 图 1
Ru-Zn催化剂的XRD图
Figure 1.
XRD pattern of the Ru-Zn catalyst
图 1
Ru-Zn催化剂的XRD图
Figure 1.
XRD pattern of the Ru-Zn catalyst
引用本文:
孙海杰, 秦会安, 黄振旭, 苏曼菲, 李永宇, 刘寿长, 刘仲毅. 反应修饰剂ZnSO4和预处理对苯选择加氢制环己烯Ru-Zn催化剂性能的影响[J]. 无机化学学报,
2017, 33(1): 73-80.
doi:
10.11862/CJIC.2016.283

Citation: SUN Hai-Jie, QIN Hui-An, HUANG Zhen-Xu, SU Man-Fei, LI Yong-Yu, LIU Zhong-Yi, LIU Shou-Chang. Effect of Reaction Modifier ZnSO4 and Pretreatment on Performance of Ru-Zn Catalyst for Selective Hydrogenation of Benzene to Cyclohexene[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(1): 73-80. doi: 10.11862/CJIC.2016.283
Citation: SUN Hai-Jie, QIN Hui-An, HUANG Zhen-Xu, SU Man-Fei, LI Yong-Yu, LIU Zhong-Yi, LIU Shou-Chang. Effect of Reaction Modifier ZnSO4 and Pretreatment on Performance of Ru-Zn Catalyst for Selective Hydrogenation of Benzene to Cyclohexene[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(1): 73-80. doi: 10.11862/CJIC.2016.283
反应修饰剂ZnSO4和预处理对苯选择加氢制环己烯Ru-Zn催化剂性能的影响
摘要:
共沉淀法制备了Ru-Zn催化剂,考察了反应修饰剂ZnSO4和预处理对苯选择加氢制环己烯Ru-Zn催化剂性能的影响。结果表明,反应修饰剂ZnSO4可以与Ru-Zn催化剂中助剂ZnO反应生成(Zn(OH)2)3(ZnSO4)(H2O)盐。随反应修饰剂ZnSO4浓度增加,(Zn(OH)2)3(ZnSO4)(H2O)盐量逐渐增加,Ru-Zn催化剂活性逐渐降低,环己烯选择性逐渐升高。因为(Zn(OH)2)3(ZnSO4)(H2O)盐中的Zn2+可以使Ru变为有利环己烯生成的缺电子的Ruδ+物种,而且还可以占据不适宜环己烯生成的强Ru活性位。但当反应修饰剂ZnSO4浓度高于0.41 mol·L-1后,继续增加ZnSO4浓度,由于Zn2+水解浆液酸性太强,可以溶解部分(Zn(OH)2)3(ZnSO4)(H2O)盐,Ru-Zn催化剂活性升高,环己烯选择性降低。环己烯选择性略微降低,是由于ZnSO4溶液中大量的Zn2+可以与生成的环己烯形成配合物,稳定生成的环己烯,抑制环己烯再吸附到催化剂表面并加氢生成环己烷。在ZnSO4最佳浓度0.61 mol·L-1下对Ru-Zn催化剂预处理15 h,Ru-Zn催化剂中助剂ZnO可以与ZnSO4完全反应生成(Zn(OH)2)3(ZnSO4)(H2O)盐,在该催化剂上25 min苯转化68.2%时环己烯选择性和收率分别为80.2%和54.7%。而且该催化剂具有良好的稳定性和重复使用性能。
English
Effect of Reaction Modifier ZnSO4 and Pretreatment on Performance of Ru-Zn Catalyst for Selective Hydrogenation of Benzene to Cyclohexene
Abstract:
A Ru-Zn catalyst was prepared by a co-precipitation method, and the effects of the modifier ZnSO4 and the pretreatment on the performance of the Ru-Zn catalyst for selective hydrogenation of benzene to cyclohexene were investigated. The results showed that the modifier ZnSO4 could react with the promoter ZnO of the Ru-Zn catalyst to form a indissoluble (Zn(OH)2)3(ZnSO4)(H2O) salt. Moreover, the amount of this salt increased with the increase of the concentration of the modifier ZnSO4, which made the activity of this catalyst increase and the selectivity to cyclohexene decrease. This is because the Zn2+ of the salt not only could transform the active sites Ru into the electron-deficient species in favor of the formation of cyclohexene, but also could occupy the strong active sites Ru unfavorable for the formation of cyclohexene. However, when the concentration of the modifier ZnSO4 was higher than 0.41 mol·L-1, the further increased concentration of ZnSO4 could dissolve part of the (Zn(OH)2)3(ZnSO4)(H2O) salt due to the strong acidity for the hydrolysis of Zn2+ of ZnSO4, thus the activity of Ru-Zn catalyst increased and the selectivity to cyclohexene decreased. But the selectivity was slightly decreased, which was because Zn2+ of ZnSO4 in the solution could form a complex with cyclohexene stabilizing cyclohexene formed and preventing cyclohexene from being adsorbed on surface of the catalyst again and being hydrogenated into cyclohexane. When the Ru-Zn catalyst was pretreated for 15 h under the condition of the optium ZnSO4 concentration of 0.61 mol·L-1, the promoter ZnO of Ru-Zn catalyst could completely react with ZnSO4 to form the (Zn(OH)2)3(ZnSO4)(H2O) salt, and this catalyst gave a selectivity of 80.2% and a cyclohexene yield of 54.7% at the benzene conversion of 68.2% at 25 min. Moreover, this catalyst had a good stability and a excellent reusability.
-
Key words:
- benzene
- / selective hydrogenation
- / cyclohexene
- / Ru
- / Zn
- / reaction modifier
- / pretreatment
-
己内酰胺和己二酸是尼龙-6和尼龙-66的单体,广泛应用纺织、汽车、电子等行业,是现代化学工业的基本原料[1-2]。从苯选择加氢制环己烯出发制备己内酰胺和己二酸具有安全、节能、环保、碳原子利用率高等优点,引起人们的广泛关注[3]。然而,热力学上苯加氢反应更倾向于生成环己烷[4]。因此,只能从动力学解决这一挑战,即开发高活性高选择的苯选择加氢制环己烯催化剂。
反应修饰剂是提高苯选择加氢制环己烯Ru基催化剂环己烯选择性的主要途径之一。苯选择加氢制环己烯Ru催化剂的反应修饰剂主要包括无机盐(如ZnSO4[5]、FeSO4或CdSO4[6])、无机碱(如NaOH[4, 7])、胺类(如乙二胺或二乙醇胺[8])和醇类(如聚乙二醇[8-9])等。其中,ZnSO4作反应修饰剂Ru催化剂上环己烯收率最高。Sun等利用ZnSO4作反应修饰剂分别在Ru-Mn催化剂[10]、Ru-Co-B/ZrO2催化剂[11]和Ru-Zn催化剂[12]上分别获得了61.3%、62.8%和59.6%的环己烯收率。然而关于反应修饰剂ZnSO4的作用机理至今没有相关文献报道。
预处理也是提高苯选择加氢制环己烯Ru基催化剂环己烯选择性的途径之一。预处理是将催化剂在反应修饰剂ZnSO4溶液中于反应条件下保持1 h或更长的时间,工业运行为22 h[13].Sun等[9]在反应修饰剂ZnSO4溶液中对Ru-Zn催化剂预处理了22 h获得了64.5%的环己烯收率。然而关于预处理在提高Ru基催化剂环己烯选择性所起的作用也没有文献报道。
本文正是在此基础上,用共沉淀法制备了Ru-Zn催化剂,考察了反应修饰剂ZnSO4和预处理对苯选择加氢制环己烯Ru-Zn催化剂性能的影响,探讨了反应修饰ZnSO4的作用机理和预处理的作用。
1 实验部分
1.1 实验试剂
水合三氯化钌(AR)购自昆明贵研铂业股份有限公司;七水合硫酸锌(AR)购自天津福晨化学试剂厂;氢氧化钠(GR)、苯(AR)均购自天津市科密欧化学试剂有限公司。
1.2 催化剂制备
Ru-Zn催化剂的制备采用共沉淀法制备,具体方法如下:取20.0 g RuCl3·3H2O和4.5 g ZnSO4·7H2O配成200 mL混合溶液,将该混合加热至80 ℃左右。然后在搅拌下快速向该溶液中倒入5%的NaOH溶液200 mL,在80 ℃左右继续搅拌30 min。将上述浆液倒入1 L的哈氏合金高压反应釜中,密封。用N2气置换3次空气后,再用H2气置换3次空气,将H2压力调节至5 MPa,温度控制在150 ℃,搅拌800 r·min-1,还原3 h。将所得黑色固体用蒸馏水洗涤至滤液至中性,真空干燥,即得Ru-Zn催化剂。
1.3 催化表征
催化剂物相分析在荷兰PAN Nalytical公司的X′Pert PRO型X射线衍射(XRD)仪上进行。Ni滤光片,滤除Kβ线,Cu Kα射线(λ=0.154 18 nm),管电压40 kV,管电流40 mA,扫描范围5°~90°,扫描步长0.03°。催化剂中各元素含量分析在德国Bruker公司的S4 Pioneer型X射线荧光仪(XRF)上进行。催化剂形貌在日本电子公司的JEOL JEM 2100型透射电子显微镜(TEM)上观察。X射线光电子能谱(XPS)在ESCALAB 250型电子能谱仪(Thermo-VG Scientific)上进行,以Al Kα的X射线为激发光源,测定催化剂表面物种中Ru3p、ZN2p、C1s和O1s电子结合能(Eb)及Zn LMM俄歇电子能谱,以表面污染碳的C1s电子结合能(284.6 eV)校正电子结合能数据。DFT理论计算采用Gaussian 09程序包中的M06-2X方法,6-31G(d,p)基组。
1.4 催化剂评价
催化剂评价在1 L哈氏合金高压反应釜中进行,具体步骤如下:取2 g上述制备的Ru-Zn催化剂,46 g ZnSO4·7H2O和280 mL水,密封于1 L的高压反应釜内。用N2气置换3次空气后,再用H2气置换3次N2气,将H2压力调节至5 MPa,搅拌800 r·min-1。升温至150 ℃后,将搅拌转速调节至1 400 r·min-1,加入140 mL苯,反应开始计时,每隔5 min取样。在杭州科晓GC-1690型气相色谱仪上分析产物组成,计算苯转化率,环己烯选择性和收率。色谱柱为AT·SE-30(30 m×0.32 mm×0.25 μm),柱箱温度为70 ℃,进样温度和检测器温度均为210 ℃,柱前压为0.1 MPa。催化剂预处理即升温至150 ℃后,继续搅拌一定时间,然后将搅拌转速调节至1 400 r·min-1,加入140 mL苯,加氢条件同上。
2 结果与讨论
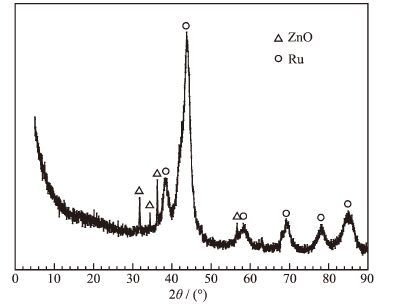
图 1给出了Ru-Zn催化剂的XRD图。可以看出,Ru-Zn催化剂上出现了金属Ru和ZnO的特征衍射峰,说明Ru-Zn催化剂中Ru和Zn分别主要以金属Ru和ZnO存在。Wang等[2]和Sun等[14]也认为Ru-Zn催化剂中Ru-Zn分别以金属Ru和ZnO存在。根据Scherrer公式计算出催化剂粒径为4.2 nm.
图 1
Ru-Zn催化剂的XRD图
Figure 1.
XRD pattern of the Ru-Zn catalyst
图 2给出了Ru-Zn催化剂的TEM照片和粒径分布图。可以看出,Ru-Zn催化剂为圆形或椭圆形的,粒径主要集中分布在4.3 nm左右,与XRD计算结果一致。
 图 2
Ru-Zn催化剂的TEM照片和粒径分布图
Figure 2.
TEM image (a) and particle size distribution (b) of the Ru-Zn catalyst
图 2
Ru-Zn催化剂的TEM照片和粒径分布图
Figure 2.
TEM image (a) and particle size distribution (b) of the Ru-Zn catalyst
图 3给出了不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的XRD图。所有样品上都出现了金属Ru的特征衍射峰。根据Scherrer公式计算出催化剂粒径为4.3 nm左右(见表 1),与催化剂粒径一致。随反应修饰剂ZnSO4浓度增加,催化剂上ZnO的特征衍射峰消失,逐渐出现了(Zn(OH)2)3(ZnSO4)(H2O)盐的特征衍射峰,且衍射峰的强度逐渐增加。这说明Ru催化剂中ZnO与反应修饰剂ZnSO4反应生成了(Zn(OH)2)3(ZnSO4)(H2O)盐,且随ZnSO4浓度增加生成的(Zn(OH)2)3(ZnSO4)(H2O)盐量增加。然而当ZnSO4浓度高于0.41 mol·L-1后,继续增加ZnSO4浓度,(Zn(OH)2)3(ZnSO4)(H2O)盐的衍射峰又消失了,说明该盐量减少。
 图 3
不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的XRD图
Figure 3.
XRD patterns of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 after hydrogenation of benzene
图 3
不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的XRD图
Figure 3.
XRD patterns of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 after hydrogenation of benzene
 表 1
不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的组成、粒径及浆液pH值
Table 1.
Composition and particle size of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 as well as pH value of the slurry after hydrogenation a Measured by pH meter at room temperature; b Measured by XRD; c Measured by XRF; d Blank: in distilled water.
表 1
不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的组成、粒径及浆液pH值
Table 1.
Composition and particle size of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 as well as pH value of the slurry after hydrogenation a Measured by pH meter at room temperature; b Measured by XRD; c Measured by XRF; d Blank: in distilled water.
 表 1 不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的组成、粒径及浆液pH值
表 1 不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的组成、粒径及浆液pH值
Table 1. Composition and particle size of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 as well as pH value of the slurry after hydrogenation a Measured by pH meter at room temperature; b Measured by XRD; c Measured by XRF; d Blank: in distilled water.表 1给出了不同浓度反应修饰剂ZnSO4中加氢后Ru-Zn催化剂的组成及pH值。可以看出,随ZnSO4浓度增加,催化剂上nZn / nRu和nS / nRu原子比逐渐增加,与XRD结果(Zn(OH)2)3(ZnSO4)(H2O)盐量增加一致。然而当ZnSO4浓度高于0.41 mol·L-1后,再增加ZnSO4浓度,催化剂上nZn / nRu和nS / nRu原子比却逐渐减小,与XRD结果(Zn(OH)2)3(ZnSO4)(H2O)盐量减少一致。随ZnSO4浓度增加,室温下pH值逐渐减小,说明ZnSO4水解程度增加,酸性增加。一般情况下,升温有利于水解。这意味着0.57 mol·L-1和0.63 mol·L-1 ZnSO4浆液在加氢温度150 ℃下有更强的酸性,可以溶解部分生成的(Zn(OH)2)3(ZnSO4)(H2O)盐,因此nZn / nRu和nS / nRu原子比逐渐减小。
图 5(a)、(b)和(c)给出了蒸馏水中苯加氢后Ru-Zn催化剂的Ru3p3/2、ZN2p XPS谱和Zn LMM AES谱。可以看出,位于461.6 eV 处的谱峰可以归属为金属Ru03p3/2,说明催化剂表面Ru主要以金属态存在,与XRD结果一致。由ZN2p3/2的电子结合能为1 022.9 eV和Zn LMM电子动能为986.5 eV,催化剂中Zn以Zn2+存在,与XRD结果Zn主要以ZnO存在一致。图 5(d)、(e)和(f)给出了0.63 mol·L-1 ZnSO4中苯加氢后Ru-Zn催化剂的Ru3p3/2、ZN2p XPS谱和Zn LMM AES谱。可以看出,位于462.2 eV处的谱峰可以归属为金属Ru03p3/2,比蒸馏水中加氢后Ru-Zn催化剂的Ru3p3/2高0.6 eV。由ZN2p3/2的电子结合能为1 021.9 eV和Zn LMM电子动能为987.8 eV,催化剂中Zn以Zn2+存在,与XRD结果Zn主要以(Zn(OH)2)3(ZnSO4)(H2O)盐存在一致。且ZN2p3/2的电子结合能比蒸馏水中加氢后Ru-Zn催化剂的低1.0 eV,Zn LMM电子动能高1.3 eV,说明金属Ru0将部分电子转移给了(Zn(OH)2)3(ZnSO4)(H2O)盐的Zn2+.
 图 5
蒸馏水和0.63 mol·L-1 ZnSO4中苯加氢后Ru-Zn催化剂的Ru3p3/2、ZN2p XPS谱和Zn LMM AES谱
Figure 5.
XPS profiles of Ru3p3/2 and ZN2p as well as AES profiles of Zn LMM of the Ru-Zn catalyst in distilled water and 0.63 mol·L-1 ZnSO4 after hydrogenation of benzene
图 5
蒸馏水和0.63 mol·L-1 ZnSO4中苯加氢后Ru-Zn催化剂的Ru3p3/2、ZN2p XPS谱和Zn LMM AES谱
Figure 5.
XPS profiles of Ru3p3/2 and ZN2p as well as AES profiles of Zn LMM of the Ru-Zn catalyst in distilled water and 0.63 mol·L-1 ZnSO4 after hydrogenation of benzene
图 6给出了15 min不同浓度反应修饰剂ZnSO4中Ru-Zn催化剂上苯转化率、环己烯选择性和收率。可以看出,随ZnSO4浓度增加,催化剂活性逐渐降低,环己烯选择性逐渐升高。当ZnSO4浓度高于0.41 mol·L-1后,再增加ZnSO4浓度,催化剂活性升高,环己烯选择性略微降低。当ZnSO4最佳浓度为0.63 mol·L-1时,15 min苯转化率为58.6%,环己烯选择性和收率分别为77.4%和45.3%。该浓度是工业Ru-Zn催化剂正常运行浓度。再增加ZnSO4浓度,浆液酸性会更强,对装置的腐蚀性就增加。
 图 6
不同浓度反应修饰剂ZnSO4中Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 6.
Performance of the Ru-Zn catalyst in the presence of the concentration of the modifier ZnSO4
图 6
不同浓度反应修饰剂ZnSO4中Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 6.
Performance of the Ru-Zn catalyst in the presence of the concentration of the modifier ZnSO4
结合以上表征,ZnSO4浓度影响催化剂上生成的(Zn(OH)2)3(ZnSO4)(H2O)盐量、浆液中的Zn2+浓度和pH值,进而影响Ru-Zn催化剂的苯选择加氢制环己烯性能。当ZnSO4浓度低于0.41 mol·L-1时,随ZnSO4浓度增加,生成的(Zn(OH)2)3(ZnSO4)(H2O)盐量逐渐增加,增加的Zn2+可以转移更多Ru电子,生成更多的Ruδ+物种。Mazzieri等[15]发现缺电子的Ruδ+物种越多,环己烯在催化剂上的吸附能力越弱,越易从催化剂表面逸出,避免它进一步加氢生成环己烷,环己烯选择性逐渐升高。同时增加的Zn2+会优先吸附在较强的Ru活性位上,苯和环己烯只能吸附在剩下吸附能力弱的Ru活性位。这也抑制了环己烯进一步加氢生成环己烷,但降低了催化剂活性。因此,当ZnSO4浓度低于0.41 mol·L-1,随ZnSO4浓度增加,催化剂活性逐渐降低,环己烯选择性逐渐升高。当ZnSO4浓度高于0.41 mol·L-1后,随ZnSO4浓度增加,浆液酸性太强,会溶解部分生成(Zn(OH)2)3(ZnSO4)(H2O)盐,(Zn(OH)2)3(ZnSO4)(H2O)盐量逐渐减小,催化剂活性又逐渐升高,环己烯选择性降低。但环己烯选择性却略有降低,这是由于随ZnSO4浓度增加浆液中Zn2+可以与环己烯形成配合物,图 7给出了Zn2+与环己烯形成配合物的优势构型。利用DFT方法计算出Zn2+分别与1个环己烯分子和2个环己烯分子形成配合物的能量为-618.7和-1 075.6 kJ·mol-1,这说明环己烯可以与Zn2+形成配合物,稳定液相中的环己烯,避免了环己烯再吸附到催化剂表面并加氢生成环己烷。
 图 7
Zn2+与环己烯形成的配合物的优化构型
Figure 7.
Optimized geometries of the complexes formed by Zn2+ and cyclohexene
图 7
Zn2+与环己烯形成的配合物的优化构型
Figure 7.
Optimized geometries of the complexes formed by Zn2+ and cyclohexene
图 8给出了不同预处理时间苯加氢后Ru-Zn催化剂的XRD图。可以看出,不同预处理时间苯加氢后Ru-Zn催化剂上都出现了金属Ru的特征衍射峰。根据Scherrer公式计算出催化剂粒径为4.3 nm左右(表 2),说明预处理不影响催化剂粒径。随预处理时间增加,催化剂上逐渐出现了(Zn(OH)2)3(ZnSO4)(H2O)盐。这可能是由于催化剂中ZnO逐渐与浆液中ZnSO4反应生成了(Zn(OH)2)3(ZnSO4)(H2O)盐。
表 2
不同预处理时间苯加氢后Ru-Zn催化剂的组成
Table 2.
Composition of the Ru-Zn catalyst under the different pretreatment time after hydrogenation
 表 2 不同预处理时间苯加氢后Ru-Zn催化剂的组成
表 2 不同预处理时间苯加氢后Ru-Zn催化剂的组成
Table 2. Composition of the Ru-Zn catalyst under the different pretreatment time after hydrogenation 图 8
不同预处理时间苯加氢后Ru-Zn催化剂的XRD图
Figure 8.
XRD patterns of the Ru-Zn catalyst under the different pretreatment time after hydrogenation of benzene
图 8
不同预处理时间苯加氢后Ru-Zn催化剂的XRD图
Figure 8.
XRD patterns of the Ru-Zn catalyst under the different pretreatment time after hydrogenation of benzene
表 2给出了不同预处理时间苯加氢后Ru-Zn催化剂的组成。可以看出,随预处理时间增加,催化剂中nZn / nRu和nS / nRu逐渐增加,与XRD图上逐渐出现(Zn(OH)2)3(ZnSO4)(H2O)盐的特征峰一致。这说明催化剂中ZnO逐渐与浆液中ZnSO4反应生成了(Zn(OH)2)3(ZnSO4)(H2O)盐。然而预处理15 h后,再增加预处理时间,nZn / nRu和nS / nRu原子比不变,说明催化剂中ZnO已经完全与浆液中ZnSO4反应生成了(Zn(OH)2)3(ZnSO4)(H2O)盐。
图 10给出了不同预处理时间Ru-Zn催化剂的苯选择加氢制环己烯性能。可以看出,随预处理时间增加,催化剂活性逐渐降低,环己烯选择性逐渐升高。然而预处理15 h后,再增加预处理时间,催化剂活性和环己烯选择性几乎不变,说明预处理的最佳时间为15 h。预处理15 h,25 min苯转化率为68.2%,环己烯选择性和收率分别为80.2%和54.7%。因为苯、环己烯和环己烷的沸点接近,工业用溶剂萃取的方法分离它们。环己烯选择性越高,分离就越容易,成本也越低,因此工业要求环己烯的选择性不低于80%。显然,预处理15 h,Ru-Zn催化剂满足了这一要求,而且获得了54.7%的环己烯收率。这超过目前工业Ru-Zn催化剂运行水平:苯转化40%时环己烯选择性和收率分别为80%和32%。
 图 10
不同预处理时间Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 10.
Performance of Ru-Zn catalyst under the different pretreatment time
图 10
不同预处理时间Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 10.
Performance of Ru-Zn catalyst under the different pretreatment time
结合表征,预处理的主要作用是使催化剂中ZnO与反应修饰剂ZnSO4充分反应生成(Zn(OH)2)3(ZnSO4)(H2O)盐。预处理时间增加,生成的 (Zn(OH)2)3(ZnSO4)(H2O)盐量增加,Ruδ+物种增加,催化剂活性降低,环己烯选择性升高。我们课题组还在相同条件下对Ru-La-B/ZrO2催化剂进行了预处理,认为预处理还可以使催化剂的结构稳定化,延长催化剂的使用寿命[16].
图 11给出了预处理后Ru-Zn催化剂的重复使用性能。可以看出,预处理15 h后,Ru-Zn催化剂重复使用6次中,苯转化率稳定66.4%以上,环己烯选择性和收率稳定79.3%和53.8%以上。这说明Ru-Zn催化剂具有良好的重复使用性和稳定性,具有良好的应用前景。
 图 11
Ru-Zn催化剂的重复使用性能
Figure 11.
Reusability of the Ru-Zn catalyst
图 11
Ru-Zn催化剂的重复使用性能
Figure 11.
Reusability of the Ru-Zn catalyst
3 结论
反应修饰剂ZnSO4可以与Ru-Zn催化剂中助剂ZnO反应生成(Zn(OH)2)3(ZnSO4)(H2O)盐。该盐中的Zn2+可以使Ru变为有利环己烯生成的缺电子的Ruδ+物种,而且还可以占据不适宜环己烯生成的强Ru活性位。因此,反应修饰剂ZnSO4浓度增加,(Zn(OH)2)3(ZnSO4)(H2O)盐量的逐渐增加,Ruδ+物种量增加,催化剂活性逐渐降低,环己烯选择性逐渐升高。但当反应修饰剂ZnSO4浓度高于0.41 mol·L-1后,继续增加ZnSO4浓度,由于Zn2+水解浆液酸性太强,可以溶解部分(Zn(OH)2)3(ZnSO4)(H2O)盐,Ru-Zn催化剂活性升高,环己烯选择性降低。环己烯选择性略有降低,是由于ZnSO4溶液中大量的Zn2+可以与生成的环己烯形成配合物,稳定生成的环己烯,抑制环己烯再吸附到催化剂表面并加氢生成环己烷。在ZnSO4最佳浓度0.61 mol·L-1下对Ru-Zn催化剂预处理15 h,Ru-Zn催化剂中助剂ZnO可以与ZnSO4完全反应生成(Zn(OH)2)3(ZnSO4)(H2O)盐,在该催化剂上25 min 苯转化68.2%时环己烯选择性和收率分别为80.2%和54.7%。而且该催化剂具有良好的稳定性和重复使用性能。
-
-
[1]
Liao H G, Ouyang D H, Zhang J, et al. Chem. Eng. J., 2014, 243:207-216 doi: 10.1016/j.cej.2014.01.014
-
[2]
王正宝,张琪,路晓飞,等.催化学报, 2015,36(3):400-407 doi: 10.1016/S1872-2067(14)60231-XWANG Zheng-Bao, ZHANG Qi, LU Xiao-Fei , et al. Chin. J. Catal., 2015,36(3):400-407 doi: 10.1016/S1872-2067(14)60231-X
-
[3]
Yan X H, Zhang Q, Zhu M Q, et al. J. Mol. Catal. A:Chem., 2016,413:85-93 doi: 10.1016/j.molcata.2015.12.013
-
[4]
Zhang P, Wu T B, Jiang T, et al. Green Chem., 2013,15:152-159 doi: 10.1039/C2GC36596K
-
[5]
Nagahara H, Ono M, Konishi M. et al. Appl. Surf. Sci., 1997, 121/122:448-451 doi: 10.1016/S0169-4332(97)00325-5
-
[6]
Struijk J, Moene R, van der Kamp T, et al. Appl. Catal. A: Gen., 1992,89:77-102 doi: 10.1016/0926-860X(92)80079-R
-
[7]
Liu H Z, Liang S G, Wang W T, et al. J. Mol. Catal. A:Chem., 2011,341:35-41 doi: 10.1016/j.molcata.2011.03.021
-
[8]
Fan G Y, Li R X, Li X J, et al. Catal. Commun., 2008,9: 1394-1397 doi: 10.1016/j.catcom.2007.11.039
-
[9]
Sun H J, Jiang H B, Li S H, et al. Chem. Eng. J., 2013,218: 415-424 doi: 10.1016/j.cej.2012.12.041
-
[10]
孙海杰,江厚兵,李帅辉,等.催化学报, 2013,34(4):684-694 doi: 10.1016/S1872-2067(11)60489-0SUN Hai-Jie, JIANG Hou-bin, LI Shuai-Hui, et al. Chin. J. Catal., 2013,34(4):684-694 doi: 10.1016/S1872-2067(11)60489-0
-
[11]
孙海杰,李帅辉,张元馨,等.催化学报, 2013,34(8):1482-1488 doi: 10.1016/S1872-2067(12)60637-8SUN Hai-Jie, LI Shuai-Hui, ZHANG Yuan-Xin, et al. Chin. J. Catal., 2013,34(8):1482-1488 doi: 10.1016/S1872-2067(12)60637-8
-
[12]
孙海杰,陈建军,黄振旭,等.无机化学学报, 2016,32(2):202-210 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20160202&journal_id=wjhxxbcnSUN Hai-Jie, CHEN Jian-Jun, HUANG Zhen-Xu, et al. Chinese J. Inorg. Chem., 2016,32(2):202-210 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20160202&journal_id=wjhxxbcn
-
[13]
孙海杰,郭伟,周小莉,等.催化学报, 2011,32(1):1-16SUN Hai-Jie, GUO Wei, ZHOU Xiao-Li, et al. Chin. J. Catal., 2011,32(1):1-16
-
[14]
Sun H J, Wang H X, Jiang H B, et al. Appl. Catal. A:Gen., 2013,450:160-168 doi: 10.1016/j.apcata.2012.10.016
-
[15]
Mazzieri V A, L'Argentire P C, Coloma-Pascual F. Ind. Eng. Chem. Res., 2003,42(11):2269-2272 doi: 10.1021/ie0209428
-
[16]
王辉,刘仲毅,师瑞娟,等.催化学报, 2005,26(5):407-411WANG Hui, LIU Zhong-Yi, SHI Rui-Juan, et al. Chin. J. Catal., 2005,26(5):407-411
-
[1]
-

图 2 Ru-Zn催化剂的TEM照片和粒径分布图
Figure 2 TEM image (a) and particle size distribution (b) of the Ru-Zn catalyst

图 3 不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的XRD图
Figure 3 XRD patterns of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 after hydrogenation of benzene

图 5 蒸馏水和0.63 mol·L-1 ZnSO4中苯加氢后Ru-Zn催化剂的Ru3p3/2、ZN2p XPS谱和Zn LMM AES谱
Figure 5 XPS profiles of Ru3p3/2 and ZN2p as well as AES profiles of Zn LMM of the Ru-Zn catalyst in distilled water and 0.63 mol·L-1 ZnSO4 after hydrogenation of benzene
(a,b,c) Blank: in distilled water; (d,e,f) 0.63 mol·L-1

图 6 不同浓度反应修饰剂ZnSO4中Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 6 Performance of the Ru-Zn catalyst in the presence of the concentration of the modifier ZnSO4

图 7 Zn2+与环己烯形成的配合物的优化构型
Figure 7 Optimized geometries of the complexes formed by Zn2+ and cyclohexene
(a) Complex formed by Zn2+ and one cyclohexene molecule; (b) Complex formed by Zn2+ and two cyclohexene molecules

图 8 不同预处理时间苯加氢后Ru-Zn催化剂的XRD图
Figure 8 XRD patterns of the Ru-Zn catalyst under the different pretreatment time after hydrogenation of benzene

图 10 不同预处理时间Ru-Zn催化剂的苯选择加氢制环己烯性能
Figure 10 Performance of Ru-Zn catalyst under the different pretreatment time
表 1 不同浓度反应修饰剂ZnSO4中苯加氢后Ru-Zn催化剂的组成、粒径及浆液pH值
Table 1. Composition and particle size of the Ru-Zn catalyst in the presence of the different concentration of the modifier ZnSO4 as well as pH value of the slurry after hydrogenation a Measured by pH meter at room temperature; b Measured by XRD; c Measured by XRF; d Blank: in distilled water.
 下载: 导出CSV
下载: 导出CSV
表 2 不同预处理时间苯加氢后Ru-Zn催化剂的组成
Table 2. Composition of the Ru-Zn catalyst under the different pretreatment time after hydrogenation
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 366
- HTML全文浏览量: 82
 下载:
下载:
