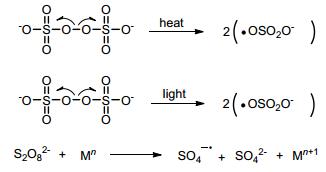
 图式1
过硫酸盐分解生成硫酸根自由基负离子
Scheme1.
Persulfates decomposed to the sulfate radical anions
图式1
过硫酸盐分解生成硫酸根自由基负离子
Scheme1.
Persulfates decomposed to the sulfate radical anions
引用本文:
赵景峰, 段新华, 郭丽娜. 过硫酸盐促进的自由基反应进展[J]. 有机化学,
2017, 37(10): 2498-2511.
doi:
10.6023/cjoc201705003

Citation: Zhao Jingfeng, Duan Xinhua, Guo Li'na. Recent Advances in Persulfates-Promoted Radical Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2498-2511. doi: 10.6023/cjoc201705003
Citation: Zhao Jingfeng, Duan Xinhua, Guo Li'na. Recent Advances in Persulfates-Promoted Radical Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2498-2511. doi: 10.6023/cjoc201705003
过硫酸盐促进的自由基反应进展
摘要:
过硫酸盐作为廉价、环境友好、易操作的强氧化剂,在有机合成中具有广泛的应用.众所周知,过硫酸盐在加热、光照或过渡金属还原条件下可分解产生硫酸根自由基负离子(SO4-·).研究表明,该自由基负离子具有很强的单电子氧化性,可以氧化各种中性分子或负离子产生相应的自由基活性中间体.这些新生成的活性中间体可以进行一系列化学转化,从而生成结构多样的、有用的化合物.对近几年过硫酸盐促进的自由基反应进行了总结.分五部分:第一、二部分着重阐述过硫酸盐促进的自由基环化以及碳-氢键官能团化反应;第三部分介绍可见光辅助的过硫酸盐促进的有机反应;第四部分简述过硫酸盐促进的其他自由基反应.最后,对过硫酸盐促进的自由基反应进行了总结和展望.
English
Recent Advances in Persulfates-Promoted Radical Reaction
Abstract:
As cheap, environmentally friendly, easily handled strong oxidants, persulfates have been widely used in organic synthesis. It is well known that persulfates may decompose to sulfate radical anions (SO4-·) under heating, light irradiation or transition-metal reductive conditions. Studies indicate that the sulfate radical anion is a powerful single-electron oxidant, which can oxidize a variety of neutral molecules and anions to give the corresponding radicals. The new generated radicals can further undergo a series of chemical transformations to provide structurally diverse and useful compounds. Recent radical reactions promoted by persulfates are summarized. The full text contains five parts. In the first and second parts, recent advance in the radical cyclization reactions and functionalization of C-H bonds promoted by persulfates is discussed. The third part introduced the persulfates-mediated photocatalytic reactions. The fourth part emphasized persulfates-promoted other free radical reactions. Finally, some perspectives on the future development of this chemistry are given.
-
Key words:
- persulfates
- / sulfate radical anions
- / radical
- / cyclization
- / C—H functionalization
-
自由基反应是有机化学中非常重要的一类反应.这类反应通过单电子转移(SET)的方式实现新化学键的形成.自由基反应因具有反应活性好、反应条件温和、官能团容忍性好等优点, 已经成为有机合成中构筑化学键的有效方法之一[1~3].近几年, 无过渡金属参与的自由基反应取得了长足的发展, 其在碳-碳键及碳-杂键的构筑中得到了广泛的应用.同时, 过硫酸盐参与的离子型反应也已经有大量综述文献报道[4].在这篇综述里面, 我们主要对过硫酸盐促进的自由基反应进行了归纳和总结.过硫酸盐作为氧化剂、辅助剂等参与的其他有机反应以及过硫酸盐促进的离子型反应不是本文讨论的范畴.
过硫酸盐包括一硫酸盐(peroxymonosulfate或oxone)和二硫酸盐(peroxydisulfate或persulfate), 一般指二硫酸盐, 是一种常见的氧化剂.过硫酸盐具有低毒、价格低廉、易于操作等优点且具有单电子氧化的特性, 因此得到了化学家们的广泛关注.过硫酸盐性质较稳定, 但是通过光照、加热或过渡金属还原等方式, 其很容易分解为活性较高的硫酸根自由基负离子(SO4-·).SO4-·的标准氧化电位为E0=2.5~3.1 V, 强于其他氧化剂且寿命较长[5](Scheme 1).过硫酸盐早期主要作为干洗漂白剂使用, 后来作为自由基引发剂被广泛地应用于工业中合成树脂和橡胶.此外, 过硫酸盐也可用于有机污染物的降解[5d].近些年来, 过硫酸盐促进的自由基反应相继被报道, 使其在各类有机小分子的合成中大放异彩.
图式1
过硫酸盐分解生成硫酸根自由基负离子
Scheme1.
Persulfates decomposed to the sulfate radical anions
20世纪70年代, Kochi等[6]对Ag(Ⅰ)/过硫酸盐体系下烷基羧酸脱羧产生烷基自由基的反应机理进行了深入的研究.在此基础上, 意大利化学家Minisci利用该体系实现了各类杂环化合物的烷基化反应(即Minisci反应).与经典的Friedel-Crafts反应相比, Minisci反应能有效地合成各类烷基取代缺电子杂环化合物.然而, Minisci反应也存在着反应条件较苛刻、昂贵金属催化剂的使用以及选择性较差等缺点.因此发展廉价、高选择性、无过渡金属参与的自由基反应无疑具有重要的意义.近几年的研究表明, 过硫酸盐本身就可促进自由基的偶联和环化反应, 无需过渡金属催化剂的参与.本综述按照反应类型将近年来过硫酸盐促进的自由基反应分为四类进行了概述.
1 过硫酸盐促进的自由基环化反应
20世纪30年代拜耳斯坦有机化学手册中记载的杂环化合物种类, 就已占当时已发现的有机化合物的三分之一. 1971年, 已知的数百万种有机化合物中有一半以上是杂环化合物.由于杂环化合物与医药、材料、生命科学等领域有密切的关系, 它们很多具有良好的生物和药理活性, 并且在合成染料、聚合物材料方面也有着广泛的应用价值.因此杂环化合物的合成一直倍受化学工作者的关注, 是有机合成研究的重要内容之一[7].近年来随着绿色化学的兴起, 人们对有机合成化学的要求也越来越高.串联反应由于其在“一锅反应中”可以连续地形成多个化学键, 成为有机合成中提高反应效率的理想策略之一.目前, 串联自由基加成/环化策略已被成功地应用到各类氮杂及氧杂环状化合物的合成中, 成为简洁、高效地合成杂环化合物的绿色新途径之一.
1.1 羟吲哚的合成
众所周知, 羟吲哚是许多天然生物碱的核心骨架如SPX-5、Horsfiline、Rhynchophylline等, 这些化合物具有良好的生物活性并被广泛地应用于医药行业(图 1).例如, Rhynchophylline能够抑制外周血管收缩, 使血管阻力和血压降低的同时, 还具有抗血小板聚集和抗血栓的作用.鉴于这类化合物独特的化学结构和广谱生物活性以及潜在药用价值, 发展高效合成羟吲哚化合物的新方法一直是有机合成化学研究的热点领域之一[7b, 7c].
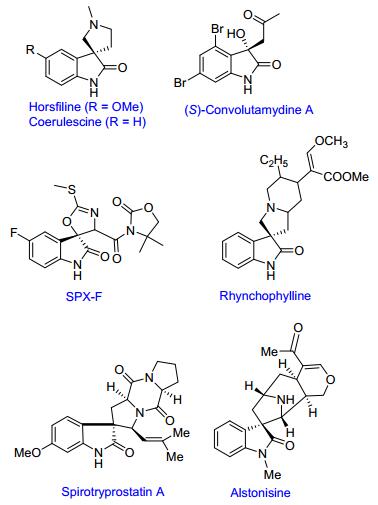 图1
含羟吲哚骨架的天然产物和药物分子
Figure1.
Natural products and pharmaceutical molecules containing oxindole skeletons
图1
含羟吲哚骨架的天然产物和药物分子
Figure1.
Natural products and pharmaceutical molecules containing oxindole skeletons
近年来, 丙烯酰胺的烯烃双官能团化反应已经发展成为合成羟吲哚化合物的有效方法之一.不同类型的自由基前体化合物均可与N-苯基丙烯酰胺高效地发生串联自由基加成/环化反应, 生成各种官能团化的羟吲哚化合物[8a~8c]. 2013年, Yang等[7d]在无过渡金属的存在下, 利用廉价易得的K2S2O8作为氧化剂, 以NaNO2作为硝基自由基(•NO2)前体, 实现了NaNO2与N-苯基丙烯酰胺的自由基环化反应, 合成一系列硝乙基取代的羟吲哚化合物(Scheme 2).该反应具有良好的底物普适性和官能团容忍性.此外, 作者还对产物中的硝基进行了衍生化, 制备出了不同官能团化的羟吲哚化合物.后来, Zhang等[8e]利用类似的策略以NaN3为叠氮源, 实现了N-苯基丙烯酰胺的自由基环化叠氮化反应, 成功地将叠氮基引入到了羟吲哚分子中(Eq. 1).
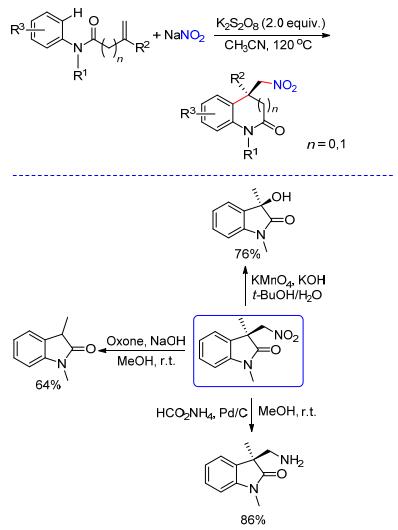 图式2
过硫酸钾促进的N-苯基丙烯酰胺与亚硝酸钠的环化反应
Scheme2.
K2S2O8-mediated nitrification of N-phenyl acrylamide
图式2
过硫酸钾促进的N-苯基丙烯酰胺与亚硝酸钠的环化反应
Scheme2.
K2S2O8-mediated nitrification of N-phenyl acrylamide
在绿色合成理念的引导下, 我们小组希望探索基于C—H键官能团化的方式来直接构建C—C键与C—杂键的新型、理想的有机反应. 2013年, 我们小组[9a]在N-苯基丙烯酰胺与1, 3-二羰基化合物的自由基环化反应的基础上, 以化学当量的K2S2O8作为氧化剂, 成功地实现了羟甲基取代丙烯酰胺与1, 3-二羰基化合物的双环化反应[9b].我们巧妙地利用二羰基化合物的烯醇异构化, 并结合自由基加成/环化反应, 轻松地合成了难以制备且具有重要应用价值的螺环吲哚-2-酮类化合物(Eq. 2).反应中, 我们一步构建了两个C—C键和一个C—O键.通过对反应机理的研究, 我们提出了自由基加成、分子内环化、分子内脱水的历程.令人满意的是, 该反应具有原料易得、反应条件温和、原子利用率高等优点, 而且反应中各种官能团如卤素、氰基、酯基等均能兼容, 这为氧杂螺羟吲哚进一步的衍生化提供了可能.
硫三氟甲基(SCF3)具有强负电子以及良好的亲油性, 在药物、农药的分子中引入SCF3能够极大地改进这些化合物的活性.因此, 发展高效的引入SCF3的反应备受合成化学家的关注. 2014年Wang等[10a]报道了K2S2O8促进的N-芳基丙烯酰胺与硫三氟甲基试剂的串联环化反应(Scheme 3).作者认为在K2S2O8的存在下, 硫三氟甲基银(AgSCF3)被氧化生成Ag(Ⅱ)SCF3中间体, 该中间体经过单电子转移(SET)产生硫三氟甲基自由基(•SCF3), 进而完成后续的环化反应. AgSCF3在该反应中不仅作为硫三氟甲基试剂使用, 还起到氧化的作用.该反应中选择AgSCF3至关重要, 类似的试剂CuSCF3并不能得到预期的目标产物.此外, Wang等[10b]利用K2S2O8单电子氧化实现了廉价易得的Langlois (CF3SO2Na)试剂与N-苯基丙烯酰胺的自由基环化, 高产率地得到三氟乙基取代的羟吲哚.
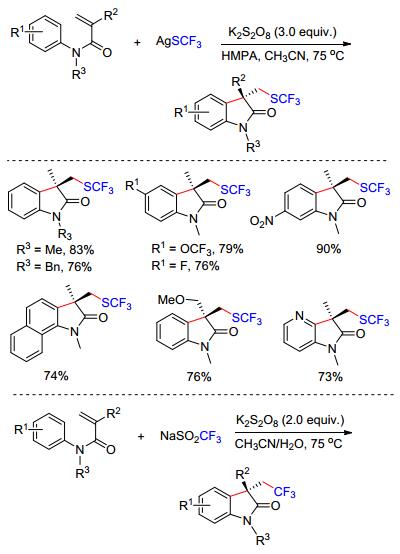 图式3
过硫酸钾促进的N-苯基丙烯酰胺与AgSCF3的环化反应
Scheme3.
K2S2O8-mediated cyclization reaction of N-phenyl acrylamide with AgSCF3
图式3
过硫酸钾促进的N-苯基丙烯酰胺与AgSCF3的环化反应
Scheme3.
K2S2O8-mediated cyclization reaction of N-phenyl acrylamide with AgSCF3
芳基亚硫酸在K2S2O8的氧化下产生的磺酰基自由基(•SO2Ar)也可以与N-苯基丙烯酰胺进行自由基加成/环化, 得到磺化的吲哚.与其他的磺化试剂如磺酰氯、磺酰肼等相比, 该磺化试剂表现出了极高的原子利用率. 2014年Wang课题组[11a]使用芳基亚硫酸盐, 在过硫酸钾的促进下高效合成了一系列磺酰基取代的羟吲哚类化合物(Scheme 4). 2016年, Guo等[11b]使用二硫化物作为磺化试剂也实现了磺化吲哚的合成. K2S2O8在反应中起到氧化二硫化物的作用, 作者通过O18标记实验证实产物中的氧来源于水和氧化剂.并且此反应能放大量到克级别, 说明该反应具有良好的实用性(Scheme 4).
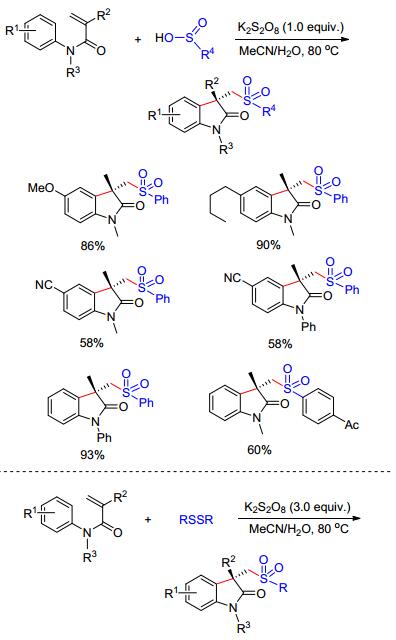 图式4
K2S2O8促进的N-苯基丙烯酰胺磺化反应
Scheme4.
K2S2O8-mediated sulfonation of N-phenyl acrylamide
图式4
K2S2O8促进的N-苯基丙烯酰胺磺化反应
Scheme4.
K2S2O8-mediated sulfonation of N-phenyl acrylamide
有机含膦化合物由于其具有不同的生物活性和光电性能, 近年来受到了人们广泛地关注. 2013年, Yang等[12a]报道了银催化的二芳基氧膦与N-苯基丙烯酰胺的自由基环化反应.后来, Li等[12b]发现仅在化学当量的K2S2O8氧化下, 该反应也可以顺利进行, 从而为合成这类具有潜在的生物活性和光电性能的含膦羟吲哚化合物提供了更为简洁和实用的方法(Eq. 3).
张力环烷醇是一类非常重要有机合成的中间体.研究表明, 在单电子氧化的条件下, 此类化合物可发生自由基的开环-重排反应, 生成含有羰基官能团的碳自由基. 2016年, 我们小组[13a]以过硫酸盐为氧化剂, 首次成功地实现了无过渡金属催化的环丙醇开环与N-苯基丙烯酰胺关环的串联反应, 得到了一系列难以制备的含羰基取代基的羟吲哚化合物.除了环丙醇外, 三级环丁醇也可发生类似的自由基开环/环化反应.然而, 1-芳基环丁醇参与的反应产率较低.这是因为其自身可发生分子内的自由基环化得到1-四氢萘酮的副产物(Eq. 4).与此同时, Yang等[13b]利用银催化的反应体系也实现了上述反应.
过硫酸盐作为廉价、易得的单电子氧化剂在羟吲哚的合成上表现出良好的活性.多种类型的化合物在无过渡金属催化下, 仅以化学当量过硫酸盐为氧化剂就可产生不同类型的活性自由基中间体.这些自由基中间体可进一步被N-苯基丙烯酰胺所捕获, 从而为合成各种不同取代基的羟吲哚环化合物提供了简洁、实用的新方法.
1.2 过硫酸盐促进的其他杂环及碳环化合物的合成
除羟吲哚外, 过硫酸盐促进的自由基环化反应还可以用来合成其他的杂环化合物. 2014年, Laha等[14a]利用过硫酸钾氧化实现二苯并二氮杂的碳-碳及碳-氮键的裂解反应, 得到一系列取代的酚嗪产物, 该反应可同时断裂C—N和C—C键, 并分子内形成新的C—N键(Eq. 5).通过对照试验, 作者认为该反应是通过过硫酸钾氧化二苯并二氮杂产生苄基自由基、碳-氮键裂解生成芳胺自由基、分子内自由基环化脱羧的历程进行的.苄基型C(sp3)—H的氮化/氧化被证明是向含苄基位置引入含氮/氧官能团的有效方法之一.随后, 该课题组使用过硫酸钾氧化N-芳基苄胺的方法, 还实现了苯甲脒和苯并噁嗪等杂环化合物的合成.对芳基亚胺或亚胺物质的亲核加成是该反应的决速步骤(Eq. 6)[14b].
在K2S2O8的氧化下, α-酮酸钾脱羧产生的酰基自由基可以与邻胺基苯甲酰胺发生缩合/环化生成各种取代的喹唑啉.在相同的条件下, α-酮酸钾还可以与邻胺基硫酚、邻氨基苯酚、邻苯二胺、邻氨基苯甲酸发生作用, 得到各类含氮杂环化合物[14c].利用该方法该课题组以商业上易得的吡唑和邻乙氧基苯甲酰甲酸盐为起始原料, 经过两步以53%的总收率制备了药物西地那非(Eq. 7).
二氢呋喃是一类非常重要的环状化合物, 不仅广泛存在于天然产物及药物分子中, 而且也是非常重要的有机合成中间体.在我们前期利用自由基反应合成羟吲哚的基础上, 我们发现在过硫酸钾的氧化下1, 3二羰基化合物也可与苯乙烯发生自由基加成-环化反应, 生成二氢呋喃类化合物[15].当使用α-甲基苯乙烯为原料时, 我们可以以较好的收率得到含有季碳中心的二氢呋喃.经过反应机理的研究, 我们认为该反应有可能是经过自由基加成/阳离子环化的历程进行的(Eq. 8).
苯并噁嗪是一类非常重要的含氧、氮的六元杂环化合物.其可发生聚合反应形成类似酚醛树脂的含氮网络结构, 这种苯并噁嗪树脂作为一种新型的热固性树脂, 具有优良的电绝缘性、高的热氧稳定性和良好的阻燃性.此外, 该骨架广泛存在于一些药物分子中, 如抗焦虑和抗惊厥的药物“Etifoxine”. 2015年, 我们小组[16a]发现在K2S2O8的氧化下, 廉价、易得的硫氰酸钾(KSCN)可以生成亲电的硫氰基自由基(•SCN), 该自由基可以高选择性的与烯基取代的酰胺发生加成/环化反应, 得到各种硫氰基取代的苯并噁嗪.利用该策略, 我们通过改变烯基酰胺的结构, 还成功地制备出一系列含亚胺基-异苯并呋喃类化合物(Scheme 5).在此基础上, 我们又实现了硫氰基取代二氢呋喃和苯并内脂类化合物的合成[16b].以上反应均表现出反应条件温和、底物适用范围广及官能团容忍性好等优点, 为合成含硫氰基的杂环化合物提供了新思路.值得一提的是, 产物分子中的SCN基团可以通过水解、还原、环加成等化学转化来制备结构多样的含硫化合物.因此, 该反应也为C—S键的构筑及结构多样性含硫化合物的合成提供了高效、绿色的新方法.
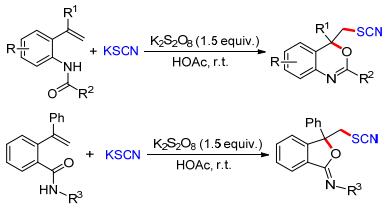 图式5
K2S2O8促进的烯基酰胺硫氰化反应
Scheme5.
K2S2O8-mediated tandem radical thiocyanooxygenation of olefinic amides
图式5
K2S2O8促进的烯基酰胺硫氰化反应
Scheme5.
K2S2O8-mediated tandem radical thiocyanooxygenation of olefinic amides
2016年, Guan和Gao等[17]利用K2S2O8促进的自由基加成环化的策略实现了双分子烯胺的偶联/环化反应, 从而得到一系列具有重要应用价值的对称多取代吡咯化合物.该反应主要是通过K2S2O8单电子氧化烯胺产生胺基自由基正离子对另一分子烯酰胺加成, 脱除质子和氨后得到预期的吡咯.令人满意的是, 将该反应放大到10 mmol后, 仍然可以以很高的分离产率得到目标产物.因此, 该反应具有良好的潜在实用性(Eq. 9).
除了杂环化合物外, Shi等[17]利用过硫酸盐促进的自由基环化的策略实现了碳环化合物的合成.在化学当量Na2S2O8的氧化下, AgSCF3产生的亲电硫三氟甲基自由基(•SCF3)可对芳基取代的甲叉基环丙烷进行选择性的加成, 随后经开环、分子内环化得到硫三氟甲基取代的二氢萘产物.遗憾的是, 由于甲叉基环丙烷不稳定且在该体系中容易被氧化, 因此产率不是十分理想.作者发现, 在当采用4 equiv. Na2S2O8为氧化剂时, 二氢萘产物可脱氢生成芳构化的产物. Na2S2O8参与了两步氧化过程:第一步氧化AgSCF3产生SCF3•自由基, 第二步氧化反应中间体, 使得该中间体易于脱氢芳构化(Scheme 6).
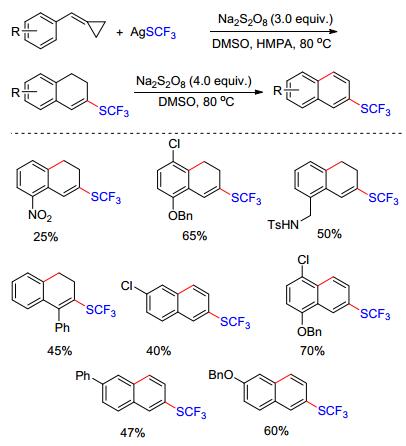 图式6
K2S2O8促进的甲叉基环丙烷的硫三氟甲基化反应
Scheme6.
K2S2O8-mediated trifluoromethylthiolation of methylenecyclopropanes
图式6
K2S2O8促进的甲叉基环丙烷的硫三氟甲基化反应
Scheme6.
K2S2O8-mediated trifluoromethylthiolation of methylenecyclopropanes
2 过硫酸盐促进的碳-氢键官能团化反应
2.1 sp2杂化碳-氢键官能团化反应
碳-氢键是有机化合物中最普遍的化学键.利用过渡金属催化的碳-氢键直接官能团化反应构筑碳-碳及碳-杂键是当今有机合成化学研究的前沿领域之一.相比传统的偶联方法, 碳-氢键官能团化反应无需预先活化反应底物, 从而提高了反应的原子利用率、缩短了反应步骤符合当前绿色化学发展的要求.在这一领域中, Minisci发展的对杂环化合物的碳-氢键直接官能团化的反应(Minisci反应)为碳-碳键的构建提供了有效的方法[6].近年来的研究表明, 无需过渡金属银催化, 过硫酸盐本身就可促进Minisci类型的反应发生.
2013年, Liu等[19]发现在K2S2O8促进下喹喔啉或苯并噻唑可以与甲醇发生自由基的偶联反应, 一步得到2-喹喔啉或2-苯并噻唑甲醛二甲缩醛(Scheme 7).该缩醛类化合物在酸性条件下, 可进一步转化为醛基, 这为在杂环化合物中引入醛基提供了简洁、方便的新方法.初步的机理研究表明, 该反应经历了关键的中间体——Minisci羟甲基化产物Ⅰ.羟甲基自由基(•CH2OH)来源于过硫酸盐对甲醇的氧化.化合物Ⅰ在K2S2O8及氧气存在下, 经多步转化形成醛Ⅱ, 其在酸性条件下进一步与甲醇作用最终形成二甲缩醛产物.
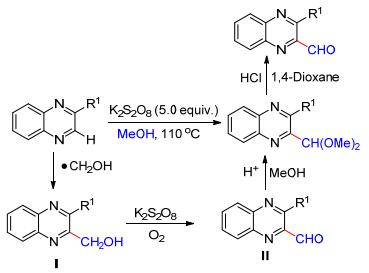 图式7
K2S2O8促进的杂环化合物与甲醇的官能团化反应
Scheme7.
K2S2O8-mediated cross-coupling of heterocycles with methanol
图式7
K2S2O8促进的杂环化合物与甲醇的官能团化反应
Scheme7.
K2S2O8-mediated cross-coupling of heterocycles with methanol
在季铵盐的催化下, 2-苯基苯甲醛在K2S2O8的氧化下可发生分子内环化反应, 生成不同取代的芴酮(Eq. 10)[20a].该反应中四乙基溴化铵(TEAB)的存在有利于使过硫酸盐产生硫酸根自由基负离子(SO4-·).该自由基负离子氧化2-苯基苯甲醛产生酰基自由基中间体, 紧接着发生分子内自由基环化得到预期的目标产物.相比于传统的过渡金属催化和酸催化的Friedel-Crafts方法, 该反应具有反应催化体系廉价、容易操作、实用性强等优点.在类似的反应体系下, Prabhu等[20b]还实现了醛与杂芳环化合物如异喹啉、喹啉及喹喔啉的交叉脱氢偶联(CDC偶联), 为杂环化合物的酰化反应提供了新方法. K2S2O8与四丁基卤化铵盐同样对该反应是缺一不可的, 并且在该廉价的体系下, 无论是芳香醛, 还是脂肪醛都能很好地参与反应(Scheme 8).
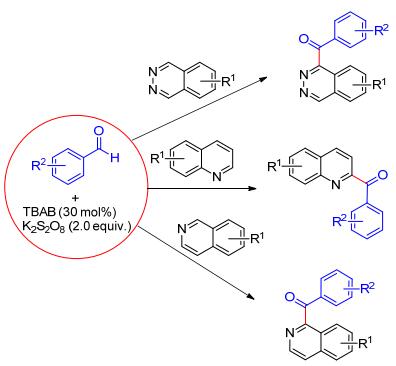 图式8
K2S2O8促进的异喹啉、喹啉、喹喔啉分子间酰化反应
Scheme8.
K2S2O8-mediated intermolecular acylation of isoquinolines, quinolines and quinazines
图式8
K2S2O8促进的异喹啉、喹啉、喹喔啉分子间酰化反应
Scheme8.
K2S2O8-mediated intermolecular acylation of isoquinolines, quinolines and quinazines
2014年Jeganmohan等[21a]发现, 在K2S2O8的促进下苄基酮可以与富电子芳烃发生偶联反应, 得到不同取代的α-二芳基苄基酮.值得一提的是, 该反应条件温和, 且不需要过渡金属催化剂.作者认为, K2S2O8在反应中首先起到了氧化苄基酮产生α-羰基苄基自由基Ⅰ的作用.该活性中间体在酸性条件下与氧气反应, 随后发生Hock重排得到相应的碳正离子Ⅱ.该碳正离子中间体再与两分子的芳烃作用的最终产物(Scheme 9).后来, 该课题组利用K2S2O8氧化还实现了两种不同酚的交叉氧化偶联反应, 高选择性地得到一系列不对称的联二酚产物[21b].该反应通过阳离子自由基中间体进行, 加入带负电荷的抗衡离子如Bu4N+HSO3-可以与阳离子酚自由基中间体配位并使其稳定, 这有助于反应朝交叉偶联方向进行而非自偶联.并且测量和比较不同类型的酚、萘化合物后发现, 只有氧化还原电位低于0.7 V的酚才能与SO4-·反应产生相应的阳离子酚自由基中间体, 高于0.7 V的酚并不能发生反应(Eq. 11).
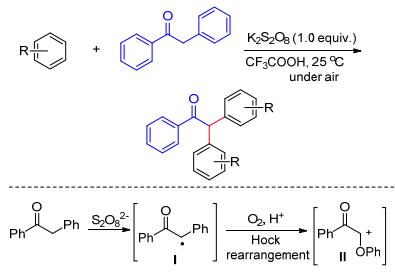 图式9
K2S2O8促进苄基酮的碳-碳键裂解反应
Scheme9.
K2S2O8-mediated carbon-carbon bond cleavage of benzyl ketones
图式9
K2S2O8促进苄基酮的碳-碳键裂解反应
Scheme9.
K2S2O8-mediated carbon-carbon bond cleavage of benzyl ketones
醌及其衍生物由于其独特的生物活性和光电性能使其在药物、染料和光敏材料等领域具有广泛应用价值.醌类衍生物最简单的制备方法是以易得的醌为起始原料, 通过碳-氢键的直接官能团化得到.在Baran工作[22a]的基础上, Ilangovan等[22b]发现只需要在过硫酸盐的氧化下, 芳基硼酸就可以与醌发生碳-氢键的直接官能团化, 得到一系列芳基取代的醌(Scheme 10).作者认为反应有可能是经过过硫酸盐氧化芳基硼酸产生芳基自由基中间体(•Ar)进行的.该反应为醌类化合物的衍生化提供了简单、实用的新方法.
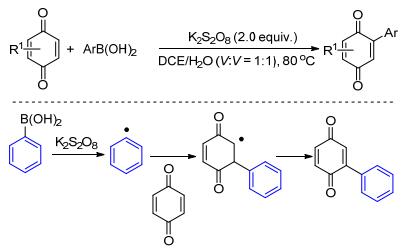 图式10
K2S2O8促进的醌与芳基硼酸的芳基化反应
Scheme10.
K2S2O8-mediated arylation of quinone and aryl bonric acid
图式10
K2S2O8促进的醌与芳基硼酸的芳基化反应
Scheme10.
K2S2O8-mediated arylation of quinone and aryl bonric acid
三氟甲基(CF3)是有机化学中独特的取代基, 将CF3引入到有机化合物中能够显著改变化合物的酸性、偶极距、极性、亲脂性以及其化学和代谢稳定性.含三氟甲基的化合物已在医药、农药和材料等领域显示出重要的应用价值.发展简洁、高效的方法将CF3引入到有机化合物中无疑具有重要的研究意义. 2016年, Gong等[23a]以Na2S2O8为氧化剂, 以廉价的Langlois’reagent (NaSO2CF3)为三氟甲基源, 在温和的条件下实现了富电子芳烃的三氟甲基化反应(Eq. 12).机理研究表明该反应经历了自由基历程.作者认为Langlois’reagent在过硫酸盐的氧化下生成了三氟甲基自由基(•CF3)继而引发了反应.需要说明的是, 反应的区域选择性与芳烃的亲电取代反应是一致的, 而且底物中烷氧基的存在对高产率的获得是至关重要的.值得一提的是, 该反应即使放大到克级, 依然能以很高的收率得到目标产物.除了Langlois’reagent, 硫氰酸氨(NH4SCN)在K2S2O8单独氧化下也可以实现对富电子苯酚、苯胺的C—H键的区域选择性硫氰化[23b].与以前的硫氰化反应相比, 该反应体系避免了有毒过渡金属、强腐蚀性的卤素及强氧化剂的使用, 因而更具有实用性.
C—S键是一种广泛存在于具有生物活性的分子中, 包括磺酰胺类抗生素和哮喘药物顺尔宁抗生素等.并且含硫有机物在化工、材料领域都有很广泛的应用, 这使得C—S键的构筑在有机合成中具有重要的地位, C—S键的构筑已经引起有机合成化学家的广泛关注. 2015年, Wang课题组[24a]报道了K2S2O8促进的咪唑并吡啶C-3位硫氰化反应, 在温和条件下以KSCN为硫源高效地形成C—S键, 反应不受电子效应和位阻效应的影响, 各种取代的原料均以较高的收率制备一系列具有生物活性的咪唑并吡啶衍生物, 该方法也可用于其他杂环化合物如噻唑、吲哚等的修饰(Scheme 11).此外, 巯基也能在氧化条件下产生硫自由基用于构建C—S键, 该课题组发现苯硫酚在过硫酸盐条件下能与苯甲酰甲酸发生脱羧偶联反应, 合成一系列硫代羧酸酯衍生物.其中, 作者推测其K2S2O8同时氧化苯硫酚和苯甲酰甲酸各自产生硫自由基和脱CO2后的苯甲酰基自由基, 然后发生自由基-自由基偶联完成转化[24b](Scheme 11).
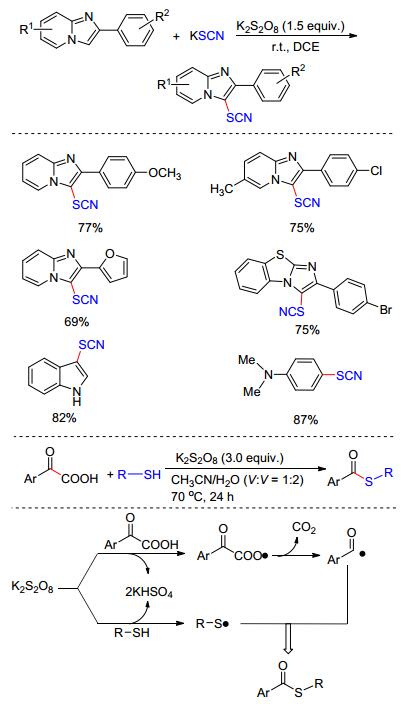 图式11
K2S2O8促进下C—S键的构筑
Scheme11.
K2S2O8 promoted construction of C—S bonds
图式11
K2S2O8促进下C—S键的构筑
Scheme11.
K2S2O8 promoted construction of C—S bonds
2.2 sp3杂化碳-氢键官能团化反应
含有sp3杂化的碳-氢键的官能团化反应是有机化学中极具挑战性的课题之一. 2016年, Shi等[25a]以Na2S2O8为氧化剂实现了胺以及氨基酸衍生物远程的脂肪碳-氢键的高选择性氧化.该反应具有良好的底物普适性, 各种含氨基的化合物, 包括一系列的伯胺、氨基酸以及二肽都可高效地发生碳-氢键氧化反应.此外, 作者对反应的机理进行了深入的研究, 通过18O标记实验发现产物中的氧并非来自氧气或过硫酸盐而是来自于水.在反应体系中加入自由基捕获剂如TEMPO后反应明显被抑制, 意味着该反应有可能是自由基历程(Scheme 12).在上述工作的基础上, 该研究小组[25b]以K2S2O8为作为氧化剂, 3-PySO2N3作为叠氮源又实现了邻苯二甲酰胺保护的胺类化合物远程碳-氢键的叠氮化反应(Scheme 12).值得一提的是, 该反应的选择性非常好, 底物中的三级C—H键的反应活性远大于二级C—H键.这一结果完全符合自由基中间体反应的特征.由于诱导效应会影响不同三级C—H键的电子云密度, 这直接导致了叠氮化容易发生在富电子的远端的三级C—H键.
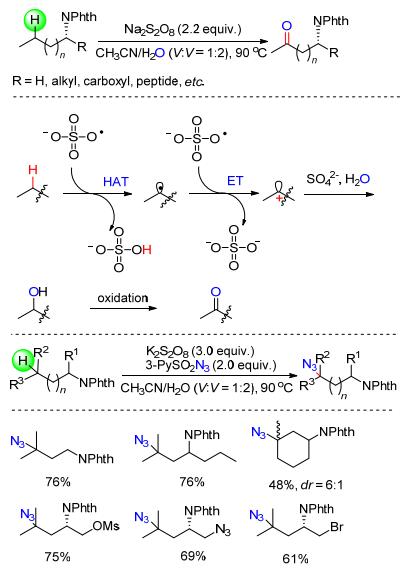 图式12
过硫酸盐促进的远程脂肪碳-氢键氧化和叠氮化反应
Scheme12.
K2S2O8 promoted oxidation and azidation of remote aliphatic C—H bonds
图式12
过硫酸盐促进的远程脂肪碳-氢键氧化和叠氮化反应
Scheme12.
K2S2O8 promoted oxidation and azidation of remote aliphatic C—H bonds
二氟亚甲基常被用于与药物分子骨架和具有生物活性分子的设计中, 这是由于二氟亚甲基具有良好的亲脂性和代谢稳定性.最近, Liang课题组[25c]报道了过硫酸盐促进的苄基C(sp3)—H的氟化反应, 制备了一系列F18取代的芳基二氟甲烷产物.甲苯衍生物在过硫酸盐的促进下产生苄基自由基, 该自由基中间体与Selectfluor产生的氟自由基通过自由基-自由基偶联得到产物(Scheme 13).利用过硫酸盐氧化的方法, Singh等[25d]也实现了甲苯衍生物自身的氧化偶联反应, 该反应也是通过苄基自由基中间体进行的.
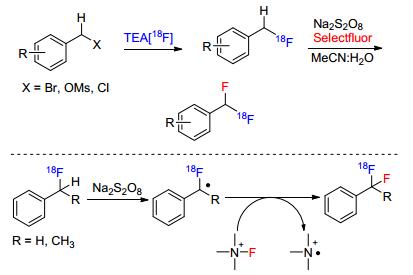 图式13
过硫酸盐促进的苄基C—H键的氟化反应
Scheme13.
Na2S2O8-mediated fluorination of benzylic C—H bonds
图式13
过硫酸盐促进的苄基C—H键的氟化反应
Scheme13.
Na2S2O8-mediated fluorination of benzylic C—H bonds
3 可见光辅助的过硫酸盐促进的有机反应
可见光促进的有机反应由于采用丰富、易得的、可再生的清洁能源, 使得这类反应完全符合绿色化学和可持续发展的基本要求.因此, 近几年备受有机合成化学家们的关注[26].针对环醚与杂环化合物发生Minisci反应中使用贵金属催化剂、强氧化剂、反应条件苛刻等局限, Shah等[27a]发现在可见光的存在下过硫酸盐可产生高活性的硫酸根自由基负离子(SO4-·), 从而高效地引发环醚与杂环化合物的Minisci反应(Scheme 14).通过对反应的深入研究, 作者认为该反应是通过杂环化合物与过硫酸负离子形成的电子给体接受体的络合物(EDA complex)中间体完成的.与此同时, Ji等[27b]在类似的反应条件下实现酰胺、吡咯烷酮以及环醚与杂环化合物的自由基偶联反应(Scheme 14).这类Minisci反应具有反应条件温和、易于操作、反应体系简单等优点, 更有利于杂环化合物的进一步修饰.
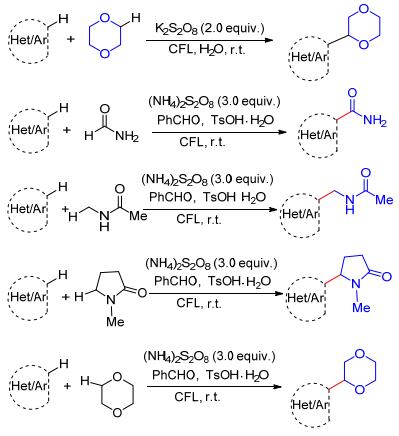 图式14
可见光辅助下过硫酸盐促进的Minisci反应
Scheme14.
K2S2O8-mediated Minisci reaction in the presence of visible light
图式14
可见光辅助下过硫酸盐促进的Minisci反应
Scheme14.
K2S2O8-mediated Minisci reaction in the presence of visible light
利用可见光辅助K2S2O8产生过硫酸自由基负离子的策略, Xia等[28a]报道了苯酚与环状芳胺的自由基偶联反应. K2S2O8在反应中起到了氧化苯酚和芳胺的双重作用, 氧化过程中产生的碳自由基和胺自由基可以高选择性地发生交叉偶联(Eq. 13).该反应是为数不多的利用可见光辅助的方法实现芳胺与酚的交叉脱氢偶联(CDC), 这为碳-氮键的形成提供了新的策略.值得一提的是, 反应中加入自由基捕获剂TEMPO后, 反应并没有被抑制, 反应产率反而有所提高, 作者认为有可能是因为TEMPO可以与反应中产生的氮自由基结合, 从而起到延长瞬态氮自由基寿命的作用.
杂芳环亚砜在天然产物、药物、除草剂和有机功能材料等领域有着广泛的应用, 因此关于这类化合物的合成在有机化学中具有重要的意义.最近, König等[28b]报道了可见光加速的过硫酸盐氧化亚磺酰胺与杂环芳烃的亚磺酰化反应(Eq. 14).与传统过渡金属催化的亚磺酰化反应相比, 该方法具有反应条件温和、氧化剂廉价易得、易于操作等优点.各类富电子杂环芳烃吡咯或吲哚在过硫酸盐的氧化下均可高效地发生碳-氢键的直接亚磺酰化, 反应中过硫酸盐在光照的条件下产生硫酸自由基负离子.通过机理的研究作者认为该自由基中间体单电子氧化亚磺酰胺产生亲电亚磺酰亚胺正离子后与富电子杂环芳烃发生亲电取代反应得到预期目标产物.
4 过硫酸盐促进的其他反应
α-酮酸的脱羧偶联反应是有机合成中构筑碳-碳键的有效途径之一, 近年来受到了有机合成化学家们的广泛关注[29a].在我们小组前期研究的基础上, 2015年, 我们成功地实现了水相中α-酮酸与高价碘炔的脱羧炔基化反应.该反应具有非常好的底物适用范围, 各种α-酮酸和高价碘炔均可高产率地生成相应的α-炔酮化合物[29b].该方法的优点在于氧化剂的用量少、反应原料易得、反应条件温和且反应在水相中进行等, 这些特点完全符合当今绿色化学发展的要求.此外, 草氨酸也能与高价碘炔试剂发生脱羧偶联, 从而轻松实现在有机合成中具有重要应用价值的丙炔酰胺的合成(Eq. 15).需要说明的是, 利用草氨酸为反应物时, 加入催化量的硝酸银可以提高反应的收率.初步的机理研究表明该反应是通过自由基历程进行的.我们认为高价碘炔基试剂在该反应中可能具有双重作用:除了作为炔基化试剂外, 还可以辅助酮酸脱羧产生相应的酰基自由基.对于产物硅基炔化物, 我们还利用“一锅法”通过脱硅-环加成及脱硅-偶联反应实现了其他功能有机分子的合成.随后, Feng和Xu小组[29c]也报道了类似工作.
此外, 我们利用过硫酸盐氧化的方法, 还实现了张力环醇与高价碘炔的氧化开环-炔基化反应(Scheme 15)[29d].该反应弥补了前面α-酮酸脱羧-炔基化反应只能合成α-炔基酮的局限性, 可以用来合成碳链较长的炔酮化合物.我们认为该反应经历如下的路径:首先K2S2O8单电子氧化环丁醇使其产生烷氧基自由基Ⅰ.该中间体发生自由基重排开环得到稳定的烷基自由基Ⅱ, 烷基自由基对高价碘炔选择性加成消除后即可得到δ-炔酮衍生物.此反应也适用于环张力较大的三元环或环张力较小的五元环, 实现一系列远程炔酮的合成.值得一提的是, 当以1, 2-二芳基取代的环丁醇1b为反应底物时, 碳-碳键的裂解发生在连有取代基较多的一端, 这与过渡金属催化的环烷醇裂解反应选择性完全不同[30], 进一步说明该反应是经过自由基中间体进行的.
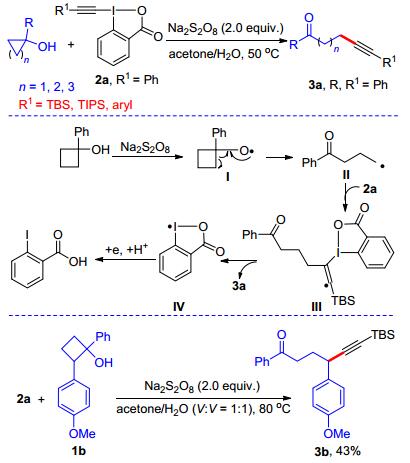 图式15
过硫酸钠促进的张力环烷醇的碳-碳键裂解/炔基化反应
Scheme15.
Na2S2O8-mediated alkynylation of tertiary cycloalkanols via radical C—C bond cleavage
图式15
过硫酸钠促进的张力环烷醇的碳-碳键裂解/炔基化反应
Scheme15.
Na2S2O8-mediated alkynylation of tertiary cycloalkanols via radical C—C bond cleavage
过硫酸盐作为单电子氧化剂, 不仅可以促进碳、氮、膦、硫自由基的生成, 也可以促进硒自由基的形成. 2016年, Sun和Liu等[31]报道了K2S2O8促进的烯烃的硒胺化反应, 高选择性地实现了含胺基的有机硒化合物的合成.反应中, 各种胺源, 如邻磺酰苯甲酰亚胺、二苯磺酰亚胺、苯并三唑、吡唑、氯代嘌呤等均可作为胺基化试剂参与反应.作者认为在过硫酸盐的氧化下联硒化物可以产生硒自由基(•SePh), 该自由基选择性对烯烃加成产生新的烷基自由基.随后, 烷基自由基被氧化成碳正离子再与各种胺发生取代即可得到目标产物.然而, 反应经历硒嗡离子中间体也不能完全的排除(Eq. 16).
5 总结与展望
综上所述, 过硫酸盐促进的自由基偶联及环化反应在过去的几年里取得了快速的发展.由于其具有廉价易得、使用方便、活性较高等优点使其在各类自由基反应中表现出良好的实用性.有理由相信随着研究的不断的深入, 更多、更新颖的过硫酸盐促进的自由基反应将被发现和报道.
与其他自由基类型的反应相比, 过硫酸盐促进的反应还存在着反应类型较少, 反应效率不高等问题.如何根据过硫酸盐活性中间体的特性, 设计新型串联的自由基反应以及发展与其他催化方式协同活化的反应无疑是今后重点发展的方向.相对于过硫酸盐在高分子材料和有机物的降解等工业方面的广泛的应用, 其在小分子的合成方面的应用还处于起步阶段, 发展适合于未来工业生产的过硫酸盐促进的功能化小分子的合成具有重要的意义.
-
-
[1]
(a) Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800.
(b) Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603.
(c) Wille, U. Chem. Rev. 2013, 113, 813.
(d) Hoffmann, R. W. Chem. Soc. Rev. 2016, 45, 577.
(e) Staveness, D.; Bosque, I.; Stephenson, C. R. J. Acc. Chem. Res. 2016, 49, 2295.
(f) Matcha, K.; Narayan, R.; Antonchick, A. P. Angew. Chem., Int. Ed. 2013, 52, 7985.
(g) Tang, S.; Liu, K.; Liu, C.; Lei, A.-W. Chem. Soc. Rev. 2015, 44, 1070. -
[2]
(a) Caron, S.; Dugger, R. W.; Ruggeri, S. G.; Ragan, J. A.; Ripin, D. H. B. Chem. Rev. 2006, 106, 2943.
(b) Xu, G.; Chance, M. R. Chem. Rev. 2007, 107, 3514.
(c) Nicolaou, K. C.; Chen, J. S. Chem. Soc. Rev. 2009, 38, 2993.
(d) Yu, L.-F.; Hu, H.-N.; Nan, F.-J. J. Org. Chem. 2011, 76, 1448.
(e) Yin, J.; Kong, L.-L.; Gao, S.-H. Chin. J. Org. Chem. 2013, 33, 259(in Chinese).
(尹军, 孔丽丽, 高栓虎, 有机化学, 2013, 33, 259.)
(f) Newcomb, E. T.; Knutson, P. C.; Pedersen, B. A.; Ferreira, E. M. J. Am. Chem. Soc. 2016, 138, 108. -
[3]
(a) Satoh, K.; Kamigaito, M. Chem. Rev. 2009, 109, 5120.
(b) Ratera, I.; Veciana, J. Chem. Soc. Rev. 2012, 41, 303.
(c) Tzirakis, M. D.; Orfanopoulos, M. Chem. Rev. 2013, 113, 5262.
(d) Zhang, N.; Samanta, S. R.; Rosen, B. M.; Percec, V. Chem. Rev. 2014, 114, 5848. -
[4]
For selected reviews, see:
(a) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(b) Bras, J. L.; Muzart, J. Chem. Rev. 2011, 111, 1170.
(c) Enthaler, S.; Company, A. Chem. Soc. Rev. 2011, 40, 4912.
(d) Sun, M.; Zhang, J.; Putaj, P.; Caps, V.; Lefebvre, F.; Pelletier, J.; Basset, J.-M. Chem. Rev. 2014, 114, 981.
(e) Xia, Y.; Wang, J. Chem. Soc. Rev. 2017, 46, 2306. -
[5]
(a) Liang, C.-J.; Bruell, C. J.; Marley, M. C.; Sperry, K. L. Soil Sediment Contam. 2003, 12, 207.
(b) House, D. A. Chem. Rev. 1962, 62, 185.
(c) Liang, C.-J.; Bruell, C. J.; Marley, M. C.; Sperry, K. L. Chemosphere 2004, 55, 1225.
(d) Wang, Y.-B.; Hong, C.-S. Water. Res. 1999, 33, 2031. -
[6]
(a) Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Tetrahedron 1971, 27, 3575.
(b) Minisci, F.; Galli, R.; Cecere, M.; Malatesta, V.; Caronna, T. Tetrahedron Lett. 1968, 9, 5609.
(c) Minisci, F.; Vismara, E.; Fontana, F. Heterocycles 1989, 28, 489. -
[7]
(a) Lipshutz, B. H. Chem. Rev. 1986, 86, 795.
(b) Wong, H. N. C.; Yu, P.; Yick, C. Y. Pure Appl. Chem. 1999, 71, 1041.
(c) Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. Chem. Soc. Rev. 2000, 29, 109.
(d) Chinchilla, R.; Najera, C.; Yus, M. Chem. Rev. 2004, 104, 2667.
(e) Lee, H.-K.; Chan, K.-F.; Hui, C.-W.; Yim, H.-K.; Wu, X.-W.; Wong, H. N. C. Pure Appl. Chem. 2005, 77, 139. -
[8]
(a) Chen, J.-R.; Yu, X.-Y.; Wen, J. Synthesis 2015, 47, 604.
(b) Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H. Synthesis 2015, 47, 1195.
(c) Li, C.-C.; Yang, S.-D. Org. Biomol. Chem. 2016, 14, 4365.
(d) Li, Y.-M.; Wei, X.-H.; Li, X.-A.; Yang, S.-D. Chem. Commun. 2013, 49, 11701.
(e) Qiu, J.; Zhang, R.-H. Org. Biomol. Chem. 2014, 12, 4329. -
[9]
(a) Wang, H.; Guo, L.-N.; Duan, X.-H. Chem. Commun. 2013, 49, 10370.
(b) Wang, H.; Guo, L.-N.; Duan, X.-H. Org. Lett. 2013, 15, 5254. -
[10]
(a) Yin, F.; Wang, X.-S. Org. Lett. 2014, 16, 1128.
(b) Wei, W.; Wen, J.-W.; Yang, D.-S.; Liu, X.-X.; Guo, M.-Y.; Dong, R.-M.; Wang, H. J. Org. Chem. 2014, 79, 4225. -
[11]
(a) Wei, W.; Wen, J.-W.; Yang, D.-S.; Du, J.; You, J.-M.; Wang, H. Green Chem. 2014, 16, 2988.
(b) Zhang, M.-Z.; Ji, P.-Y.; Liu, Y.-F.; Xu, J.-W.; Guo, C.-C. Adv. Synth. Catal. 2016, 358, 2976. -
[12]
(a) Li, Y.-M.; Sun, M.; Wang, H.-L.; Tian, Q.-P.; Yang, S.-D. Angew. Chem., Int. Ed. 2013, 52, 3972.
(b) Li, Y.-M.; Shen, Y.-H.; Chang, K.-J.; Yang, S.-D. Tetrahedron 2014, 70, 1991(in Chinese). -
[13]
(a) Guo, L.-N.; Deng, Z.-Q.; Wu, Y.; Hu, J. RSC Adv. 2016, 6, 27000.
(b) Li, Y.-H.; Wang, J.-J.; Wei, X.-H.; Yang, S.-D. Chin. J. Org. Chem. 2015, 35, 638(in Chinese).
(李永红, 王君姣, 魏小红, 杨尚东, 有机化学, 2015, 35, 638.) -
[14]
(a) Laha, J. K.; Tummalapalli, K. S. S.; Gupta. A. Org. Lett. 2014, 16, 4392.
(b) Laha, J. K.; Tummalapalli, K. S. S.; Nair, A.; Patel, N. J. Org. Chem. 2015, 80, 11351.
(c) Laha, J. K.; Patel, K. V.; Tummalapalli, K. S. S.; Dayal, N. Chem. Commun. 2016, 52, 10245. -
[15]
Wang, S.; He, L.-Y.; Guo, L.-N. Synthesis 2015, 47, 3191. doi: 10.1055/s-00000084
-
[16]
(a) Yang, H.; Duan, X.-H.; Zhao, J.-F.; Guo, L.-N. Org. Lett. 2015, 17, 1998.
(b) Guo, L.-N.; Gu, Y.-R.; Yang, H.; Hu, J. Org. Biomol. Chem. 2016, 14, 3098. -
[17]
Gao, P.; Wang, J.; Bai, Z.-J.; Shen, L.; Yan, Y.-Y.; Yang, D.-S.; Fan, M.-J.; Guan, Z.-H. Org. Lett. 2016, 18, 6074. doi: 10.1021/acs.orglett.6b03060
-
[18]
Chen, M.-T.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2017, 4, 86. doi: 10.1039/C6QO00536E
-
[19]
Liu, Y.-K.; Jiang, B.; Zhang, W.; Xu, Z.-Y. J. Org. Chem. 2013, 78, 966. doi: 10.1021/jo302450f
-
[20]
(a) Shi, Z.-Z.; Glorius, F. Chem. Sci. 2013, 4, 829.
(b) Siddaraju, Y.; Lamani, M.; Prabhu, K. R. J. Org. Chem. 2014, 79, 3856. -
[21]
(a) More, N. Y.; Jeganmohan, M. Org. Lett. 2014, 16, 804.
(b) More, N. Y.; Jeganmohan, M. Org. Lett. 2015, 17, 3042. -
[22]
(a) Fujiwara, Y.; Domingo, V.; Seiple, I. B.; Gianatassio, R.; Bel, M. D.; Baran, P. S. J. Am. Chem. Soc. 2011, 133, 3292.
(b) Ilangovan, A.; Polu, A.; Satish, G. Org. Chem. Front. 2015, 2, 1616. -
[23]
(a) Wang, D.-G.; Deng, G.-J.; Chen, S.-Y.; Gong, H. Green Chem. 2016, 18, 5967.
(b) Mete, T. B.; Khopade, T. M.; Bhat, R. G. Tetrahedron Lett. 2017, 58, 415. -
[24]
(a) Yang, D.-S.; Yan, K.-L.; Wei, W.; Li, G.-Q.; Lu, S.-L.; Zhao, C.-X.; Tian, L.-J.; Wang, H. J. Org. Chem. 2015, 80, 11073.
(b) Yan, K.-L.; Yang, D.-S.; Wei, W.; Zhao, J.; Shuai, Y.-Y.; Tian, L.-J.; Wang, H. Org. Biomol. Chem. 2015, 13, 7323. -
[25]
(a) Li, X.; Che, X.; Chen, G.-H.; Zhang, J.; Yan, J.-L.; Zhang, Y.-F.; Zhang, L.-S.; Hsu, C.-P.; Gao, Y.-Q.; Shi, Z.-J. Org. Lett. 2016, 18, 1234.
(b) Li, X.; Shi, Z.-J. Org. Chem. Front. 2016, 3, 1326.
(c) Yuan, G.-Y.; Wang, F.; Stephenson, N. A.; Wang, L.; Rotstein, B. H.; Vasdev, N.; Tang, P.-P.; Liang, S. H. Chem. Commun. 2017, 53, 126.
(d) Kumar, P.; Guntreddi, T.; Singh, R.; Singh, K. N. Org. Chem. Front. 2017, 4, 147. -
[26]
(a) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(b) Ravelli, D.; Protti, S.; Fagnoni, M. Chem. Rev. 2016, 116, 9850.
(c) Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828. -
[27]
(a) Devari, S.; Shah, B.-A. Chem. Commun. 2016, 52, 1490.
(b) Zhang, Y.-Q.; Teuscher, K. B.; Ji, H.-T. Chem. Sci. 2016, 7, 2111. -
[28]
(a) Zhao, Y.-T.; Huang, B.-B.; Yang, C.; Xia, W.-J. Org. Lett. 2016, 18, 3326.
(b) Meyer, A. U.; Alexander, W.; König, B. Angew. Chem., Int. Ed. 2017, 56, 409. -
[29]
(a) Guo, L.-N.; Wang, H.; Duan, X.-H. Org. Biomol. Chem. 2016, 14, 7380.
(b) Wang, H.; Guo, L.-N.; Wang, S.; Duan, X.-H. Org. Lett. 2015, 17, 3054.
(c) Wang, P.-F.; Feng, Y.-S.; Cheng, Z.-F. J. Org. Chem. 2015, 80, 9314.
(d) Wang, S.; Guo, L.-N.; Wang, H.; Duan, X.-H. Org. Lett. 2015, 17, 4798. -
[30]
(a) Nishimura, T.; Ohe, K.; Uemura, S. J. Am. Chem. Soc. 1999, 121, 2645.
(b) Nishimura, T.; Uemura, S. J. Am. Chem. Soc. 1999, 121, 11010. -
[31]
Sun, K.; Wang, X.; Lv, Y.-H.; Li, G.; Jiao, H.-Z.; Dai, C.-W.; Li, Y.-Y.; Zhang, C.; Liu, L. Chem. Commun. 2016, 52, 8471. doi: 10.1039/C6CC04225B
-
[1]
-

图 1 含羟吲哚骨架的天然产物和药物分子
Figure 1 Natural products and pharmaceutical molecules containing oxindole skeletons

图式2 过硫酸钾促进的N-苯基丙烯酰胺与亚硝酸钠的环化反应
Scheme 2 K2S2O8-mediated nitrification of N-phenyl acrylamide

图式3 过硫酸钾促进的N-苯基丙烯酰胺与AgSCF3的环化反应
Scheme 3 K2S2O8-mediated cyclization reaction of N-phenyl acrylamide with AgSCF3

图式5 K2S2O8促进的烯基酰胺硫氰化反应
Scheme 5 K2S2O8-mediated tandem radical thiocyanooxygenation of olefinic amides

图式6 K2S2O8促进的甲叉基环丙烷的硫三氟甲基化反应
Scheme 6 K2S2O8-mediated trifluoromethylthiolation of methylenecyclopropanes

图式7 K2S2O8促进的杂环化合物与甲醇的官能团化反应
Scheme 7 K2S2O8-mediated cross-coupling of heterocycles with methanol

图式8 K2S2O8促进的异喹啉、喹啉、喹喔啉分子间酰化反应
Scheme 8 K2S2O8-mediated intermolecular acylation of isoquinolines, quinolines and quinazines

图式9 K2S2O8促进苄基酮的碳-碳键裂解反应
Scheme 9 K2S2O8-mediated carbon-carbon bond cleavage of benzyl ketones

图式10 K2S2O8促进的醌与芳基硼酸的芳基化反应
Scheme 10 K2S2O8-mediated arylation of quinone and aryl bonric acid

图式12 过硫酸盐促进的远程脂肪碳-氢键氧化和叠氮化反应
Scheme 12 K2S2O8 promoted oxidation and azidation of remote aliphatic C—H bonds

图式14 可见光辅助下过硫酸盐促进的Minisci反应
Scheme 14 K2S2O8-mediated Minisci reaction in the presence of visible light
-


 下载:
下载:















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 719
- 文章访问数: 30894
- HTML全文浏览量: 11780
 下载:
下载:
