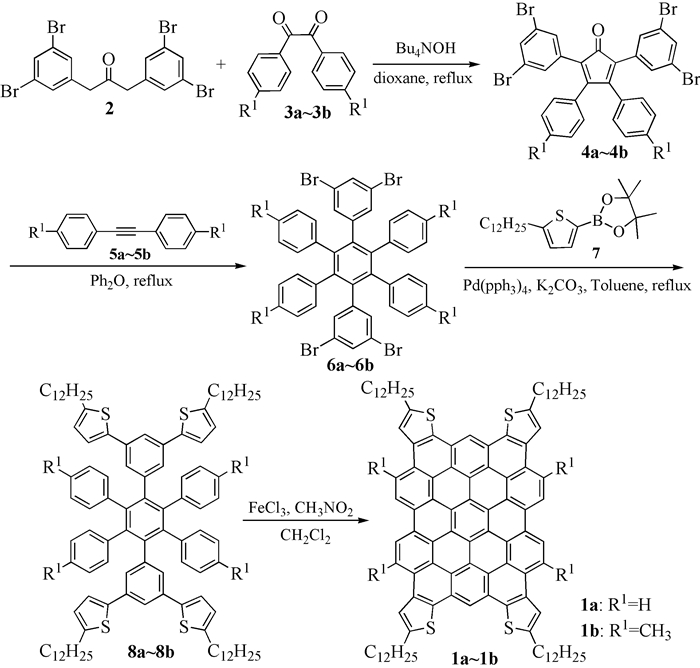
 图式1
化合物1的合成路线
图式1.
Synthetic route of compound 1
图式1
化合物1的合成路线
图式1.
Synthetic route of compound 1
引用本文:
殷江辉, 徐娅, 向芸颉, 张灯青, 李贤英, 金武松. 矩形多环芳烃的合成[J]. 化学通报,
2017, 80(8): 772-776.

Citation: Yin Jianghui, Xu Ya, Xiang Yunjie, Zhang Dengqing, Li Xianying, Jin Wusong. Syntheses of Rectangle Type Polycyclic Aromatic Hydrocarbons[J]. Chemistry, 2017, 80(8): 772-776.
Citation: Yin Jianghui, Xu Ya, Xiang Yunjie, Zhang Dengqing, Li Xianying, Jin Wusong. Syntheses of Rectangle Type Polycyclic Aromatic Hydrocarbons[J]. Chemistry, 2017, 80(8): 772-776.
矩形多环芳烃的合成
摘要:
本文用3,4-二苯基-2,5-二(3,5-二溴苯基)环戊二烯酮(4a)与二苯乙炔(5a)通过Diels-Alder环加成反应得到1,2,4,5-四苯基-3,6-二(3,5-二溴苯基)苯(6a)。化合物6a通过经典的Suzuki偶联反应得到1,2,4,5-四苯基-3,6-二(3,5-二(4-十二烷基噻吩))苯基苯(8a),再利用FeCl3作为氧化剂发生Scholl氧化脱氢关环反应,得到目标化合物1a。采用类似合成方法,得到目标化合物1b。化合物的结构均通过1H NMR和MALDI-TOF MS表征,并对其光谱特征、热性能及电学性能进行了初步研究。
-
关键词:
- 矩形
- / 多环芳烃
- / Scholl氧化关环
- / 噻吩
English
Syntheses of Rectangle Type Polycyclic Aromatic Hydrocarbons
Abstract:
1, 2, 4, 5-tetraphenyl-3, 6-di(3, 5-dibromophenyl)benzene (6a) was synthesized by Diels-Alder cycloaddition reaction of 2, 5-bis(3, 5-dibromophenyl)-3, 4-diphenylcyclopenta-2, 4-dienone (4a) with 1, 2-diphenylethyne (5a). 1, 2, 4, 5-tetraphenyl-3, 6-di(3, 5-di(4-dodecylthiophene))phenylbenzene (8a) was obtained from 6a by classical Suzuki coupling reaction, then the target compound 1a was obtained for 8a by Scholl cyclodehydrogenation using FeCl3 as catalyst. The similar synthesis method was used to obtain the target compound 1b. The structures of these compounds were characterized by 1H NMR and MALDI-TOF MS, and their spectral characteristics, thermal and electrical properties were investigated.
-
π电子材料由于其独特的电子、光电子和磁性能而在分子电子学方面有着潜在的应用[1, 2]。相比π共轭的聚合物,多环芳烃(PAH)具有独特的物理化学特性和优异的自组装性能,在场效应晶体管、发光二极管、太阳能电池和超分子材料领域有着重要应用前景[3]。与全碳的多环芳烃相比,当骨架中含有氮[4]、硫[5]、氧[6]等富电子或电负性强的杂原子时,这些多环芳烃的电子密度及极性通常发生改变[7]。因此对杂环多环芳烃的合成及性能的研究已经成为一个热门领域。其中,对含噻吩单元的研究已经有了长足的发展。目前为止,诸如苯[8]、萘[9]、蒽[10, 11]、菲[12]、并四苯[13]、蔻[14]等骨架中含硫原子的多环芳烃衍生物已经被广泛研究[15]。例如,Zhang等[16]采用共价自组装的合成策略,在三氯化铁促进下实现π-扩展硫杂三噻吩-三苯并蒄(TBTTCs)的合成;利用环化脱氢反应,Chen等[17]合成的六噻吩并蒄(HTC)显示出优异的自组装性能和电子特性等。因此我们着眼于开发新型功能性含杂原子的多环芳烃,设计合成了一类新型噻吩并多环芳烃衍生物1,并对其基本物性进行了初步探讨。
本文以3, 5-二溴苄基溴和对甲基苯磺基甲基异氰为起始原料,合成了1, 3-二(3, 5-二溴苄基)丙酮(2),化合物2和3a在四叔丁基氢氧化铵的作用下发生缩合反应,得到3, 4-二苯基-2, 5-二(3, 5-二溴苯基)环戊二烯酮(4a);然后其在二苯醚的作用下与二苯乙炔发生Diels-Alder环加成得到1, 2, 4, 5-四苯基-3, 6-二(3, 5-二溴苯基)苯(6a);6a在Pd(PPh3)4催化下与化合物7发生Suzuki偶联反应制得1, 2, 4, 5-四苯基-3, 6-二(3, 5-二(4-十二烷基噻吩))苯基苯(8a);化合物8a在FeCl3作用下发生Scholl氧化关环得到目标化合物1a。采用类似合成方法得到目标化合物1b(图式 1)。
图式1
化合物1的合成路线
图式1.
Synthetic route of compound 1
1 实验部分
1.1 仪器与试剂
Brucker Model Avance DMX 400 (400MHz)型核磁共振谱仪,CDCl3作溶剂;ABSciex 4800型基质辅助激光电离飞行时间(MALDI-TOF)质谱仪;JAI model LC-9201型循环制备高效液相色谱仪,CHCl3作为流动相;普析通用TU-1901型紫外可见分光光度计;HORIBA model Fluoromax-4型荧光光谱仪;TG-2091F1型(德国耐驰仪器有限公司)热重分析仪;CHI600E系列(上海辰华仪器有限公司)电化学工作站。
所用试剂均为分析纯,分别购自北京依诺凯、百灵威或国药集团有限公司。化合物2参考文献[18]方法合成。
1.2 化合物4a的合成
参考文献[19]的方法,在氩气保护下,向装有磁力搅拌子的干燥两口瓶中加入252mg(0.479mmol)化合物2、101mg(0.48mmol)化合物3a和3mL新蒸的1, 4-二氧六环,加热至125℃回流,加入0.24mL 1.0 mol/L四丁基氢氧化铵的甲醇溶液,反应10min后,溶液变为酒红色,TLC追踪至无原料点,停止反应。待反应冷却到室温后,用CH2Cl2萃取(50mL×3),合并有机相,蒸馏水洗(50mL×3),无水硫酸镁干燥,过滤,旋蒸除去溶剂,剩余物用硅胶柱层析提纯(洗脱剂石油醚/CH2Cl2,体积比10:1),得紫红色固体100mg,产率30%。1H NMR(400MHz,CDCl3)δ:7.56 (d,J=2.2Hz,2H),7.35 (d,J=7.3Hz,2H),7.33~7.30 (m,4H),7.26 (d,J=7.4Hz,4H),6.93 (d,J=7.6Hz,4H);13C NMR(101MHz,CDCl3)δ:197.96,156.48,133.78,133.14,131.58,131.55,129.55,129.01,128.45,122.96,122.50;MALDI-TOF MS m/z:699.94 [M]+。
1.3 化合物4b的合成
用与以上相同的方法合成化合物4b,产率47%。1H NMR(400MHz,CDCl3)δ:7.52 (t,J=1.7Hz,2H),7.29(d,J=1.7Hz,4H),7.04 (d,J=7.9Hz,4H),6.78(d,J=8.1Hz,4H),2.35(s,6H);13C NMR(101MHz,CDCl3)δ:198.00,156.58,139.76,134.16,132.94,131.56,129.13,129.06,128.63,128.63,122.71,122.43,21.52;MALDI-TOF MS m/z:727.97 [M]+。
1.4 化合物6a的合成
参考文献[20]的方法,将100mg(0.143mmol)4a和26mg(0.144mmol)5a置于50mL Schlenk管中,抽真空充Ar气三次,加入2mL二苯醚,回流反应16h,反应溶液由酒红色变为棕黄色并伴有浅黄色固体析出。待反应瓶冷却到室温后,将反应液依次用CH2Cl2(5mL×3)、THF(5mL×3) 和乙醇(5mL×3) 洗涤,收集滤饼,得浅黄色固体90mg,产率74%。MALDI-TOF MS m/z:849.96 [M]+。
1.5 化合物6b的合成
用与以上相同的方法合成化合物6b,产率64%。MALDI-TOF MS m/z:906.11 [M]+。
1.6 化合物8a的合成
参考文献[21]的方法,在氩气保护下,向装有磁力搅拌子和回流装置的50 mL两口瓶中依次加入80mg(0.094mmol) 6a、425mg(1.12mmol) 7、35mg (0.0282mmol)Pd(PPh3)4、0.7mL (2mol/L,冷冻除氧) K2CO3溶液和35mL甲苯(冷冻除氧),100℃回流72h。冷至室温后,CH2Cl2萃取(50mL×3),合并有机相,蒸馏水洗(50mL×3),无水硫酸镁干燥,过滤,旋蒸蒸除溶剂,剩余物用硅胶层析柱提纯(洗脱剂石油醚/CH2Cl2,体积比5:1),得白色固体55mg,产率38%。1H NMR(400MHz,CDCl3)δ:7.22(s,2H),6.99~6.91(m,24H),6.87(d,J=6.6Hz,4H),6.76(d,J=3.1Hz,4H),6.65(s,4H),2.77(t,J=7.5Hz,9H),1.71~1.62(m,10H),1.29(s,89H),0.90(t,J=6.4Hz,14H);13C NMR(101MHz,CDCl3)δ:145.48,141.62,141.19,140.52,140.37,139.77,133.45,131.37,127.98,126.88,125.63,124.72,122.51,119.86,31.95,31.71,30.24,29.70,29.67,29.60,29.43,29.39,29.16,22.73,14.16;MALDI-TOF MS m/z:1536.16 [M+H]+。
1.7 化合物8b的合成
用与以上相同的方法合成化合物8b,产率53%。1H NMR(400MHz,CDCl3)δ:7.19(s,2H),6.88(d,J=1.3Hz,4H),6.79(d,J=8.0Hz,8H),6.71(t,J=5.8Hz,12H),6.63(d,J=3.5Hz,4H),2.76(t,J=7.5Hz,8H),2.07(s,12H),1.75~1.59(m,8H),1.45~1.18(m,72H),0.88(t,J=6.8Hz,12H);13C NMR(101MHz,CDCl3)δ:145.36,141.92,141.55,140.39,139.96,137.56,134.63,133.30,131.33,128.31,127.56,124.64,122.46,119.88,31.94,31.72,30.25,29.69,29.66,29.58,29.42,29.37,29.14,22.71,21.06,14.14;MALDI-TOF MS m/z:1592.18 [M+H]+。
1.8 化合物1a的合成
参考文献[22]的方法,向装有磁力搅拌子的250mL两口烧瓶中,加入80mg (0.0521mmol) 8a,体系抽真空鼓氩气三次,加入120mL新蒸的CH2Cl2,氩气鼓泡5min,将360mg(2.22mmol)无水氯化铁溶解在3.5mL硝基甲烷中,缓慢加入到两口瓶中,45min后MALDI-TOF MS追踪显示反应完全。1h后向反应液中加入甲醇猝灭反应,析出沉淀,抽滤,滤饼用CH2Cl2(10mL×3)、THF(10mL×3) 洗涤,得砖红色固体60mg,产率76%。HRMS(MALDI-TOF),C106H114S4[M]+,m/z:实测值1516.3181,计算值1516.3180。
1.9 化合物1b的合成
用与以上相同的方法合成化合物1b,产率75%。HRMS(MALDI-TOF),C110H122S4[M]+,m/z:实测值1572.4250,计算值1572.4260。
2 结果与讨论
2.1 HRMS(MALDI-TOF)表征
由于所合成的目标化合物1a、1b各自分子之间有很大的π共轭体系,堆积作用很强,分子以某种动态聚集体形式存在,无法用核磁验证,所以我们通过HRMS(MALDI-TOF)来确定。化合物1a、1b的HRMS分别与理论值相符,如图 1所示,证实了所得到的化合物结构正确。
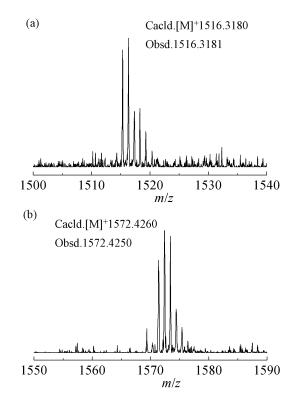 图 1
化合物1a、1b的HRMS(MALDI-TOF)
Figure 1.
HRMS (MALDI-TOF) of (a) 1a and (b) 1b
图 1
化合物1a、1b的HRMS(MALDI-TOF)
Figure 1.
HRMS (MALDI-TOF) of (a) 1a and (b) 1b
2.2 紫外吸收光谱
化合物1a、1b的紫外吸收光谱图如图 2所示。在浓度均为1×10-5mol/L时,化合物1a的甲苯溶液的最大吸收波长为413nm,为π-π*特征吸收,同时在波长为524和563 nm也有吸收;化合物1b的甲苯中溶液的最大吸收波长为419nm,为π-π*特征吸收,同时在波长为533和558 nm处也有吸收。这两种化合物的紫外可见光谱形状相似且波长位移较小,可能是因为母体环相同所致。
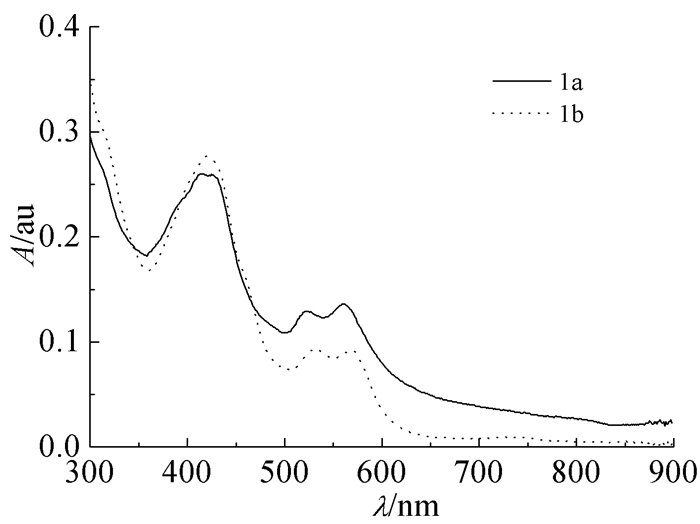 图 2
化合物1a、1b的甲苯溶液1×10-5mol/L)的紫外可见光谱
Figure 2.
UV-Vis spectra of 1a and 1b (1×10-5mol/L) in toluene
图 2
化合物1a、1b的甲苯溶液1×10-5mol/L)的紫外可见光谱
Figure 2.
UV-Vis spectra of 1a and 1b (1×10-5mol/L) in toluene
2.3 荧光光谱
化合物1a、1b的甲苯溶液(1×10-5mol/L)分别在其最大吸收波长413、419 nm激发下测得的荧光光谱图如图 3所示。化合物1a、1b的最大发射波长分别为643、655 nm,属于近红外区域;且在该浓度下得到的均为宽峰,这进一步说明化合物1a、1b在溶液中极易通过π-π堆积作用形成聚集体,表明这类化合物具有一定的荧光性能,为以后作为发光材料等方面的研究奠定了基础。
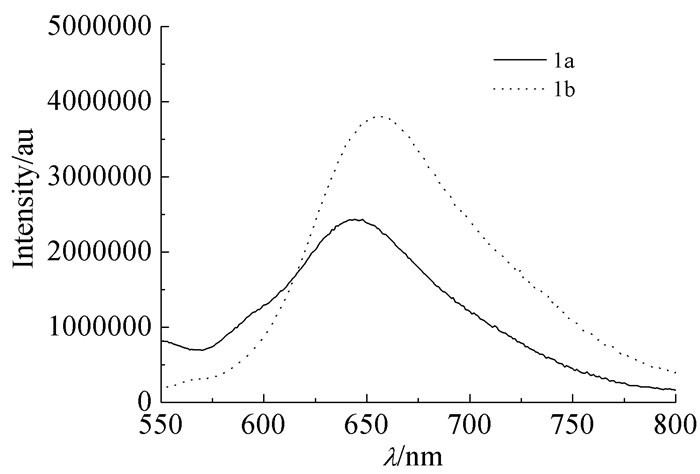 图 3
化合物1a、1b的甲苯溶液1×10-5mol/L)的荧光光谱
Figure 3.
Fluorescence spectra of 1a and 1b (1×10-5mol/L) in toluene
图 3
化合物1a、1b的甲苯溶液1×10-5mol/L)的荧光光谱
Figure 3.
Fluorescence spectra of 1a and 1b (1×10-5mol/L) in toluene
2.4 热性能研究
为了探究其热性能,我们对化合物1a、1b进行热重分析,热重测试以30℃为起始测试温度,在氮气保护下,升温速率为10℃/min,一直升温至900℃,结果如图 4所示。1a的热分解温度约为350℃,到900℃时约有40%的残留;1b的热分解温度也约为350℃,到900℃时约有50%的残留。由此表明该类化合物具有良好的热稳定性,有望应用于对热稳定性要求比较高的有机纳米器件。
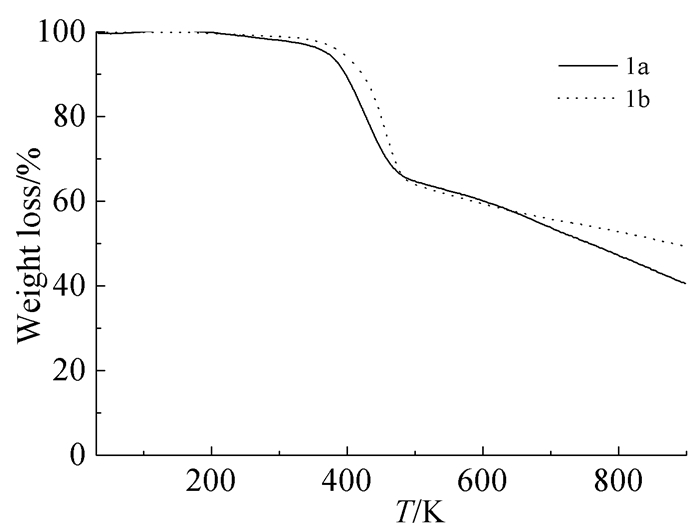 图 4
化合物1a、1b的TG曲线
Figure 4.
TGA curves of 1a and 1b
图 4
化合物1a、1b的TG曲线
Figure 4.
TGA curves of 1a and 1b
2.5 电学性能研究
有机电致发光材料能带的准确测定对于有机电致发光器件的研究至关重要,通过循环伏安法能给出有机光电材料的全部能带结构参数。在测定氧化还原电位时采取的试验条件为:玻碳电极为工作电极,铂丝电极为辅助电极,银电极(Ag/AgNO3-乙腈溶液)为参比电极,避光密封;考虑到其溶解性,选择邻二氯苯为溶剂;以四丁基四氟硼酸铵为支持电解质。循环伏安曲线如图 5所示,最终测得化合物1a、1b的氧化电位分别是0.382、0.377 V,通过进一步计算,可知化合物1a、化合物1b的HOMO能级分别为-4.918、-4.913 eV。由此可见,化合物1a和1b是强电子供体,在光伏器件中有潜在应用价值。
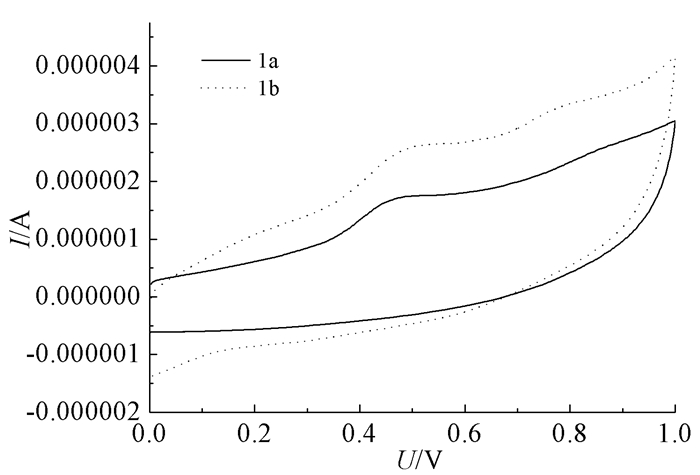 图 5
化合物1a、1b的循环伏安曲线
Figure 5.
Cyclic voltammetry curves of 1a and 1b
图 5
化合物1a、1b的循环伏安曲线
Figure 5.
Cyclic voltammetry curves of 1a and 1b
3 结论
本文以1, 3-二(3, 5-二溴苄基)丙酮为起始原料,利用其和苯偶酰反应得到3, 4-二苯基-2, 5-二(3, 5-二溴苯基)环戊二烯酮,再与对苯乙炔通过D-A反应得到2, 3, 4, 5-四苯基-1, 4-二(3, 5-二溴苯基)苯,再和噻吩硼酸酯类化合物进行Suzuki偶联反应得到氧化前驱体,再经三氯化铁氧化关环,得到目标化合物1a。由于化合物1a的溶解性不是很好,我们在其矩形两边分别引入两个甲基,得到了化合物1b,不仅溶解性大为改善,也为测试和分离带来很多方便。紫外和荧光光谱研究表明,这类化合物在溶液中发出红色荧光,可作为一种新型的红色主体发光材料用于有机发光二极管(OLED)、荧光传感器等领域。并且这两个化合物具有良好的热稳定性,有望应用于对热稳定性要求比较高的有机纳米器件。另外,循环伏安研究表明此类矩形多环芳烃是强电子供体,在光伏器件中有着潜在的应用。
-
-
[1]
S Laschat, A Baro, N Steinke et al. Angew. Chem. Int. Ed., 2007, 46(26):4832~4887. http://europepmc.org/abstract/MED/17568461
-
[2]
A Tsuda, A Osuka. Science, 2001, 293(5527):79~82. http://europepmc.org/abstract/MED/11441176
-
[3]
H E Katz, Z Bao, S L Gilat. Acc. Chem. Res., 2001, 34(5):359~369. http://europepmc.org/abstract/MED/11352714
-
[4]
M Matena, M Stohr, T Riehm et al. Chem. Eur. J., 2010, 16(7):2079~2091. http://europepmc.org/abstract/med/20077537
-
[5]
K Y Chernichenko, V V Umerin, R V Shpanchenko et al. Angew. Chem. Int. Ed., 2006, 45(44):7367~7370. doi: 10.1002/anie.200602190/suppinfo
-
[6]
D Wu, W Pisula, M C Haberecht et al. Org. Lett., 2009, 11(24):5686~5689. http://europepmc.org/abstract/MED/19919078
-
[7]
P J Stang, B Olenyuk. Acc. Chem. Res., 1997, 30(12):502~518.
-
[8]
Y Nicolas, P Blanchard, E Levillain et al. Org. Lett., 2004, 6(2):273~276.
-
[9]
I Osaka, T Abe, S Shinamura et al. J. Am. Chem. Soc., 2010, 132(14):5000~5001. http://europepmc.org/abstract/med/20297819
-
[10]
M Mamada, T Minamiki, H Katagiri et al. Org. Lett., 2012, 14(16):4062~4065. http://europepmc.org/abstract/MED/22845870
-
[11]
D Lehnherr, R Hallani, R Mcdonald et al. Org. Lett., 2012, 14(1):62~65. http://europepmc.org/abstract/MED/22126701
-
[12]
M Endou, Y Ie, Y Aso. Heterocycles, 2008, 76(2):1043~1048.
-
[13]
M L Tang, T Okamoto, Z Bao. J. Am. Chem. Soc., 2006, 128(50):16002~16003. doi: 10.1002/adfm.200701529/pdf
-
[14]
C Y Chiu, B Kim, A A Gorodetsky et al. Chem. Sci., 2011, 2(8):1480~1486. http://pubs.rsc.org/en/content/articlehtml/2011/sc/c1sc00156f
-
[15]
L Chen, S R Puniredd, Y Z Tan et al. J. Am. Chem. Soc., 2012, 134(43):17869~17872. http://europepmc.org/abstract/MED/23061521
-
[16]
Q Zhang, H Peng, J Wei et al. J. Am. Chem. Soc., 2014, 136(13):5057~5064. http://europepmc.org/abstract/med/24564649
-
[17]
L Chen, K S Mali, S R Puniredd et al. J. Am. Chem. Soc., 2013, 135(36):13531~13537. http://europepmc.org/abstract/med/25523150
-
[18]
O Possel, A M Van Leusen. Heterocycles, 1977, 7(1):77~80.
-
[19]
Y Liang, M G Schwab, L Zhi et al. J. Am. Chem. Soc., 2010. 132(42):15030~15037. doi: 10.1021/ja106612d
-
[20]
W Jin, Y Yamamoto, T Fukushima et al. J. Am. Chem. Soc., 2008. 130(29):9434~9440. http://europepmc.org/abstract/MED/18576635
-
[21]
N Miyaura, T Yanagi, A Suzuki. Synth. Commun., 1981. 11(7):513~519.
-
[22]
Z B Shifrina, M S Averina, A L Rusanov et al. Macromolecules, 2000. 33(10):3525~3529. doi: 10.1021/ma991369f
-
[1]
-

图 2 化合物1a、1b的甲苯溶液1×10-5mol/L)的紫外可见光谱
Figure 2 UV-Vis spectra of 1a and 1b (1×10-5mol/L) in toluene

图 3 化合物1a、1b的甲苯溶液1×10-5mol/L)的荧光光谱
Figure 3 Fluorescence spectra of 1a and 1b (1×10-5mol/L) in toluene
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
