Login In
Login In
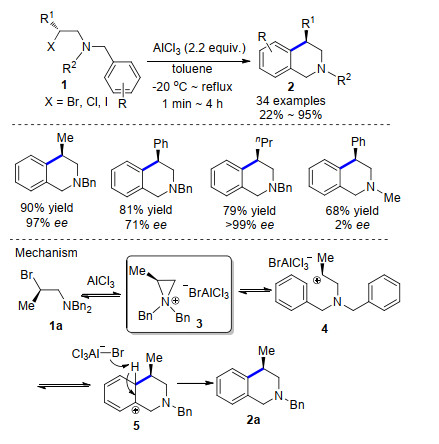
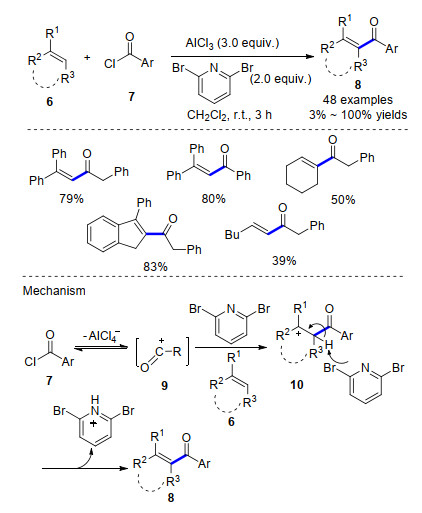
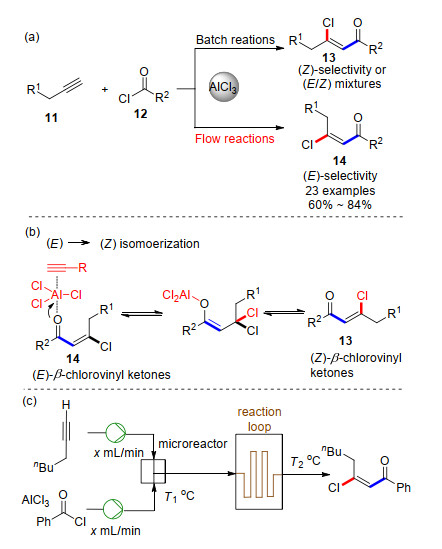
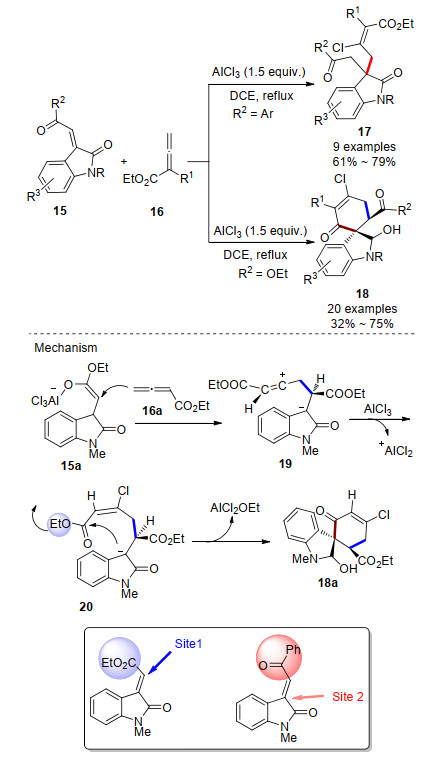
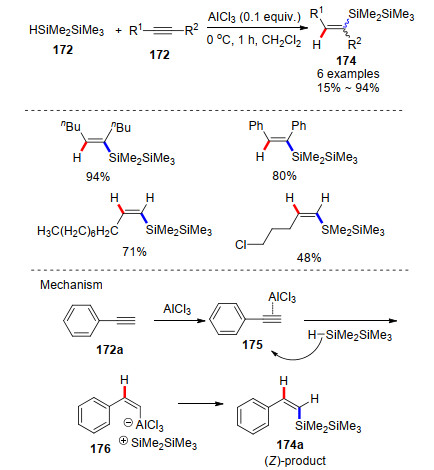
Recent Advances in AlCl3-Promoted Organic Reactions
- Corresponding author: Gao Wenchao, gaowenchao@tyut.edu.cn
Figures(36)
Citation:
Yuan Kangning, Zhao Yuying, Chang Honghong, Tian Jun, Gao Wenchao. Recent Advances in AlCl3-Promoted Organic Reactions[J]. Chinese Journal of Organic Chemistry,
;2020, 40(9): 2607-2625.
doi:
10.6023/cjoc202004042

Figures(36)





Chen, Z.; Qiu, X. L.; Yan, W.; Yang, H. N.; Ji, S. C.; Chen, M. H. Adv. Earth Sci. 2003, 18, 545 (in Chinese).
Ashkenazi, D. Technol. Forecast. Soc. Change 2019, 143, 101.
doi: 10.1016/j.techfore.2019.03.011
(a) Curtiss, L. A. Int. J. Quantum Chem. 1978, 14, 709.
(b) Bigelow, M. J. J. Chem. Educ. 1969, 46, 495.
(c) Aarset, K.; Shen, Q.; Thomassen, H.; Richardson, A. D.; Hedberg, K. J. Phys. Chem. A 1999, 103, 1644.
Pearson, R. G. J. Am. Chem. Soc. 1963, 85, 3533.
doi: 10.1021/ja00905a001
Yamamoto, Y. J. Org. Chem. 2007, 72, 7817.
doi: 10.1021/jo070579k
Zhao, Y.; Yang, Z.; Tang, L. Chin. J. Org. Chem. 2003, 23, 1219 (in Chinese).
Kürti, L.; Czakó, B. Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, San Diego, CA, 2005.
Chong, H. S.; Chen, Y. W. Org. Lett. 2013, 15, 5912.
doi: 10.1021/ol4013537
Rossiter, B. E.; Swingle, N. M. Chem. Rev. 1992, 92, 771.
doi: 10.1021/cr00013a002
Swaminathan, S.; Narayanan, K. V. Chem. Rev. 1971, 71, 429.
doi: 10.1021/cr60273a001
Beak, P.; Berger, K. R. J. Am. Chem. Soc. 1980, 102, 3848.
doi: 10.1021/ja00531a029
Shono, T.; Nishiguchi, I.; Sasaki, M.; Ikeda, H.; Kurita, M. J. Org. Chem. 1983, 48, 2503.
doi: 10.1021/jo00163a015
Hoffmann, H. M. R.; Tsushima, T. J. Am. Chem. Soc. 1977, 99, 6008.
doi: 10.1021/ja00460a028
Tanaka, S.; Kunisawa, T.; Yoshii, Y.; Hattori, T. Org. Lett. 2019, 21, 8509.
doi: 10.1021/acs.orglett.9b02688
Koo, H.; Kim, H. Y.; Oh, K. Org. Chem. Front. 2019, 6, 1868.
doi: 10.1039/C9QO00217K
Xu, S. B.; Li, C. J.; Jia, X. S.; Li, J. J. Org. Chem. 2014, 79, 11161.
doi: 10.1021/jo502209f
Devi, N. S.; Singh, S. J.; Devi, L. R.; Singh, O. M. Tetrahedron Lett. 2013, 54, 183.
doi: 10.1016/j.tetlet.2012.10.126
Chen, L.; Teng, W.; Geng, X. L.; Zhu, Y. F.; Guan, Y. H.; Fan, X. H. Appl. Organomet. Chem. 2017, 31, 3863.
doi: 10.1002/aoc.3863
Xu, X. M.; Lei, C. H.; Tong, S.; Zhu, J. P.; Wang, M. X. Org. Chem. Front. 2018, 5, 3138.
doi: 10.1039/C8QO00839F
Aleksić, M.; Bertoša, B.; Nhili, R.; Uzelac, L.; Jarak, I.; Depauw, S.; David-Cordonnier, M. H.; Kralj, M.; Tomić, S.; Karminski-Zamola, G. J. Med. Chem. 2012, 55, 5044.
doi: 10.1021/jm300505h
Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Chem. Rev. 2014, 114, 2587.
doi: 10.1021/cr400441m
Paul, S.; Shrestha, R.; Edison, T. N. J. I.; Lee, Y. R.; Kim, S. H. Adv. Synth. Catal. 2016, 358, 3050.
doi: 10.1002/adsc.201600429
Wang, Z.; Xue, L.; He, Y.; Weng, L.; Fang, L. J. Org. Chem. 2014, 79, 9628.
doi: 10.1021/jo501753p
Gao, W. C.; Liu, T.; Cheng, Y. F.; Chang, H. H.; Li, X.; Zhou, R.; Wei, W. L.; Qiao, Y. J. Org. Chem. 2017, 82, 13459.
doi: 10.1021/acs.joc.7b02498
Gao, W. C.; Cheng, Y. F.; Chang, H. H.; Li, X.; Wei, W. L.; Yang, P. J. Org. Chem. 2019, 84, 4312.
doi: 10.1021/acs.joc.9b00256
Yu, X. Z.; Shang, Y. Z.; Cheng, Y. F.; Tian, J.; Niu, Y.; Gao, W. C. Org. Biomol. Chem. 2020, 18, 1806.
doi: 10.1039/D0OB00050G
Zhang, L.; Li, X. J.; Wang, Z. W.; Zhao, J. W.; Wang, J. J.; Han, J. W.; Zhu, S. Z. Tetrahedron 2013, 69, 7975.
doi: 10.1016/j.tet.2013.07.004
Wang, X. N.; Krenske, E. H.; Johnston, R. C.; Houk, K. N.; Hsung, R. P. J. Am. Chem. Soc. 2015, 137, 5596.
doi: 10.1021/jacs.5b02561
Zhu, Y. Y.; Zhang, M. L.; Li, T.; Song, X. X. ChemistrySelect 2019, 4, 10838.
doi: 10.1002/slct.201903330
Cao, D. P.; Zhang, K. P.; An, R.; Xu, H.; Hao, S.; Yang, X. G.; Hou, Z.; Guo, C. Org. Lett. 2019, 21, 8948.
doi: 10.1021/acs.orglett.9b03260
Tskhovrebov, A. G.; Naested, L. C. E.; Solari, E.; Scopelliti, R.; Severin, K. Angew. Chem., Int. Ed. 2015, 54, 1289.
doi: 10.1002/anie.201410067
Zhu, Z. Q.; Bao, P.; Wang, T. T.; Huang, Z. Z. Chin. J. Chem. 2014, 32, 1176.
doi: 10.1002/cjoc.201400516
(a) Zhou, J. H.; Jiang, B.; Meng, F. F.; Xu, Y. H.; Loh, T. P. Org. Lett. 2015, 17, 4432.
(b) Yepes, D.; Pérez, P.; Jaquea, P.; Fernández, I. Org. Chem. Front. 2017, 4, 1390.
Masson, G; Lalli, C.; Benohoud, M.; Dagousset, G. Chem. Soc. Rev. 2013, 42, 902.
doi: 10.1039/C2CS35370A
Jian, W. J.; Qian, B.; Bao, H. L.; Li, D. L. Tetrahedron 2017, 73, 4039.
doi: 10.1016/j.tet.2016.10.049
Yang, G.; Shen, Y.; Li, K.; Sun, Y.; Hua, Y. J. Org. Chem. 2011, 76, 229.
doi: 10.1021/jo1020773
Yang, G.; Sun, Y.; Shen, Y.; Chai, Z.; Zhou, S.; Chu, J.; Chai, J. J. Org. Chem. 2013, 78, 5393.
doi: 10.1021/jo400554a
Shen, Y.; Chai, J.; Yang, G.; Chen, W.; Chai, Z. J. Org. Chem. 2018, 83, 12549.
doi: 10.1021/acs.joc.8b01798
Yang, G.; Wang, T.; Chai, J.; Chai, Z. Eur. J. Org. Chem. 2015, 2015, 1040.
doi: 10.1002/ejoc.201403325
Augustin, A. U.; Sensse, M.; Jones, P. G.; Werz, D. B. Angew. Chem., Int. Ed. 2017, 56, 14293.
doi: 10.1002/anie.201708346
Ge, J. J.; Yao, C. Z.; Wang, M. M.; Zheng, H. X.; Kang, Y. B.; Li, Y. D. Org. Lett. 2016, 18, 228.
doi: 10.1021/acs.orglett.5b03367
Wang, Z.; Yuan, Z. H.; Han, X. Y.; Weng, Z. Q. Adv. Synth. Catal. 2018, 360, 2078.
(a) Beck, B.; Magnin-Lachaux, M. Herdtweck, E.; Dömling, A. Org. Lett. 2001, 3, 2875.
(b) Kaim, L. E.; Gizolme, M.; Grimaud, L. Org. Lett. 2006, 8, 5021.
Lyu, L. Y.; Xie, H.; Mu, H. X.; He, Q. J.; Bian, Z. X.; Wang, J. Org. Chem. Front. 2015, 2, 815.
doi: 10.1039/C5QO00106D
Hu, Q. Q.; Liu, Y.; Deng, X. C.; Li, Y. J.; Chen, Y. F. Adv. Synth. Catal. 2016, 358, 1689.
doi: 10.1002/adsc.201600098
Dimitrov, P.; Emert, J.; Faust, R. Macromolecules 2012, 45, 3318.
doi: 10.1021/ma3003856
Chen, J.; Mao, J.-C.; He, Y.; Shi, D. Q.; Zou, B. Y.; Zhang, G. Q. Tetrahedron 2015, 71, 9496.
doi: 10.1016/j.tet.2015.10.030
Zhai, J. J.; Yao, Z. G.; Xu, F. Chin. J. Org. Chem. 2014, 34, 1639 (in Chinese).
Liu, G. Q.; Cui, B.; Xu, R.; Li, Y. M. J. Org. Chem. 2016, 81, 5144.
doi: 10.1021/acs.joc.6b00725
Kumar, K. S.; Meesa, S. R.; Rajeshama, B.; Bhaskera, B.; Ashfaq, M. A.; Khan, A. A.; Rao, S. S.; Pal, M. Bioorg. Med. Chem. 2012, 20, 1711.
doi: 10.1016/j.bmc.2012.01.012
Nakhi, A.; Archana, S.; Seerapu, G. P. K.; Chennubhotla, K. S.; Kumar, K. L.; Kulkarni, P.; Haldar, D.; Pal, M. Chem. Commun. 2013, 49, 6268.
doi: 10.1039/c3cc42840k
Shiro, D.; Fujiwara, S.; Tsuda, S.; Iwasaki, T.; Kuniyasu, H.; Kambe, N. Tetrahedron Lett. 2015, 56, 1531.
doi: 10.1016/j.tetlet.2015.01.096
Hachiya, I.; Nakamura, K.; Hara, M.; Sato, K.; Shimizu, M. J. Org. Chem. 2019, 84, 14770.
doi: 10.1021/acs.joc.9b02364
Tadeusz. S.; Jagodziski, T. S. Chem. Rev. 2003, 103, 197.
doi: 10.1021/cr0200015
Alla, S. K.; Sadhu, P.; Punniyamurthy, T. J. Org. Chem. 2014, 79, 7502.
doi: 10.1021/jo501216h
Kumar, K.; Konar, D.; Goyal, S.; Gangar, M.; Chouhan, M.; Rawal, R. K.; Nair, V. A. ChemistrySelect 2016, 1, 3228.
doi: 10.1002/slct.201600601
Liu, X.; Zhang, S. B.; Dong, Z. B. Eur. J. Org. Chem. 2018, 39, 5406.
Yamamoto, Y. J. Org. Chem. 2007, 72, 7817.
doi: 10.1021/jo070579k
Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2017, 139, 6074.
doi: 10.1021/jacs.7b02553
Nikonov, G. I. ACS Catal. 2017, 7, 7257.
doi: 10.1021/acscatal.7b02460
Kato, N.; Tamura, Y.; Kashiwabara, T.; Sanji, T.; Tanaka, M. Organometallics 2010, 29, 5274.
doi: 10.1021/om100376d
Yongqing Kuang , Jie Liu , Jianjun Feng , Wen Yang , Shuanglian Cai , Ling Shi . Experimental Design for the Two-Step Synthesis of Paracetamol from 4-Hydroxyacetophenone. University Chemistry, 2024, 39(8): 331-337. doi: 10.12461/PKU.DXHX202403012
Xinxin YU , Yongxing LIU , Xiaohong YI , Miao CHANG , Fei WANG , Peng WANG , Chongchen WANG . Photocatalytic peroxydisulfate activation for degrading organic pollutants over the zero-valent iron recovered from subway tunnels. Chinese Journal of Inorganic Chemistry, 2025, 41(5): 864-876. doi: 10.11862/CJIC.20240438
Ning CHEN , Jingle CHEN , Hongyuan ZHU , Huali CHEN , Liguang WU , Ting WANG . Mechanism and performance regulation of Co/Zr-doped mesoporous TiO2 catalysts in activating sodium persulfate for tetracycline degradation. Chinese Journal of Inorganic Chemistry, 2026, 42(3): 507-518. doi: 10.11862/CJIC.20250275
Dan LUO , Xingcheng LIU , Dong LI , Tong CHANG . Metal-support interaction effects on CO activation over Con/SiO2 catalysts. Chinese Journal of Inorganic Chemistry, 2025, 41(11): 2337-2344. doi: 10.11862/CJIC.20250003
Hui Wang , Abdelkader Labidi , Menghan Ren , Feroz Shaik , Chuanyi Wang . Recent Progress of Microstructure-Regulated g-C3N4 in Photocatalytic NO Conversion: The Pivotal Roles of Adsorption/Activation Sites. Acta Physico-Chimica Sinica, 2025, 41(5): 100039-0. doi: 10.1016/j.actphy.2024.100039
Anqi LI , Wenjing YANG , Xueming LI , Yanfong REN . Performance and mechanism of a foam Ti/FeCo-Fe2O3-CoFe2O4/SnO2-Sb anode for synergistic activation of peroxymonosulfate toward degradation of organic pollutants. Chinese Journal of Inorganic Chemistry, 2026, 42(5): 944-958. doi: 10.11862/CJIC.20250323
Yongxin LIU , Xingchen LI , Hongjia LIU , Danni LI , Tao ZHANG , Xi CHEN . Enhancement effect of Fe3O4 conversion to MIL-100(Fe) on activation of persulfate for degradation of antibiotic. Chinese Journal of Inorganic Chemistry, 2025, 41(12): 2503-2513. doi: 10.11862/CJIC.20250169
Jingping Li , Suding Yan , Jiaxi Wu , Qiang Cheng , Kai Wang . Improving hydrogen peroxide photosynthesis over inorganic/organic S-scheme photocatalyst with LiFePO4. Acta Physico-Chimica Sinica, 2025, 41(9): 100104-0. doi: 10.1016/j.actphy.2025.100104
.
CCS Chemistry | 超分子活化底物为自由基促进高效选择性光催化氧化
. CCS Chemistry, 2025, 7(10.31635/ccschem.025.202405229): -.Yuanqing Wang , Yusong Pan , Hongwu Zhu , Yanlei Xiang , Rong Han , Run Huang , Chao Du , Chengling Pan . Enhanced Catalytic Activity of Bi2WO6 for Organic Pollutants Degradation under the Synergism between Advanced Oxidative Processes and Visible Light Irradiation. Acta Physico-Chimica Sinica, 2024, 40(4): 2304050-0. doi: 10.3866/PKU.WHXB202304050
Xiaogang YANG , Xinya ZHANG , Jing LI , Huilin WANG , Min LI , Xiaotian WEI , Xinci WU , Lufang MA . Synthesis, structure, and photoelectric properties of Zinc(Ⅱ)-triphenylamine based metal-organic framework. Chinese Journal of Inorganic Chemistry, 2025, 41(10): 2078-2086. doi: 10.11862/CJIC.20250167
Hui-Ying Chen , Hao-Lin Zhu , Pei-Qin Liao , Xiao-Ming Chen . Integration of Ru(Ⅱ)-Bipyridyl and Zinc(Ⅱ)-Porphyrin Moieties in a Metal-Organic Framework for Efficient Overall CO2 Photoreduction. Acta Physico-Chimica Sinica, 2024, 40(4): 2306046-0. doi: 10.3866/PKU.WHXB202306046
Peiran ZHAO , Yuqian LIU , Cheng HE , Chunying DUAN . A functionalized Eu3+ metal-organic framework for selective fluorescent detection of pyrene. Chinese Journal of Inorganic Chemistry, 2024, 40(4): 713-724. doi: 10.11862/CJIC.20230355
Chengpeng Liu , Yinxia Fu . Design and Practice of Ideological and Political Education for the Public Elective Course “Life Chemistry Experiment” in Universities. University Chemistry, 2024, 39(10): 242-248. doi: 10.12461/PKU.DXHX202404064
Fa Wang , Yu Chen , Hui Chao . Ruthenium(II) Complexes as Photoactivated Chemo-Prodrugs for Hypoxic Tumor Therapy. University Chemistry, 2025, 40(7): 200-212. doi: 10.12461/PKU.DXHX202410024
Xue-Peng Zhang , Yuchi Long , Yushu Pan , Jiding Wang , Baoyu Bai , Rui Ding . 定量构效关系方法学习探索:以钴卟啉活化氧气为例. University Chemistry, 2025, 40(8): 345-359. doi: 10.12461/PKU.DXHX202410107
Xuejie Wang , Guoqing Cui , Congkai Wang , Yang Yang , Guiyuan Jiang , Chunming Xu . Research Progress on Carbon-based Catalysts for Catalytic Dehydrogenation of Liquid Organic Hydrogen Carriers. Acta Physico-Chimica Sinica, 2025, 41(5): 100044-0. doi: 10.1016/j.actphy.2024.100044
Xiaohang JIN , Qi LIU , Jianping LANG . Room‑temperature solid‑state synthesis, structure, and third‑order nonlinear optical properties of phosphine‑ligand‑protected silver thiolate clusters. Chinese Journal of Inorganic Chemistry, 2025, 41(8): 1505-1512. doi: 10.11862/CJIC.20250125
Chenxu Gong , Weizhen Wang , Ruiying Zhang , Wenfeng Wang , Yuanming Li , Yaofeng Yuan , Keyin Ye . Computational Chemistry-Assisted Organic Structure Analysis (CCAOSA): A Case Study of Propeller-Shaped Hexabenzotriphenylene. University Chemistry, 2026, 41(4): 438-446. doi: 10.12461/PKU.DXHX202503076
Xing Yan , Yetai Cheng , Yixun Shu , Luyao Yang , Weidong Wang , Xinlu Bai , Ya-Nan Chen , Hao Lu , Zhishan Bo , Yahui Liu . Highly efficient and stable organic solar cells based on dimeric non-fused ring acceptors as the third component. Acta Physico-Chimica Sinica, 2026, 42(7): 100228-. doi: 10.1016/j.actphy.2025.100228
 DownLoad:
DownLoad: