu, K. -Q. Yang, et al., Inorg. Chem. Commun. 90 (2018) 61-64. doi: 10.1016/j.inoche.2018.02.008
Y. Jin, X. Tian, L. Jin, et al., Anal. Chem. 90 (2018) 3276-3283. doi: 10.1021/acs.analchem.7b04813
L. Guo, Y. Liu, F. Qu, et al., Microchimica Acta 186 (2019) 740. doi: 10.1007/s00604-019-3806-x
M. Wang, L. Guo, D. Cao, Anal. Chem. 90 (2018) 3608-3614. doi: 10.1021/acs.analchem.8b00146
Y. Wan, D. Zou, Y. Cui, Y. Yang, G. Qian, J. Solid State Chem. 266 (2018) 70-73. doi: 10.1016/j.jssc.2018.07.013
L. Guo, Y. Liu, R. Kong, et al., Anal. Chem. 91 (2019) 12453-12460. doi: 10.1021/acs.analchem.9b03143
M. Zheng, H. Tan, Z. Xie, et al., ACS Appl. Mater. Interfaces 5 (2013) 1078-1083. doi: 10.1021/am302862k
G.G. Hou, Y. Liu, Q.K. Liu, J.P. Ma, Y.B. Dong, Chem. Commun. 47 (2011) 10731-10733. doi: 10.1039/c1cc14115e
H. Xu, J. Gao, X. Qian, et al., J. Mater. Chem. A 4 (2016) 10900-10905. doi: 10.1039/C6TA03065C
W. Xie, M. Tian, X. Luo, et al., Sensor. Actuators B: Chem. 302 (2020) 127180.
L. Guo, M. Wang, D. Cao, Small 14 (2018) 1703822.
B. Shi, Y. Su, L. Zhang, et al., ACS Appl. Mater. Interfaces 8 (2016) 10717-10725. doi: 10.1021/acsami.6b01325
X. Ma, S. Lin, Y. Dang, et al., Anal. Bioanal. Chem. 411 (2019) 6645-6653. doi: 10.1007/s00216-019-02038-z
L. Chen, J. Ou, H. Wang, et al., ACS Appl. Mater. Interfaces 8 (2016) 20292-20300. doi: 10.1021/acsami.6b06225
X. Gao, X. Zhou, Y. Ma, C. Wang, F. Chu, New J. Chem. 42 (2018) 14751-14756. doi: 10.1039/C8NJ01805G
J. Wang, Y. Fan, H.W. Lee, et al., ACS Appl. Nano Mater. 1 (2018) 3747-3753. doi: 10.1021/acsanm.8b01083
F. Vermoortele, R. Ameloot, A. Vimont, C. Serrec, D.D. Vos, Chem. Commun. 47 (2011) 1521-1523. doi: 10.1039/C0CC03038D
T. Ahnfeldt, D. Gunzelmann, T. Loiseau, et al., Inorg. Chem. 48 (2009) 3057-3064. doi: 10.1021/ic8023265

Scheme 1 Schematic illustration the "on-off-on" fluorescent detection of Fe3+, AA using UiO-66-(OH)2 as sensor.

Figure 1 SEM images of UiO-66-(OH)2. Indication scale is 1 μm (a) and 200 nm (b). TEM images of UiO-66-(OH)2 with the ruler of 1 μm (c) and 500 nm (d).

Figure 2 (a) Fluorescence spectra of UiO-66-(OH)2 at 540 nm under different Fe3+ concentrations. (b) Linear relationship of fluorescence intensity at 540 nm as a function of Fe3+ concentration. (c) The photos of the detection system of UiO-66-(OH)2 (0.025 g/L) with response to different concentrations of Fe3+ (0−200 μmol/L) for 30 s under UV light at 365 nm.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: