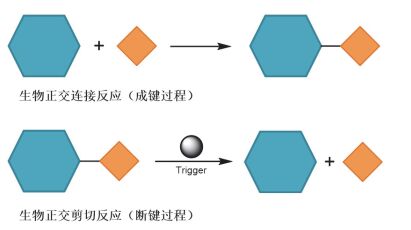
 图1
生物正交连接反应与生物正交剪切反应
Figure1.
Bioorthogonal ligation reaction and bioorthogonal cleavage reaction
图1
生物正交连接反应与生物正交剪切反应
Figure1.
Bioorthogonal ligation reaction and bioorthogonal cleavage reaction
引用本文:
王杰, 陈鹏. 生物正交剪切反应及其应用[J]. 化学学报,
2017, 75(12): 1173-1182.
doi:
10.6023/A17090419

Citation: Wang Jie, Chen Peng. Development and Applications of Bioorthogonal Cleavage Reactions[J]. Acta Chimica Sinica, 2017, 75(12): 1173-1182. doi: 10.6023/A17090419
Citation: Wang Jie, Chen Peng. Development and Applications of Bioorthogonal Cleavage Reactions[J]. Acta Chimica Sinica, 2017, 75(12): 1173-1182. doi: 10.6023/A17090419
生物正交剪切反应及其应用
摘要:
生物正交反应在化学生物学的研究中发挥着越来越重要的作用.传统的生物正交反应以新化学键生成的连接反应为主,其在实现生物分子的“标记”、“示踪”和“捕捉”等研究中发挥着重要作用.近年来,一类新兴的反应类型——以化学键断裂为基础的生物正交剪切反应逐渐发展起来,并在分子的“释放”、“激活”和“操控”等方面得到了越来越广泛的应用.本文首先重点介绍了生物正交剪切反应,总结了这些反应的特点、适用范围和已经实现的用途.随后通过具体的例子介绍了这些反应在化学生物学中的应用,包括小分子前药的激活、蛋白质功能的调控、细胞的工程化等.最后文章对生物正交剪切反应的发展趋势进行了展望.
English
Development and Applications of Bioorthogonal Cleavage Reactions
Abstract:
Bioorthogonal reactions enable us to study and manipulate biological processes under living conditions. As widely used and powerful tools, biorthogonal reactions are largely defined as "ligation reactions" that are used for labeling, tracing and capturing biomolecules. Recently, an emerging collection of biorthogonal "bond-cleavage reactions" have been developed and applied for biological studies, especially in releasing, activating and manipulating biomolecules. In this review, we will first summarize the characteristics and applications of these biorthogonal cleavage reactions. We will then focus on introducing diverse applications of biorthogonal cleavage reactions, including activation of prodrugs, rescue of intracellular protein activity, engineering of cell surface, among other interesting applications. Finally, the outlook of future development and applications of biorthogonal cleavage reactions will be discussed.
-
1 引言
化学生物学为研究者在复杂生物体系中对某一个或者某一类分子进行精确、快速、高效的标记与调控提供了强大的工具.许多化学生物学技术和方法的实现, 都高度依赖于生物正交官能团的引入和生物正交反应的发展.生物正交反应是指能够在活体环境下进行且不与生命过程相互干扰的化学反应.这一概念自2003年被美国的Bertozzi教授[1, 2]提出以来, 作为一类重要的化学生物学工具, 被广泛地应用到各个研究领域.在过去十几年间, 研究者发展出了诸如施陶丁格连接反应、环张力诱导的叠氮-炔基环加成反应、配体-铜复合物催化的叠氮-炔基环加成反应、逆电子需求的Diels-Alder反应以及过渡金属催化的偶联反应等一系列基于新化学键生成的生物正交连接反应[3~5].这些反应被广泛地应用于生物分子的“标记”、“捕捉”和“示踪”, 在生物相容材料、生物医学工程和纳米生物医学上也得到了极大的应用[6~8].
近年来, 一类新型的生物正交反应——基于化学键断裂的生物正交剪切反应逐渐发展起来, 并被越来越广泛地应用于化学生物学的研究中[9].传统的生物正交反应以“连接”功能为主, 将各类功能分子连接到靶标分子上.其中代表性的工作是Bertozzi课题组发展的活体动物的糖代谢标记及成像技术.例如, 他们通过给斑马鱼饲喂含有叠氮官能团的单糖类似物, 利用斑马鱼自身的糖代谢途径使这些含有生物正交官能团的糖变为其体内多糖的组成部分[10].随后, 他们使用连有荧光探针的二氟环辛炔试剂来标记多糖, 实现荧光成像研究.通过这种方法, 作者实现了在斑马鱼中观察多糖的分布图谱, 不仅如此, 作者还对斑马鱼中特定位置的多糖变化进行了追踪, 也将斑马鱼与糖相关的发育过程进行了可视化观察, 这些都是传统研究手段难以实现的[10].
与传统的生物正交连接反应相比, 生物正交剪切反应利用断键的化学过程, 实现了靶标分子中不同部分时空可控的“解离”(图 1).这一新兴的反应策略为目标分子在活细胞和活体环境下的“释放”、“激活”与“操控”等应用奠定了基础.
图1
生物正交连接反应与生物正交剪切反应
Figure1.
Bioorthogonal ligation reaction and bioorthogonal cleavage reaction
在下面的章节中, 我们将首先介绍生物正交剪切反应的类型和特点, 并分别阐述其在小分子前药激活、蛋白质功能调控、细胞工程化等领域中的应用.
2 生物正交剪切反应的开发
有机化学为我们提供了丰富的反应类型, 其中不乏可以实现对分子进行“剪切”的断键反应.例如, 有机合成中经常用到的保护基脱除反应就是一类典型的“剪切”反应.用来作为保护基脱除的反应通常都具有简单、高效和快速的特点, 因此基于这类反应来发展生物正交剪切反应看起来是水到渠成的想法.然而在实际情况中, 发展适用于生物体系的剪切反应并非易事.一方面, 应用于生物体系中的反应必须要求能在常温、常压、水相和中性的条件下发生, 同时还需要在多种亲核试剂和配体的干扰下进行.这一限制, 基本排除了大部分在有机合成中使用的脱保护化学.另一方面, 用于生物体系中的正交反应, 其反应物浓度通常都很低, 一般要求在纳摩尔每升至微摩尔每升的浓度范围, 在这样低浓度的情况下还要求保证反应高效性和专一性, 也具有很大的挑战性[11].
在生物体系中最早使用的剪切反应, 大多是基于光介导的脱保护反应[12].这些光脱除的保护基团种类多样, 其中以邻硝基苄基(ONB)衍生物的使用最为广泛.从最初作为小分子药物、核苷酸等分子的保护基, 到后来作为蛋白质侧链中特定残基的保护基, 这一基团在化学生物学的研究中得到了广泛的应用[13~18].然而, 随着研究的深入, 人们发现这一类保护基存在一些局限性:一方面, 其紫外光照的反应条件存在光毒性和低组织穿透性等问题; 另一方面, 该反应也并非完全生物正交, 其醛类副产物会进一步和生物体系中的亲核试剂反应, 从而对生物体系产生干扰, 甚至毒性[12, 19, 20].双光子介导的脱除反应的发展部分解决了组织穿透性和光毒性的问题, 在这一方面, Liu和Qi课题组[21]实现了利用长波长光源调控多肽和蛋白的功能.除此之外, 由于硝基很容易被硝基还原酶还原成氨基, 而硝基还原酶在众多原核生物(例如大肠杆菌)和某些乏氧的哺乳动物细胞中广泛存在, 因此在这样的体系中, 光介导的脱除反应中常用的邻硝基苄基这类官能团, 很容易被还原成邻氨基苄基, 并进一步发生脱除反应从而造成保护基的提前脱除[16, 22].因此, 发展新的生物正交剪切反应类型, 尤其是化学小分子介导的生物正交剪切反应, 是对传统生物正交反应的有效扩充, 也是该领域的重要发展方向之一.
图 2中列出了在化学生物学研究中, 常见的剪切反应的发生环境与难易程度.其中较最容易实现的是缓冲溶液中的生物正交剪切反应, 而最难实现的要数在活体动物中进行这类反应.
 图2
不同体系下的生物正交剪切反应
Figure2.
Bioorthogonal cleavage reactions in different conditions
图2
不同体系下的生物正交剪切反应
Figure2.
Bioorthogonal cleavage reactions in different conditions
如图 3所示, 表格中列出了部分近年来开发的具有代表性的生物正交剪切反应.
 图3
不同类型的生物正交剪切反应
Figure3.
Collection of bioorthogonal cleavage reactions
图3
不同类型的生物正交剪切反应
Figure3.
Collection of bioorthogonal cleavage reactions
过渡金属介导的生物正交剪切反应是相对较早被广泛研究的一类剪切反应. 2006年, Meggers等[23]首先报道了活细胞内金属Ru介导的脱烯丙氧羰基(氨基甲酸烯丙酯)反应(图 3, 反应1), 并利用这一反应在活细胞中激活了小分子荧光染料.随后的工作中, 该课题组对催化剂中的配体进行了优化, 实现了反应效率和生物相容性的提升[24, 25].针对同样的烯丙氧羰基保护基团, Koide课题组[26]在2009年报道了金属钯介导的脱保护反应, 并利用这一反应设计了针对在复杂环境中检测Pd的荧光探针.随后, Unciti-Broceta课题组[27]首次报道了钯介导的活细胞内小分子化合物上的烯丙氧羰基的剪切反应. 图 3的反应2是金属介导的炔丙氧羰基(氨基甲酸炔丙酯)的脱除反应.对于金属钯而言, 炔丙基具有较好的反应活性, 而炔丙氧羰基的反应活性则更高[28~30].在2010年, Ahn课题组[31]报道了Pd介导的炔丙基脱除反应, 并利用该反应实现了斑马鱼中的小分子荧光染料的激活.之后, 我们课题组首次将钯介导的脱除反应拓展到了活细胞内的生物大分子——蛋白质上, 并首次提出了生物正交剪切反应这一概念[27, 28].利用这一工具, 我们实现了活细胞内的蛋白质特异激活, 并以此研究了一种病原菌效应蛋白的生物学效应, 揭示了该蛋白产生毒性的分子机制[28].随后的研究中, 介导断键过程的过渡金属类型得到了丰富和拓展: 2013年, Finn课题组[32]报道了铜介导的二甲基取代的炔丙氧羰基的剪切反应; 2017年, Unciti-Broceta课题组[33]报道了金介导的炔丙氧羰基的剪切反应.
逆电子需求D-A反应(iDA)是一类发展迅速的生物正交剪切反应(图 3, 反应4).目前来看, 其反应的生物相容性最好, 反应速率最快, 应用范围也最广.反式环辛烯和四嗪的iDA反应由来已久, 2008年Fox课题组[34]则首先报道了该反应可作为生物正交连接反应. 2013年, Robillard课题组[35]第一次报道了该反应的连接产物可以自发的通过进一步重排的方式实现脱除. 2014年, 我们课题组拓展并优化了该反应在生物大分子——蛋白质上的应用.基于该反应高效、快速和正交的特点, 可以在活细胞内实现酶的快速激活.我们在研究中发现, 加入小分子激活剂后的几分钟内, 90%以上的酶都被特异性激活, 这一时间尺度上的飞跃, 使得该方法成为研究细胞信号转导等较快速生物过程的有力工具[36].
除了上述介绍的两大类生物正交剪切反应外, 其他各具特色的生物正交剪切反应也相继出现. 2015年Bertozzi课题组[37]报道的反应5同样也很具有启发意义, 该反应利用了通常认为不稳定的氧化胺结构作为三级胺的保护基团, 利用其对联硼酸频哪醇酯的氧化, 实现了氧的转移, 从而释放了三级胺的结构, 实现了对荧光探针的激活, 并提出了“条件性生物正交反应”的概念. 2016年Pluth课题组[38]报道了反应6, 这是一类可以“自促进”的生物正交剪切反应, 该反应首先由硫化氢将叠氮还原为氨基而引发1, 6消除过程, 而此过程中再次生成的硫化氢产物可以反过来继续还原叠氮官能团, 从而进一步促进剪切反应的发生.于此同时, 不同类型的剪切反应被越来越多地开发出来, 包括图 3中的反应7~11等[39~45].
将生物正交剪切反应的拓展到哺乳动物体内是该领域研究的另一个重要方面.在2016年, 我们课题组将iDA反应拓展到了活动物体内[46]. 2017年, Weissleder[47]和吴水珠课题组[48]利用钯催化剂介导的烯丙氧羰基的脱保护反应, 在活体动物(小鼠)中实现了针对小分子化合物的生物正交剪切反应.
值得注意的是, 上述反应1~9中, 化学家们虽然开发出了众多的生物正交剪切反应, 但大多数都是集中在释放氨基的反应, 而用于释放其他官能团的生物正交剪切反应种类较少. Koide课题组[26]和Ahn课题组[31]在2009年和2010年分别报道了利用钯催化剂释放小分子荧光探针上酚羟基的反应.我们课题组报道的反应10, 则将释放酚羟基的剪切反应拓展至大分子层面, 并将该反应用于激活以酪氨酸为关键残基的蛋白质酶[43]. 2016年, Devaraj课题组[44]报道了利用四嗪与乙烯基醚的iDA反应释放酚羟基的工作(反应11). 2017, Bernardes课题组[45]也报道了四嗪与乙烯基苯酚醚的生物正交剪切反应.
在随后的章节中, 我们将通过实例详细介绍生物正交剪切反应的开发与应用.
3 生物正交剪切反应用于活性小分子的激活
生物正交剪切反应, 最早被用于小分子化合物活性的可控释放, 主要包括小分子探针的光物理性质和小分子前药的活性.该策略的核心思想是将分子的活性基团先保护起来, 暂时“关闭”分子的功能, 待需要的时候再利用生物正交剪切反应移除保护基团, 实现功能的重新激活.我们课题组将这种利用化学断键反应恢复分子活性的策略称为“化学脱笼”, 受到了广泛的关注[49~51].
“化学脱笼”策略的一类重要应用就是前药的激活(图 4-a), 到目前为止, 一系列的前药激活的方法被发展出来, 并用以在癌细胞的周围或内部富集足够浓度的活性小分子药物.这些新方法既可以极大地降低药物的毒副作用, 同时在药代动力学和药物吸收利用方面也存在优势.利用生物正交剪切反应激活前药的策略包括两个步骤, 第一步是将前药载带到合适的位置, 第二步利用脱保护反应, 释放前药的活性.这里重点介绍第二部分, 也即用于释放小分子前药活性的生物正交剪切反应.
 图4
利用生物正交剪切反应激活小分子前药
Figure4.
Prodrug activation via bioorthogonal cleavage reactions
图4
利用生物正交剪切反应激活小分子前药
Figure4.
Prodrug activation via bioorthogonal cleavage reactions
过渡金属介导的生物正交剪切反应在前药激活中应用广泛(图 4-b), 其最大的特点包括两个: (1)理论上讲, 过渡金属可以以催化剂的形式持续不断的激活前药. (2)过渡金属介导的剪切反应可以是异相反应体系. 2014年, Unciti-Broceta课题组[29]和Bradley课题组[30]报道利用负载有纳米钯的树脂, 实现了对5-氟尿嘧啶前药和吉西他滨前药的激活(异相反应体系).在论文中, 他们写到“通过手术的方法将固载有零价Pd催化剂的树脂植入病灶部位, 使得相应的前药只能在对应的病灶部位激活, 可以实现疾病的原位治疗, 也能降低药物对其他组织和器官的毒副作用”.这一观点很好地阐明了固载的过渡金属催化剂和异相反应体系作为激活剂的优势, 固载的催化剂可以通过局部植入的方式实现对肿瘤组织的精准靶向和原位治疗[27].针对上述工作, Couvreur也在其撰写的综述表达了类似的观点: “利用固载在树脂上的钯催化剂介导的异相脱保护反应, 可以很好地控制反应反生的具体位置, 这一特点是其他生物正交反应难以实现的.这种空间可控的反应, 可以很好地实现在特定区域内对高毒性药物的原位激活, 从而提供较宽的治疗窗口, 大大拓展很多因安全问题而受限的药物在临床的应用范围”[52].相比于异相催化剂, 均相金属催化剂可以通过纳米包封和被动靶向的方式实现对肿瘤区域的特异富集.在这一方面, Weissleder[47]和吴水珠课题组[48]做出了重要的工作.他们利用脂质体或水凝胶, 将催化剂制备成纳米级的包封颗粒, 利用被动靶向效应实现对肿瘤的靶向.此外, 过渡金属介导的前药激活又被拓展到了金催化的反应体系中.利用该反应, Unciti-Broceta课题组在体外培养的细胞上验证了对前药的激活[33].
IDA反应是另一类在前药激活中应用广泛的生物正交剪切反应. 2013年, Robillard课题组[35]首次报道了利用iDA反应激活小分子前药的工作(图 4-c).在该工作中, 作者在反式环辛烯的烯丙位引入羟基, 并进一步与药物分子阿霉素的活性氨基连接, 对阿霉素的氨基进行保护, 从而大大降低了阿霉素的毒副作用.在四嗪的作用下, 保护基团可以被高效快速地脱除, 药物的活性得到恢复. 2016年, Robillard课题组[53]在提高药物靶向性上做了更进一步的突破.他们将前药连接在抗体上, 得到抗体——前药偶联物(ADC).一方面, 由于抗体的主动靶向效应相比于被动靶向效应来说, 对肿瘤区域的富集效果更明显, 所以该偶联物可以将药物更精准的载带到预期的位置.另一方面, 因为前药的活性氨基被用来与抗体偶联, 该小分子药物暂时失去活性, 也降低了该偶联物使用过程中的副作用.在实现抗体对肿瘤的靶向之后, 加入介导剪切反应的小分子四嗪, 药物就会从抗体上释放并同时激活, 从而实现了对肿瘤的选择性杀伤. 2016年, Royzen课题组[54]将介导剪切反应的四嗪固载在纳米颗粒上, 并利用该纳米颗粒, 实现了对反式环辛烯保护的阿霉素的激活, 文章虽然只在细胞层面进行了概念验证, 但是文中提出了在动物模型中, 可利用纳米颗粒的被动靶向效应实现肿瘤的靶向, 利用剪切反应激活前药并实现肿瘤杀伤.同年, Royzen又在活体动物层面上, 对上述提出的概念进行了很好的验证.该课题组[55]利用水凝胶制备成纳米颗粒, 同时在水凝胶上共价连接上四嗪分子, 利用被动靶向效应实现了四嗪修饰的水凝胶在肿瘤部位的富集.这样一来, 只有通过血液循环到达肿瘤部位的前药分子才会被激活, 提高了对肿瘤杀伤的选择性.
上述研究中, 虽然用于激活前药的剪切反应种类繁多, 但是反应大多数集中在对氨基的释放, 这在一定程度上限制了可应用的前药的种类. 2017年, Bernardes课题组[45]报道了四嗪与乙烯基苯酚醚的脱保护反应(图 4-d), 该反应虽然速度较慢, 但却是有效的针对酚羟基脱保护的生物正交剪切反应, 拓展了剪切反应在激活前药领域的应用.值得注意的是, 在脱保护反应发生之后, 药物分子的活性并不会马上被激活, 而是会发生一个分子内的重排反应, 随后才形成具有活性的药物小分子.这种双前药策略在理论上可以增加药物分子的作用有效期, 极具启发性.
此外, 利用荧光探针在“化学脱笼”前后光学性质的变化, 可以很方便的对剪切反应的效果进行评价.例如, Weissleder课题组[47]利用小分子荧光探针, 第一次在活体动物水平对钯介导的烯丙基脱除反应(图 3, 反应1)进行了系统的表征和筛选, 为活体动物上的应用提供了指导意义.不仅如此, 利用剪切反应前后的小分子探针光学性质的变化, 还能实现对内源信号分子的示踪.例如, Chang课题组在2016报道了活体动物内的铜介导的脱保护反应.该反应利用钳形三氮唑配体作为保护基团, 保护了荧光素酶底物的酚羟基.这一保护基团设计十分巧妙, 其钳形配体可以螯合金属铜离子, 之后在氧气的作用下该保护基团和酚羟基之间可发生断裂, 释放荧光素酶底物, 实现对铜离子的化学发光检测.利用这一剪切反应, Chang课题组[56]实现了活体动物(小鼠)内金属铜离子的成像研究并揭示了这一金属离子与非酒精性脂肪肝病的关系. 2016年, Devaraj课题组[44]利用四嗪与乙烯基醚的iDA反应释放酚羟基的工作, 则实现了对特定序列核酸的检测.四嗪与乙烯基醚的反应速率很慢, 然而该工作却十分巧妙地利用了这一点, 将两段DNA探针分别连上四嗪和乙烯基保护的荧光素.正常情况下, 即使这两者混合在一起, 也会因反应极慢而无法观测到乙烯基脱保护后产生的荧光.只有当待检测的核酸存在时, 因为碱基的互补配对, 这两条DNA因与待检测核酸配对而互相靠近, 从而加速了反应的发生, 荧光素也因为反应后乙烯基被“剪切”而发出荧光.利用这一原理, 作者实现了对特定核酸的快速特异性检测.
4 生物正交剪切反应用于细胞内蛋白质的激活
生物正交剪切反应的另一类重要的应用是实现蛋白质活性的化学激活.蛋白质是绝大多数生物功能的直接执行者, 通过功能获得性(gain-of-function)的方式研究蛋白质的作用机制, 比传统的功能抑制性的方式(loss-of-function)更为直接和有效[9].化学生物学家设计了多种蛋白质激活的策略:包括通过别构效应激活蛋白质的小分子化合物, 基于化学补救策略的蛋白质激活方法, 基于化学诱导二聚化的小分子激活剂, 基于光控构象变化的小分子激活剂等等[57~61].虽然方法众多, 但都存在普适性不够, 工程化过程复杂等局限性.因此发展新的蛋白质激活策略显得非常必要, 生物正交剪切反应在这方面具有极大的优势.
蛋白质的结构虽然复杂, 但是在多数蛋白质中都存在关键残基, 这些残基对蛋白质的活性起着至关重要的作用.如果对这些残基做任何改变或者修饰, 蛋白质的功能将会丧失; 而如果能够将这些改变或修饰再还原为原先的残基, 蛋白质的功能又会得到恢复.基于这一策略, 研究者通过非天然氨基酸定点插入的技术将带有保护基的氨基酸引入到蛋白质的关键位点, 替代原有的残基, 从而使蛋白质的活性丧失.然后再通过生物正交剪切反应将保护基去除, 实现蛋白质的激活.这一策略虽然清晰明了, 但是在很长一段时间内, 利用“保护-脱保护”来激活蛋白质的策略仅限于利用光介导的剪切反应.究其原因一方面是由于发展生物正交的脱保护反应相对困难, 另一方面, 带保护基的氨基酸需要通过基因编码的方式引入到蛋白质中, 因此对保护基的大小、结构等等都会有诸多的限制.这些因素都成为开发用于激活蛋白质功能的生物正交剪切反应的挑战.
2014年, 我们课题组报道了第一例利用化学小分子介导的剪切反应激活蛋白质功能的工作. OspF是志贺氏菌侵染宿主时分泌的效应蛋白, 具有磷酸苏氨酸裂解酶活性, 在被分泌到宿主细胞中后对MAPK家族激酶(ErK1/2和p38)不可逆去磷酸化, 进而对宿主细胞炎症因子的转录造成影响.利用这一方法, 我们成功实现了细胞内OspF功能的原位激活[28](图 5).我们将OspF重要的催化残基K134替换为被保护的赖氨酸后, 发现酶活完全丧失; 继而加入钯催化剂脱除炔丙氧羰基, 被抑制的OspF又恢复到野生型状态.这样, 钯催化剂就成为了工程化OspF的小分子激活剂.通过特异性的激活OspF, 我们研究了活细胞内Erk激酶在被OspF不可逆去磷酸化后细胞定位的变化, 进而得出OspF对Erk的不可逆去磷酸化发生在细胞核内, 直接影响了Erk的进核功能这一结论[28].
 图5
利用“化学脱笼”策略研究致病菌效应蛋白相关信号通路
Figure5.
Studying the effector protein of pathogenic bacteria and related signaling pathway of host via chemical decaging strategy
图5
利用“化学脱笼”策略研究致病菌效应蛋白相关信号通路
Figure5.
Studying the effector protein of pathogenic bacteria and related signaling pathway of host via chemical decaging strategy
钯介导的脱保护反应很好地展示了剪切反应介导的蛋白质激活策略在研究细胞信号通路等生物学问题中的应用价值和前景[49, 62].此外, 我们还发展了基于iDA反应的蛋白质激活方法(图 6):在该方法中, 反式环辛烯保护的赖氨酸(TCOK)首先被定点引入到酶的活性位点, 导致酶活性的丧失.随后在四嗪的作用下, 发生环加成反应及后续的氢迁移和电子迁移两次互变异构反应, 实现保护基团的脱除和游离氨基的释放[36].相比于钯介导的蛋白质激活反应, 这一iDA生物正交剪切反应的最大优势在于其高效快速.经过优化, 可以在分钟级别的时间尺度上实现活细胞内酶的高效激活[63].更为重要的是, 在2016年的工作中, 我们将这一反应拓展到了活体动物当中.该工作发展了一个通用的, 能够在活细胞及活体动物环境下激活特定激酶的方法, 为蛋白质的化学生物学研究和基于蛋白质的药物开发提供了一套全新的研究工具[46].
 图6
利用生物正交剪切反应在活体动物水平特异性激活激酶
Figure6.
Specific activation of kinase in living animal via bioorthogonal cleavage reactions
图6
利用生物正交剪切反应在活体动物水平特异性激活激酶
Figure6.
Specific activation of kinase in living animal via bioorthogonal cleavage reactions
2016年, Deiters课题组[41]报道了基于小分子膦试剂与叠氮的还原反应的生物正交剪切反应, 并借助该还原反应, 实现了对绿色荧光蛋白、核定位序列、重组酶和基因编辑酶的激活(图 7, 编号6~9).
 图7
利用生物正交剪切反应调控各类蛋白质功能
Figure7.
Manipulation of protein functions via bioorthogonal cleavage reactions
图7
利用生物正交剪切反应调控各类蛋白质功能
Figure7.
Manipulation of protein functions via bioorthogonal cleavage reactions
除了上述针对赖氨酸侧链的脱保护反应, 我们课题组[43]2016年报道了第一例利用化学小分子介导激活酪氨酸侧链的剪切反应, 利用钯介导的联烯脱除反应, 实现了对激酶翻译后修饰的调控, 对DNA聚合酶活性和炭疽杆菌致死因子(蛋白酶)等蛋白质的活性也实现了可控化学调控.
如图 7所示, 生物正交剪切反应已经能够实现对众多类型的蛋白质功能的特异调控, 相信随着新的非天然氨基酸的成功引入和新的剪切反应的开发, 这一策略将成为在活体环境下对目标蛋白质进行功能获得性研究的普适方法.
5 生物正交剪切反应用于细胞表面的工程化
利用剪切反应对生物分子关键基团的脱保护, 还能用于实现细胞表面的工程化[9]. 2015年Kasteren课题组[42]通过膦试剂对叠氮的还原反应, 调控了抗原交叉呈递过程, 实现了对T细胞抗原表位的可控性激活(图 8).
 图8
利用生物正交剪切反应在调控T细胞对抗原表位的识别
Figure8.
Manipulation the recognition between T cell and epitope via bioorthogonal cleavage reactions
图8
利用生物正交剪切反应在调控T细胞对抗原表位的识别
Figure8.
Manipulation the recognition between T cell and epitope via bioorthogonal cleavage reactions
在此设计中, 作者用叠氮官能团取代了抗原表位上关键赖氨酸的ε-氨基, 并设想通过该取代, 在不影响抗原表位交叉呈递的同时, 阻断T细胞对该表位的识别.根据结果, 掩蔽的表位与天然表位在主要组织相容性复合体Ⅰ类(MHC-1)受体上以相同的效率、相同的方式交叉显现, 并且不被其同源的CD8+T细胞识别.这表明叠氮化物在胞内加工过程中是稳定的, 并且可以在不影响抗原表位呈现的情况下, 阻断T细胞受体(TCR)对抗原表位的识别.在用TCEP处理时, 掩蔽在表位中的叠氮基团在树突状细胞表面上快速转化为氨基(也即将侧链恢复为天然的赖氨酸), 导致该表位完全活化, 进而被T细胞识别.如上所述, 通过还原剂介导的叠氮化物还原反应实现了免疫细胞的激活, 该工作为实现免疫细胞的特异化学调控提供了有力工具.
2015年, 我们与陈兴课题组[64]使用钯介导的去酰化反应来释放细胞表面聚糖上的唾液酸的关键氨基(图 9).在该工作中, N-炔丙氧羰基-神经氨酸(Neu5Poc)可以以唾液酸类似物的形式, 通过代谢途径展示在细胞表面聚糖的最外端.然后, 原位生成的钯纳米颗粒有效地将Neu5Poc上的炔丙氧羰基移除, 从而产生神经氨酸(Neu).鉴于神经氨酸在细胞内的不稳定性, 目前还没有报道能够通过代谢途径将其直接引入细胞的糖蛋白中.由于唾液酸上的羧基的负电荷是细胞表面负电荷的重要来源, 因此过量表达唾液酸的癌细胞表面通常会带有较多的负电荷, 而这些负电荷是造成癌细胞之间互相排斥和易扩散的重要原因之一.因此, 通过生物正交剪切反应, 可以将神经氨酸展示在细胞表面, 其5位氨基的释放部分地中和了这些负电荷, 我们发现这可以加速K20细胞的聚集.通过生物正交剪切反应, 我们验证了唾液酸的原位重塑可以用于操纵细胞表面电荷和聚类状态的可选策略之一[9, 51].
 图9
利用生物正交剪切反应调控细胞的聚集行为
Figure9.
Manipulation the cell clustering via bioorthogonal cleavage reactions
图9
利用生物正交剪切反应调控细胞的聚集行为
Figure9.
Manipulation the cell clustering via bioorthogonal cleavage reactions
6 剪切反应在化学生物学中的其他应用
生物正交剪切反应除了在释放分子的功能方面得到了广泛应用, 在其他化学生物学的研究中也发挥着重要作用.这里以遗传密码子拓展技术和基因编码的光交联探针为例, 介绍剪切反应在其他方面的应用.虽然这两例剪切反应都是在体外的大分子上实现, 暂时还不能称为生物正交的剪切反应, 但是由于其独特的应用前景和有望在活细胞中实现的潜力, 这里也进行简要的介绍.
遗传密码子拓展技术, 是一类在蛋白质中定点引入非天然氨基酸的重要技术, 在化学生物学领域的应用十分广泛.然而在实际过程中, 经常存在某些期待引入的非天然氨基酸因带有某些特殊的官能团, 而造成其细胞通透性、稳定性或者其他性质的缺陷, 使得定点引入这类氨基酸变得十分困难.如果能够将这些影响定点插入的活性官能团先保护起来, 以增加定点引入的效率, 随后再脱除该保护基, 最终通过剪切反应实现这类氨基酸的定点插入.
2017年, Wang课题组[65]报道了在蛋白质中定点引入磷酸化酪氨酸的工作, 在该工作中, 作者正是使用了这一“化学脱笼”策略(图 10-a).因为磷酸化酪氨酸带有较多的负电荷, 一方面其透膜性较差, 一方面通过定向进化得到识别该氨基酸的氨酰tRNA合成酶难度也较大, 这就造成了这一非天然氨基酸在蛋白质中的定点插入具有很大的挑战性.在该工作中, 作者首先是利用二甲氨基将磷酸化酪氨酸的磷酸羟基保护起来, 随后通过进化合成酶识别这一保护形式的氨基酸, 随后通过一定的化学转化, 脱除保护基, 从而将磷酸化酪氨酸定点插入目标蛋白质中, 这一工作具有很强的启发意义.
 图10
利用剪切反应在蛋白质中引入特定修饰的氨基酸
Figure10.
Site specific incorporation of amino acids with unique modifications into proteins via bioorthogonal cleavage reactions
图10
利用剪切反应在蛋白质中引入特定修饰的氨基酸
Figure10.
Site specific incorporation of amino acids with unique modifications into proteins via bioorthogonal cleavage reactions
2017年, 我们课题组[66]报道了在组蛋白中定点引入光亲和赖氨酸探针(photolysine)的工作(图 10-b).定点引入具有光亲和性质的赖氨酸是用于在特定位点捕捉组蛋白赖氨酸修饰酶的有力工具.然而, 由于这种类型的赖氨酸与天然赖氨酸的结构极为相似, 通过进化得到特异性识别光亲和赖氨酸的氨酰tRNA合成酶难度很大.因此, 我们先合成了带有对硝基苄氧羰基保护的光亲和赖氨酸前体, 并较容易地获得了识别这一非天然氨基酸的氨酰tRNA合成酶突变体, 进而相对容易的将带有保护基的光亲和赖氨酸前体定点引入到蛋白质中.随后通过硝基还原酶促进的剪切反应, 将硝基还原为氨基, 引发1, 6消除反应, 实现保护基脱除, 从而实现了光亲和赖氨酸在组蛋白中的定点插入.
可以想到的是, 通过这一策略我们还能在蛋白质中定点引入许多有用的氨基酸类似物.如图 10-c所示, 2-羟基赖氨酸、硒代半胱氨酸和硝基酪氨酸这些带有特定修饰的氨基酸, 通常都很难直接定点引入到蛋白质中.而如果是带有保护基的氨基酸, 因为已经有结构类似的非天然氨基酸被氨酰tRNA合成酶识别, 筛选到识别这些前体的合成酶相对容易.因此, 通过引入前体和脱除保护基的“两步法”实现这些特定修饰氨基酸的定点插入, 其可能性将大大增加.不仅仅是氨基酸, 通过该“保护-脱保护”策略, 可以将一些通常较难引入的砌块(例如非天然糖), 引入到生物大分子中[60].
基于遗传密码子拓展技术的光交联探针是一类捕捉蛋白-蛋白相互作用的重要工具, 在诱饵蛋白中定点引入光交联探针, 可以捕捉与诱饵蛋白相互作用的底物, 进而通过质谱鉴定, 获得诱饵蛋白相互作用底物的信息.然而在实际的研究过程中, 大量的诱饵蛋白并未能交联到底物, 这些未发生交联的诱饵蛋白的肽段会对底物肽段的质谱信号产生很大干扰, 造成较多的底物信息丢失[67].
2016年, 我们课题组[67~69]报道了可转移质谱标签的光交联探针(图 11).在该工作中, 我们利用氧化反应将探针切断, 将交联后的诱饵蛋白与底物蛋白分开, 随后利用亲和柱色谱除去诱饵蛋白, 排除了诱饵蛋白的干扰, 使鉴定到的底物蛋白数量大大增加.此外, 图 11-b中的光交联探针DiZHSeC在氧化切割的条件下能够产生丙烯酰胺的结构, 可作为质谱标签来增加底物鉴定的可信度[68].该探针一方面通过可切割的C—Se键, 增加了底物鉴定的数量, 又通过切割后产生的质谱标签, 大大降低了底物鉴定的假阳性, 并可提供蛋白-蛋白相互作用的界面信息.
 图11
利用剪切反应设计可切割光交联探针
Figure11.
Rational design of a cleavable photocross linker via bioorthogonal cleavage reactions
图11
利用剪切反应设计可切割光交联探针
Figure11.
Rational design of a cleavable photocross linker via bioorthogonal cleavage reactions
7 展望
不难看出, 生物正交剪切反应将使一些生物学问题的研究变得更加容易和有效.同时, 新的剪切反应的开发和应用, 也将进一步丰富化学生物学工具箱.
以“化学脱笼”策略为例, 上文提到了利用金属介导的剪切反应激活小分子前药.与其他类型的剪切反应不同的是, 介导剪切反应的过渡金属扮演着催化剂的角色, 而非“消耗品”, 因此如果将固载有催化剂的树脂通过手术植入肿瘤附近, 那么金属催化剂就可以在这一特定区域内连续激活前药分子.这一特点避免了其他类型的剪切反应和激活策略需要连续使用激活剂的缺点.类似的方法也可以适用于抗体偶联药物中接头的断裂和药物的释放[30].
不仅如此, “化学脱笼”策略在原位研究蛋白酶底物方面也有很大的优势.以caspase家族蛋白为例, 它们是一类与细胞凋亡和焦亡密切相关的蛋白酶.由于在细胞凋亡的过程中先后有多种caspase被激活, 在鉴定某种caspase特定底物的时候, 利用剪切反应原位激活特定caspase的方式就显得较为独特.因为当通过诱导产生细胞凋亡的时候, 细胞内多种caspase均是处于被激活的状态, 无法将鉴定到的底物与某一种caspase对应起来.
类似的, 当过表达某些原癌基因酶时, 或者细胞受到某些信号的刺激时, 下游的磷酸化水平通常会上升.然而, 磷酸化信号的传递, 通常是级联的方式, 也即在信号传递的过程中, 有多种激酶被激活.常规方法难以区分这些磷酸化水平上升的蛋白质是某一激酶的直接底物还是间接底物.相比之下, 利用生物正交剪切反应来控制的蛋白质激活(“化学脱笼”)可以高时空分辨地激活某一特定酶的活性, 从而观测下游信号、底物的变化, 鉴定出其直接的底物, 得出更为精确的信息.这一策略对于级联信号通路的研究是非常重要的.
另一方面, 虽然这一蛋白质激活策略在多种类型的蛋白质上得到了验证, 然而其反应类型仅局限于对氨基和酚羟基的“脱笼”, 细胞中还有很多蛋白质是以丝氨酸、组氨酸、谷氨酸等氨基酸作为关键位点.发展新的剪切反应类型, 对于激活以这些残基为关键位点的蛋白质至关重要.
成键反应与断键反应是化学转化的两类主要途经.然而, 与生物正交连接反应相比, 生物正交剪切反应的开发与应用才刚刚起步.我们在这里展示了这类新兴反应的现状和发展趋势.相信随着这些新反应的不断开发, 生物正交剪切反应在化学生物学中的应用会越来越广泛.
-
-
[1]
Lemieux, G. A.; de Graffenried, C. L.; Bertozzi, C. R. J. Am. Chem. Soc. 2003, 125, 4708. doi: 10.1021/ja029013y
-
[2]
Prescher, J. A.; Bertozzi, C. R. Nat. Chem. Biol. 2005, 1, 13. doi: 10.1038/nchembio0605-13
-
[3]
Patterson, D. M.; Nazarova, L. A.; Prescher, J. A. ACS Chem. Biol. 2014, 9, 592. doi: 10.1021/cb400828a
-
[4]
李劼, 王杰, 陈鹏, 化学学报, 2012, 70, 1439. doi: 10.3866/PKU.WHXB201203142Li, J.; Wang, J.; Chen, P. Acta Chim. Sinica 2012, 70, 1439. doi: 10.3866/PKU.WHXB201203142
-
[5]
杨麦云, 陈鹏, 化学学报, 2015, 73, 783. doi: 10.3866/PKU.WHXB201502062Yang, M. Y.; Chen, P. Acta Chim. Sinica 2015, 73, 783. doi: 10.3866/PKU.WHXB201502062
-
[6]
Azagarsamy, M. A.; Anseth, K. S. ACS Macro Lett. 2013, 2, 5. doi: 10.1021/mz300585q
-
[7]
Rogozhnikov, D.; O'Brien, P. J.; Elahipanah, S.; Yousaf , M. N. 2016, 6, 39806.
-
[8]
Koo, H.; Lee, S.; Na, J. H.; Kim, S. H.; Hahn, S. K.; Choi, K.; Kwon, I. C.; Jeong, S. Y.; Kim, K. Angew. Chem., Int. Ed. 2012, 51, 11836. doi: 10.1002/anie.201206703
-
[9]
Li, J.; Chen, P. R. Nat. Chem. Biol. 2016, 12, 129. doi: 10.1038/nchembio.2024
-
[10]
Laughlin, S. T.; Baskin, J. M.; Amacher, S. L.; Bertozzi, C. R. Science 2008, 320, 664. doi: 10.1126/science.1155106
-
[11]
Hao, Z.; Hong, S.; Chen, X.; Chen, P. R. Acc. Chem. Res. 2011, 44, 742. doi: 10.1021/ar200067r
-
[12]
Pelliccioli, A. P.; Wirz, J. Photochem. Photobiol. Sci. 2002, 1, 441. doi: 10.1039/b200777k
-
[13]
Cruz, F. G.; Koh, J. T.; Link, K. H. J. Am. Chem. Soc. 2000, 122, 8777. doi: 10.1021/ja001804h
-
[14]
Lenox, H. J.; McCoy, C. P.; Sheppard, T. L. Org. Lett. 2001, 3, 2415. doi: 10.1021/ol016255e
-
[15]
Wu, N.; Deiters, A.; Cropp, T. A.; King, D.; Schultz, P. G. J. Am. Chem. Soc. 2004, 126, 14306. doi: 10.1021/ja040175z
-
[16]
Chen, P. R.; Groff, D.; Guo, J.; Ou, W.; Cellitti, S.; Geierstanger, B. H.; Schultz, P. G. Angew. Chem., Int. Ed. 2009, 48, 4052. doi: 10.1002/anie.v48:22
-
[17]
Zhao, J.; Lin, S.; Huang, Y.; Zhao, J.; Chen, P. R. J. Am. Chem. Soc. 2013, 135, 7410. doi: 10.1021/ja4013535
-
[18]
Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J. W. J. Am. Chem. Soc. 2012, 134, 11912. doi: 10.1021/ja3046958
-
[19]
Jayakumar, M. K. G.; Idris, N. M.; Zhang, Y. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8483. doi: 10.1073/pnas.1114551109
-
[20]
Liu, J.; Yang, D.; Minemoto, Y.; Leitges, M.; Rosner, M. R.; Lin, A. Mol. Cell 2006, 21, 467. doi: 10.1016/j.molcel.2005.12.020
-
[21]
Chen, X.; Tang, S.; Zheng, J.; Zhao, R.; Wang, Z.; Shao, W.; Chang, H.; Cheng, J; Zhao, H.; Liu, L.; Qi, H. Nat. Commun. 2015, 6, 7220. doi: 10.1038/ncomms8220
-
[22]
Virdee, S.; Kapadnis, P. B.; Elliott, T.; Lang, K.; Madrzak, J.; Nguyen, D. P.; Riechmann, L.; Chin, J. W. J. Am. Chem. Soc. 2011, 133, 10708. doi: 10.1021/ja202799r
-
[23]
Streu, C.; Meggers, E. Angew. Chem., Int. Ed. 2006, 45, 5645. doi: 10.1002/(ISSN)1521-3773
-
[24]
Sasmal, P. K.; Carregalromero, S.; Parak, W. J.; Meggers, E. Organometallics 2012, 31, 5968. doi: 10.1021/om3001668
-
[25]
V lker, T.; Meggers, E. ChemBiochem 2017, 18, 1083. doi: 10.1002/cbic.v18.12
-
[26]
Garner, A. L.; Song, F.; Koide, K. J. Am. Chem. Soc. 2009, 131, 5163. doi: 10.1021/ja808385a
-
[27]
Yusop, R. M.; Unciti-Broceta, A.; Johansson, E. M. V.; Sánchez-Martín, R. M.; Bradley, M. Nat. Chem. 2011, 3, 239.
-
[28]
Li, J.; Yu, J.; Zhao, J.; Wang, J.; Zheng, S.; Lin, S.; Chen, L.; Yang, M.; Jia, S.; Zhang, X.; Chen, P. R. Nat. Chem. 2014, 6, 352. doi: 10.1038/nchem.1887
-
[29]
Weiss, J. T.; Dawson, J. C.; Macleod, K. G.; Rybski, W.; Fraser, C.; Torres-Sánchez, C.; Patton, E. E.; Bradley, M.; Carragher, N. O.; Unciti-Broceta, A. Nat. Commun. 2014, 5, 3277.
-
[30]
Weiss, J. T.; Dawson, J. C.; Fraser, C.; Rybski, W.; Torres-Sánchez, C.; Bradley, M.; Patton, E. E.; Carragher, N. O.; Unciti-Broceta, A. J. Med. Chem. 2014, 57, 5395. doi: 10.1021/jm500531z
-
[31]
Santra, M.; Ko, S.-K.; Shin, I.; Ahn, K. H. Chem. Commun. 2010, 46, 3964. doi: 10.1039/c001922d
-
[32]
Kislukhin, A. A.; Hong, V. P.; Breitenkamp, K. E.; Finn, M. G. Bioconjugate Chem. 2013, 24, 684. doi: 10.1021/bc300672b
-
[33]
Pérez-López, A. M.; Rubio-Ruiz, B.; Sebastián, V.; Hamilton, L.; Adam, C.; Bray, T. L.; Irusta, S.; Brennan, P. M.; Lloyd-Jones, G.; Sieger, D.; Santamaría, J.; Unciti-Broceta, A. Angew. Chem., Int. Ed. 2017, DOI: 10.1002/anie.201705609
-
[34]
Blackman, M. L.; Royzen, M.; Fox, J. M. J. Am. Chem. Soc. 2008, 130, 13518. doi: 10.1021/ja8053805
-
[35]
Versteegen, R. M.; Rossin, R.; ten Hoeve, W.; Janssen, H. M.; Robillard, M. S. Angew. Chem., Int. Ed. 2013, 52, 14112. doi: 10.1002/anie.201305969
-
[36]
Li, J.; Jia, S.; Chen, P. R. Nat. Chem. Biol. 2014, 10, 1003. doi: 10.1038/nchembio.1656
-
[37]
Kim, J.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2015, 54, 15777. doi: 10.1002/anie.201508861
-
[38]
Steiger, A. K.; Pardue, S.; Kevil, C. G.; Pluth, M. D. J. Am. Chem. Soc. 2016, 138, 7256. doi: 10.1021/jacs.6b03780
-
[39]
Matikonda, S. S.; Orsi, D. L.; Staudacher, V.; Jenkins, I. A.; Fiedler, F.; Chen, J.; Gamble, A. B. Chem. Sci. 2015, 6, 1212. doi: 10.1039/C4SC02574A
-
[40]
Ge, Y.; Fan, X.; Chen, P. R. Chem. Sci. 2016, 7, 7055. doi: 10.1039/C6SC02615J
-
[41]
Luo, J.; Liu, Q.; Morihiro, K.; Deiters, A. Nat. Chem. 2016, 8, 1027. doi: 10.1038/nchem.2573
-
[42]
Pawlak, J. B.; Gential, G. P. P.; Ruckwardt, T. J.; Bremmers, J. S.; Meeuwenoord, N. J.; Ossendorp, F. A.; Overkleeft, H. S.; Filippov, D. V.; van Kasteren, S. I. Angew. Chem., Int. Ed. 2015, 54, 5628. doi: 10.1002/anie.201500301
-
[43]
Wang, J.; Zheng, S.; Liu, Y.; Zhang, Z.; Lin, Z.; Li, J.; Zhang, G.; Wang, X.; Li, J.; Chen, P. R. J. Am. Chem. Soc. 2016, 138, 15118. doi: 10.1021/jacs.6b08933
-
[44]
Wu, H.; Alexander, S. C.; Jin, S.; Devaraj, N. K. J. Am. Chem. Soc. 2016, 138, 11429. doi: 10.1021/jacs.6b01625
-
[45]
Jiménez-Moreno, E.; Guo, Z.; Oliveira, B. L.; Albuquerque, I. S.; Kitowski, A.; Guerreiro, A.; Boutureira, O.; Rodrigues, T.; Jiménez-Osés, G.; Bernardes, G. J. L. Angew. Chem., Int. Ed. 2017, 56, 243. doi: 10.1002/anie.v56.1
-
[46]
Zhang, G.; Li, J.; Xie, R.; Fan, X.; Liu, Y.; Zheng, S.; Ge, Y.; Chen, P. R. ACS Central Science 2016, 2, 325. doi: 10.1021/acscentsci.6b00024
-
[47]
Miller, M. A.; Askevold, B.; Mikula, H.; Kohler, R. H.; Pirovich, D.; Weissleder, R. Nat. Commun. 2017, 8, 15906. doi: 10.1038/ncomms15906
-
[48]
Li, B.; Liu, P.; Wu, H.; Xie, X.; Chen, Z.; Zeng, F.; Wu, S. Biomaterials 2017, 138, 57. doi: 10.1016/j.biomaterials.2017.05.036
-
[49]
Doerr, A. Nat. Meth. 2014, 11, 472.
-
[50]
Anon. Nat. Meth. 2015, 12, 16.
-
[51]
He, C. Nat. Sci. Rev. 2015, 2, 250. doi: 10.1093/nsr/nwv030
-
[52]
Dumas, A.; Couvreur, P. Chem. Sci. 2015, 6, 2153. doi: 10.1039/C5SC00070J
-
[53]
Rossin, R.; van Duijnhoven, S. M. J.; ten Hoeve, W.; Janssen, H. M.; Kleijn, L. H. J.; Hoeben, F. J. M.; Versteegen, R. M.; Robillard, M. S. Bioconjugate Chem. 2016, 27, 1697. doi: 10.1021/acs.bioconjchem.6b00231
-
[54]
Khan, I.; Agris, P. F.; Yigit, M. V.; Royzen, M. Chem. Commun. 2016, 52, 6174. doi: 10.1039/C6CC01024E
-
[55]
Mejia Oneto, J. M.; Khan, I.; Seebald, L.; Royzen, M. ACS Central Science 2016, 2, 476. doi: 10.1021/acscentsci.6b00150
-
[56]
Heffern, M. C.; Park, H. M.; Au-Yeung, H. Y.; Van de Bittner, G. C.; Ackerman, C. M.; Stahl, A.; Chang, C. J. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14219. doi: 10.1073/pnas.1613628113
-
[57]
Zorn, J. A.; Wells, J. A. Nat. Chem. Biol. 2010, 6, 179. doi: 10.1038/nchembio.318
-
[58]
Qiao, Y.; Molina, H.; Pandey, A.; Zhang, J.; Cole, P. A. Science 2006, 311, 1293. doi: 10.1126/science.1122224
-
[59]
Schwartz, E. C.; Saez, L.; Young, M. W.; Muir, T. W. Nat. Chem. Biol. 2007, 3, 50. doi: 10.1038/nchembio832
-
[60]
Tsai, Y.-H.; Essig, S.; James, J. R.; Lang, K.; Chin, J. W. Nat. Chem. 2015, 7, 554. doi: 10.1038/nchem.2253
-
[61]
Tian, T.; Song, Y.; Wang, J.; Fu, B.; He, Z.; Xu, X.; Li, A.; Zhou, X.; Wang, S.; Zhou, X. J. Am. Chem. Soc. 2016, 138, 955. doi: 10.1021/jacs.5b11532
-
[62]
Qian, K.; Zheng, Y. G. Nat. Chem. Biol. 2014, 10, 328. doi: 10.1038/nchembio.1507
-
[63]
Fan, X.; Ge, Y.; Lin, F.; Yang, Y.; Zhang, G.; Ngai, W. S. C.; Lin, Z.; Zheng, S.; Wang, J.; Zhao, J.; Li, J.; Chen, P. R. Angew. Chem., Int. Ed. 2016, 55, 14046. doi: 10.1002/anie.v55.45
-
[64]
Wang, J.; Cheng, B.; Li, J.; Zhang, Z.; Hong, W.; Chen, X.; Chen, P. R. Angew. Chem., Int. Ed. 2015, 54, 5364. doi: 10.1002/anie.201409145
-
[65]
Hoppmann, C.; Wong, A.; Yang, B.; Li, S.; Hunter, T.; Shokat, K. M.; Wang, L. Nat. Chem. Biol. 2017, 13, 842. doi: 10.1038/nchembio.2406
-
[66]
Xie, X.; Li, X.-M.; Qin, F.; Lin, J.; Zhang, G.; Zhao, J.; Bao, X.; Zhu, R.; Song, H.; Li, X. D.; Chen, P. R. J. Am. Chem. Soc. 2017, 139, 6522. doi: 10.1021/jacs.7b01431
-
[67]
Lin, S.; He, D.; Long, T.; Zhang, S.; Meng, R.; Chen, P. R. J. Am. Chem. Soc. 2014, 136, 11860. doi: 10.1021/ja504371w
-
[68]
Yang, Y.; Song, H.; He, D.; Zhang, S.; Dai, S.; Lin, S.; Meng, R.; Wang, C.; Chen, P. R. Nat. Commun. 2016, 7, 12299. doi: 10.1038/ncomms12299
-
[69]
Zhang, S.; He, D.; Lin, Z.; Yang, Y.; Song, H.; Chen, P. R. Acc. Chem. Res. 2017, 50, 1184. doi: 10.1021/acs.accounts.6b00647
-
[1]
-

图 1 生物正交连接反应与生物正交剪切反应
Figure 1 Bioorthogonal ligation reaction and bioorthogonal cleavage reaction

图 4 利用生物正交剪切反应激活小分子前药
Figure 4 Prodrug activation via bioorthogonal cleavage reactions

图 5 利用“化学脱笼”策略研究致病菌效应蛋白相关信号通路
Figure 5 Studying the effector protein of pathogenic bacteria and related signaling pathway of host via chemical decaging strategy

图 6 利用生物正交剪切反应在活体动物水平特异性激活激酶
Figure 6 Specific activation of kinase in living animal via bioorthogonal cleavage reactions

图 7 利用生物正交剪切反应调控各类蛋白质功能
Figure 7 Manipulation of protein functions via bioorthogonal cleavage reactions

图 8 利用生物正交剪切反应在调控T细胞对抗原表位的识别
Figure 8 Manipulation the recognition between T cell and epitope via bioorthogonal cleavage reactions

图 9 利用生物正交剪切反应调控细胞的聚集行为
Figure 9 Manipulation the cell clustering via bioorthogonal cleavage reactions

图 10 利用剪切反应在蛋白质中引入特定修饰的氨基酸
Figure 10 Site specific incorporation of amino acids with unique modifications into proteins via bioorthogonal cleavage reactions
-


 下载:
下载:










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 542
- 文章访问数: 15932
- HTML全文浏览量: 5373
 下载:
下载:
