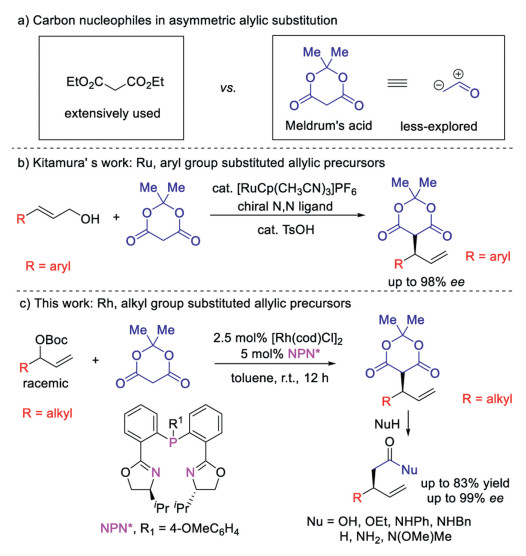
Scheme 1.
Asymmetric allylic substitution with Meldrum's acid.

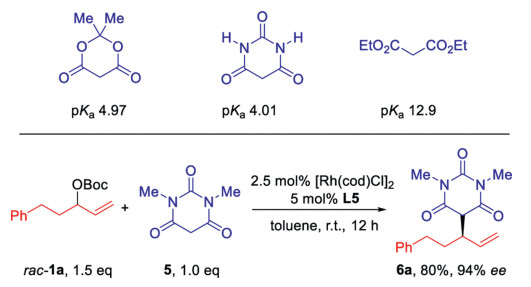
Meldrum's acid is a very useful molecule in organic synthesis for its versatile transformation by its strong nucleophilicity and electrophilicity [1]. Due to the easy transformation to acetic acid derivatives by losing CO2 and acetone, many asymmetric carbon-carbon bond formation reactions have been realized with Meldrum's acid as the carbon nucleophile [2]. Nevertheless, the utilization of Meldrum's acid in the asymmetric allylic substitution reactions is much underexplored [3], which is on sharp contrast to extensively application of malonates as carbon nucleophiles (Scheme 1a). To the best of our knowledge, only two examples have been reported in literature. Kitamura developed a Ru(Ⅱ)-catalyzed highly branch- and enantioselective allylic alkylation of Meldrum's acid, in which aryl group substituted allylic alcohols could be successfully used (Scheme 1b) [4a, b]. Trost reported a single example with Morita-Baylis-Hillman adduct as substrate[4c]. The reactions from aliphatic group substituted allylic precursors have never been reported.
Our group has developed the earth-abundant first-row cobalt-catalyzed highly regio- and enantioselective allylic amination reaction [5]. The racemic allylic carbonates bearing simple alkyl groups are successfully used in this transformation. The applying of chiral bisoxazolinephosphine ligand is the key to control the selectivities. Recently, we found that the replacement of cobalt by rhodium could further extend the scope to other carbon, oxygen, nitrogen and sulphur pronucleophiles and provide ageneral reaction platform for different nucleophiles [6]. Herein, we report a Rh(Ⅰ)-catalysed regio- and enantio-selective allylicalkylation of Meldrum acid with racemic allylic substrates bearing alkyl groups. β-Alkyl-γ, δ-unsaturated carboxylic acid and its derivatives could be synthesized in up to 83% yield and 99% ee (Scheme 1c).
We commenced the study with racemic allylic t-butyl carbonate 1a and Meldrum's acid 2a as the model substrates (Table 1). A variety of chiral bisoxazolinephosphine ligands were examined in the presence of 2.5 mol% of [Rh(cod)Cl]2 in toluene at room temperature. The R2 group on the oxazoline ring has a profound effect on the reactivity and enantioselectivity of the product 3a. Reaction with L1 bearing a small methyl group leads to 29% yield and 53% ee (entry 1). The ee increases to 96% when the methyl was replace by t-butyl or phenyl group, while the conversions remain low (L2 and L3, entries 2 and 3). To our delight, L4 with an isopropyl group on the oxazoline ring could help give a 70% yield and 97% ee for 3a (entry 4). The reactions in other solvents gave lower yields, which indicates less polar solvents are benefit for the conversion (entries 5–8). We then examined the substituent effect of R1 in the ligands. Applying of L5 with an electron-donating 4-MeOC6H4 group could further improve the yield to 85% (entry 9). While, L6 and L7 with 4-NMe2C6H4 and 4-MeO-3,5-tBu2C6H2 groups respectively are not as good as the L5, probably due to the relatively bigger size (entries 10 and 11). As predicted, a lower 48% yield was obtained when L8 with an electron-withdrawing CF3-C6H4 group was used (entry 12). The reaction with 5 mol% Rh(cod)2BF4 gave no any product 3a, which suggests the chloride anion plays a key role in the reaction (entry 13). In all cases, no trace of the linear isomer of 3a or diallylation product could be observed [7]. The absolute configuration of 3a was assigned to be R by single crystal X-ray diffraction analysis of 3a (CCDC: 1987520).
With the optimal condition in hand (entry 9, Table 1), we examine the scope of allylic carbonates in this highly regio- and enantioselective allylic alkylation reaction (Scheme 2). For the easy determination of the ee values of the products, all the synthesized Meldrum's acid derivatives react further with aniline to give the β-alkyl-γ, δ-unsaturated amide, in which a UV-active phenyl group exists [8]. Allylic carbonates bearing phenylethyl, n-propyl, methyl and n-pentyl groups could be converted to allylation products 4a to 4d smoothly. Lower yield of 4e bearing β-branched methyl group could be obtained. 4f and 4g bearing α-branch cyclopropyl and isopropyl groups respectively were synthesized in high yields. The reaction efficiency of 1h and 1i with big cyclohexyl and 3-pentyl groups drops, while the enantioselectivities increase to 99% (4h and 4i). Alkyl group with a sulphide function could be well tolerated in this reaction (4j). The reaction of phenyl group substituted racemic allylic carbonates gave 70% yield and 53% ee.
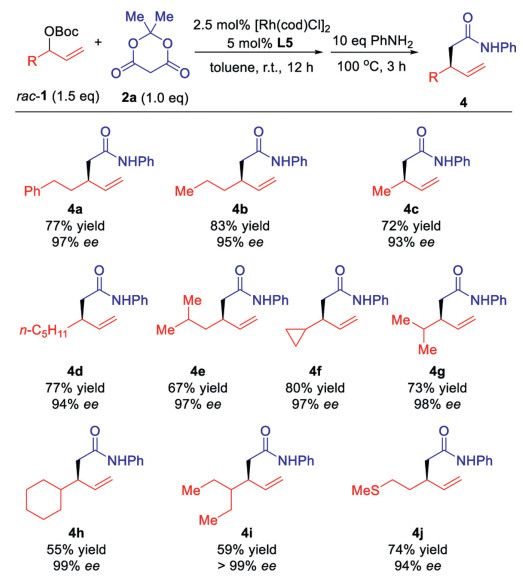
The scope of the nucleophile was then checked. 5-Substituted Meldrum's acid is not active in this reaction for the steric reason. Diethyl malonate, the open chain analogue of Meldrum's acid, cannot react as a nucleophile under the identical condition, probably due to its much lower acidity. Considering the similarity of barbituric acid's pKa value (4.01), 1,3-dimethylbarbituric acid 5 readily reacts to give 6a in 80% yield and 94% ee (Scheme 3) [9].
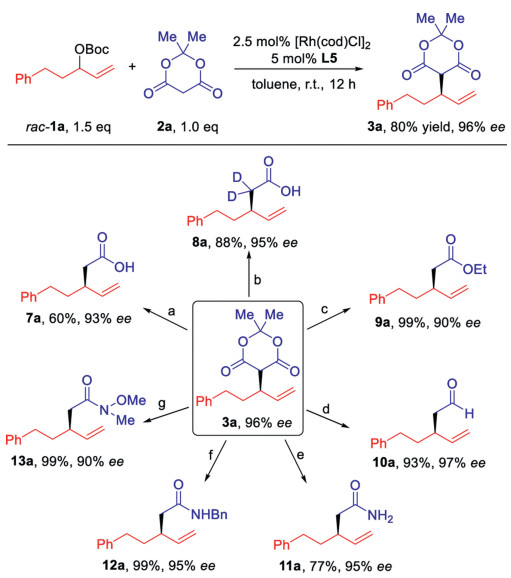
The Meldrum's acid derivative 3a bearing a chiral allyl group was synthesized in 2 mmol scale in 80% yield (Scheme 4). 3a could be transformed into the free carboxylic acid 7a [8] and the α-deuterated 8a [10] easily by treatment with Et3N or pyridine in water (D2O) (routes a and b). The corresponding ethyl ester 9a was synthesized similarly by reaction with ethanol and Et3N in 99% yield (route c) [2d]. The substituted Meldrum's acid 3a was readily reduced by Et3N and PhSiH3 to aldehyde 10a in 93% yield, without any erosion of the enantioselectivity (route d) [11]. Not only aniline, NH3·H2O and aliphatic benzylamine could also react with 3a to prepare the primary and secondary amide 11a [12] and 12a [13]. Finally, Weinreb amide 13a was synthesized in high yield, which allows the ketone synthesis by reaction further with organometallic reagents [8].
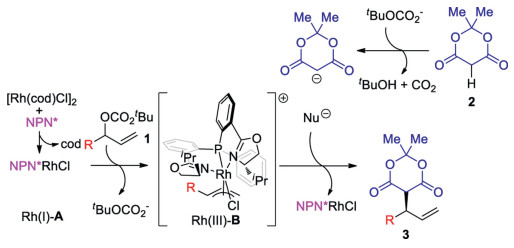
Based on the experiments above and the literature reports, aplausible mechanism was proposed as shown in Scheme 5. The in-situ formed NPN*RhCl (Rh(Ⅰ)-A) reacts with the allylic t-butyl carbonate 1 to get the chiral cationic intermediate Rh(Ⅲ)-B as a 18 electron complex. At the same time t-butyl carbonate anion was released as the counter anion [14]. The catalytic amount of basic t-butyl carbonate anion removes the acidic proton in the pronucleophile 2 to generate the Meldrum's acid anion and release neutral t-butanol and CO2, which enables the reaction conduction in base-free condition, while the less acidic diethyl malonate could not be deprotonated and react under the same condition. When Rh(cod)2BF4 was used as the rhodium precursor, t-butyl carbonate anion binds with rhodium and no free base is available to activate the Meldrum's acid. Finally, the nucleophile attacks the chiral Rh(Ⅲ)-B intermediate from the bottom side regio- and enantio-selectively via an outer-sphere mechanism to give 3 and release NPN*RhCl catalyst.
In conclusion, we have developed the Rh/bisoxazoline-phosphine-catalysed highly branched and enantioselective allylic alkylation of Meldrum's acid. Simple alkyl group based allylic precursors are utilized for the first time in the allylic alkylation of Meldrum's acid. The chiral Meldrum's acid derivatives could be converted to β-alkyl-γ, δ-unsaturated carboxylic acid and its derivatives [15]. Further extension of novel nucleophiles in this Rh-catalysed regio- and enantioselective allylic alkylation reaction is under investigation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Natural Science Foundation of China (NSFC, Nos. 21602130 and 21961023) and the startup funding from Shanghai Jiao Tong University.
Supplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.04.009.
A.M. Dumas, E. Fillion, Acc. Chem. Res. 43 (2010) 440-454. doi: 10.1021/ar900229z
(a) B.M. Trost, C. Jäkel, B. Plietker, J. Am. Chem. Soc. 125 (2003) 4438-4439;
(b) B.M. Trost, A.B.C. Simas, B. Plietker, C. Jäkel, J. Xie, Chem. Eur. J. 11 (2005) 7075-7082;
(c) N.G. Adamson, K.C.E. Wilbur, S.J. Malcolson, J. Am. Chem. Soc. 140 (2018) 2761-2764;
(d) T.M. Khopade, T.B. Mete, J.S. Arora, R.G. Bhat, Chem. Eur. J. 24 (2018) 6036-6040;
(e) K.L. Kimmel, J.D. Weaver, J.A. Ellman, Chem. Sci. 3 (2012) 121-125;
(f) C. Wang, L.A. Chen, H. Huo, et al., Chem. Sci. 6 (2015) 1094-1100.
(a) U. Kazmaier, Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Springer Verlag, Berlin and Heidelberg, 2012;
(b) B.M. Trost, D.L. Van Vranken, Chem. Rev. 96 (1996) 395-422;
(c) B.M. Trost, M.L. Crawley, Chem. Rev. 103 (2003) 2921-2943;
(d) Z. Lu, S. Ma, Angew. Chem. Int. Ed. 47 (2008) 258-297;
(e) Q. Cheng, H.F. Tu, C. Zheng, et al., Chem. Rev. 119 (2019) 1855-1969.
(a) K. Miyata, H. Kutsuna, S. Kawakami, M. Kitamura, Angew. Chem. Int. Ed. 50 (2011) 4649-4653;
(b) K. Miyata, M. Kitamura, Synthesis 44 (2012) 2138-2146;
(c) B.M. Trost, M.K. Brennan, Org. Lett. 9 (2007) 3961-3964.
S. Ghorai, S.S. Chirke, W.B. Xu, J.F. Chen, C. Li, J. Am. Chem. Soc. 141 (2019) 11430-11434. doi: 10.1021/jacs.9b06035
(a) W.B. Xu, S. Ghorai, W. Huang, C. Li, ACS Catal. 10 (2020) 4491-4496;
(b) S.B. Tang, X. Zhang, H.F. Tu, S.L. You, J. Am. Chem. Soc. 140 (2018) 7737-7742;
(d) M.B. Thoke, Q. Kang, Synthesis 51 (2019) 2585-2631;
(e) P. Koschker, B. Breit, Acc. Chem. Res. 49 (2016) 1524-1536.
(a) K. Manabe, S. Kobayashi, Org. Lett. 5 (2003) 3241-3244;
(b) F.X. Felpin, Y. Landais, J. Org. Chem. 70 (2005) 6441-6446.
S. Mishra, J. Liu, A. Aponick, J. Am. Chem. Soc. 139 (2017) 3352-3355. doi: 10.1021/jacs.7b00363
(a) M. Rombola, C.S. Sumaria, T.D. Montgomery, V.H. Rawal, J. Am. Chem. Soc. 139 (2017) 5297-5300;
(b) Y. Liu, Y. Zhang, H.X. Duan, D.Y. Wanyan, Y.Q. Wang, Org. Biomol. Chem. 15 (2017) 8669-8679.
S.P. Andrews, M. Ladlow, J. Org. Chem. 68 (2003) 5525-5533. doi: 10.1021/jo034340p
C.G. Frost, B.C. Hartley, J. Org. Chem. 74 (2009) 3599-3602. doi: 10.1021/jo900390d
X. Lei, J.A. Porco Jr., Org. Lett. 6 (2004) 795-798. doi: 10.1021/ol036502+
S.A. Adediran, D. Cabaret, J.F. Lohier, M. Wakselman, R.F. Pratt, Bioorg. Med. Chem. 18 (2010) 282-291. doi: 10.1016/j.bmc.2009.10.056
(a) J. Tsuji, I. Shimazu, I. Minami, Y. Ohashi, Tetrahedron Lett. 23 (1982) 4809-4812;
(b) J. Tsuji, I. Shimazu, I. Minami, et al., J. Org. Chem. 50 (1985) 1523-1529.
(a) Y. Sempere, E.M. Carreira, Angew. Chem. Int. Ed. 57 (2018) 7654-7658;
(b) Y. Sempere, J.L. Alfke, S.L. Rossler, E.M. Carreira, Angew. Chem. Int. Ed. 58 (2019) 9537-9541.

Scheme 4 The synthetic transformation of the Meldrum's acid derivatives. (a) Et3N (3.0 equiv.), H2O (10 equiv.), toluene, 110 ℃, 3 h; (b) D2O (10 equiv.), pyridine, 100 ℃, 5 h; (c) Et3N (3.0 equiv.), EtOH (5 equiv.), toluene, 110 ℃, 3 h; (d) Et3N (4.0 equiv.), PhSiH3 (3 equiv.), THF, r.t., 2 h; (e) NH3·H2O (5.0 equiv.), toluene, 110 ℃, 3 h; (f) BnNH2 (5.0 equiv.), toluene, 110 ℃, 3 h; (g) HCl·HN(OMe)Me (2.0 equiv.), toluene, 110 ℃, 3 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载: