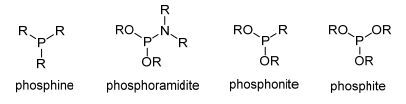
 图1
单膦配体的结构示意图
Figure1.
Structures of chiral monodentate phosphorus ligands
图1
单膦配体的结构示意图
Figure1.
Structures of chiral monodentate phosphorus ligands
引用本文:
张凤, 刘祥华, 刘玮, 邓国军. 钯-单膦催化剂在烯烃不对称硅氢化反应中的应用[J]. 有机化学,
2017, 37(10): 2555-2568.
doi:
10.6023/cjoc201704011

Citation: Zhang Feng, Liu Xianghua, Liu Wei, Deng Guojun. Application of Pd-Monodentate Phosphorus Catalysts in the Asymmetric Hydrosilylation Reactions of Alkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2555-2568. doi: 10.6023/cjoc201704011
Citation: Zhang Feng, Liu Xianghua, Liu Wei, Deng Guojun. Application of Pd-Monodentate Phosphorus Catalysts in the Asymmetric Hydrosilylation Reactions of Alkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2555-2568. doi: 10.6023/cjoc201704011
钯-单膦催化剂在烯烃不对称硅氢化反应中的应用
English
Application of Pd-Monodentate Phosphorus Catalysts in the Asymmetric Hydrosilylation Reactions of Alkenes
Abstract:
Asymmetric hydrosilylation of alkenes, which has been recognized as an important method for the preparations of optically active secondary alcohols, deserves widespread attention over the world. It is reported that such reaction can be catalyzed by Pd-monodentate phosphorus catalysts with excellent reactivity and enantioselectivity. In the past decades, a wide variety of chiral monodentate phosphorus ligands have been developed because of their stable structure, facile synthesis, convenient modification, unique efficiency. Among them, there are three predominant classes of ligands-phosphines based on an axially chiral biaryl scaffold, phosphines based on a planar chiral ferrocene scaffold and chiral phosphoramidites. Herein, the recent advances in asymmetric hydrosilylation of alkyl-substituted alkenes, styrene derivatives, 1, 3-dienes and other carbon-carbon double bond compounds catalyzed by palladium monodentate phosphorus catalysts are summarized. The perspective is also discussed.
-
Key words:
- Pd catalyzed
- / monodentate phosphorus ligands
- / alkenes
- / asymmetric hydrosilylation
-
钯催化剂具有独特的性质, 成为有机合成中不可或缺的工具[1, 2].钯催化的烯烃的不对称反应是合成多种手性化合物的重要方法之一[3, 4].手性仲醇是一类非常重要的有机合成中间体, 广泛应用于医药、农药及食品添加剂等合成中[5].烯烃的不对称硅氢化反应是氢硅烷对碳-碳双键的立体选择性加成, 碳-硅键经过Fleming-Tamao氧化反应在手性中心完全保持不变的前提下转化成碳-氧键, 从而成为制备手性仲醇的有效途径之一.在合成手性仲醇的众多方法中, 钯催化的烯烃的不对称硅氢化反应由于反应条件温和、反应效率高、环境友好、无需耐高压设备和操作简便等优点引起了科研工作者的普遍关注.
手性配体是手性催化剂产生不对称诱导和控制的源泉, 不对称催化反应的关键之一是手性配体的设计合成.虽然早在20世纪70年代, Kumada等[6]就报道了首例钯催化的烯烃的不对称硅氢化反应, 但是催化剂的选择性很低.直到20世纪90年代初, 采用钯金属前体及单膦配体所形成的手性配合物作为催化剂后, 烯烃的不对称硅氢化反应才取得了突破性的进展.单膦配体具有易于合成、结构灵活、催化性能优异等特点.此外, 当中心金属离子只能提供一个配位轨道时, 单膦配体就能够以一个配体同中心金属离子络合, 进而催化不对称反应.正是由于这些优点, 越来越多的单膦配体被合成出来.目前, 单膦配体已成为不对称催化领域中研究热点, 成功应用于不对称催化共轭加成反应、不对称催化烯丙基取代反应、不对称催化氢化反应等[7, 8].
单膦配体按照磷原子成键的种类主要可以分为膦配体(phosphine)、亚磷酰胺酯配体(phosphoramidite)、亚膦酸酯配体(phosphonite)、亚磷酸酯配体(phosphite)以及其他类型的单膦配体(图 1).其中, 平面手性单齿膦配体、轴手性单齿膦配体以及手性单齿亚磷酰胺配体在烯烃的不对称硅氢化反应中取得了优异的催化效果.鉴于单膦配体对不对称催化硅氢化反应的重要性, 且对烯烃的不对称硅氢化反应系统评述较少[9], 本文按照单膦配体的结构类型, 对钯催化的烯烃的不对称硅氢加成的研究进展加以综述, 以便于人们对该领域进行全面的了解, 更好地设计配体及催化剂从而推进该领域的应用发展.
图1
单膦配体的结构示意图
Figure1.
Structures of chiral monodentate phosphorus ligands
1 手性单齿膦配体在钯催化的烯烃的不对称硅氢化反应中的应用
1.1 平面手性单齿膦配体
在不对称催化领域中, 平面手性二茂铁类配体以其特殊的刚性骨架结构和良好的电子效应从众多手性配体中脱颖而出.其中, 许多含膦、氮的二茂铁配体(例如Josiphos、Ferriphos、Walphos等)由于兼具平面手性及中心手性, 在多种不对称催化反应中体现出了优异的不对称诱导能力, 有着广泛的应用前景[10].钯催化的不对称硅氢化反应中, 表现较为突出的二茂铁骨架手性配体则是单齿膦配体(图 2).这类配体往往含有杂原子或者芳环, 与金属钯配位时, 除了磷原子作为主要的配位点外, 金属与杂原子或芳基可能存在着次级相互作用.次级相互作用的强弱影响催化剂的活性及选择性.目前, 平面手性的二茂铁类单膦配体已成功应用于钯催化的苯乙烯、1, 3-二烯烃等底物的不对称硅氢化反应中.
 图2
用于烯烃不对称硅氢化反应的平面手性膦配体
Figure2.
Planar chiral phosphines used in the asymmetric hydrosilylation of alkenes
图2
用于烯烃不对称硅氢化反应的平面手性膦配体
Figure2.
Planar chiral phosphines used in the asymmetric hydrosilylation of alkenes
1980年, Kumada等[11]报道了首例二茂铁骨架的单齿膦配体在烯烃的不对称硅氢化反应中的性能研究.他们将配体L1与钯形成的络合物(Cat. 1)用于苯乙烯与三氯硅氢的不对称硅氢化反应中, 当催化剂的用量为0.01 mol%, 70 ℃下, 反应40 h, 底物才能完全转化, 所得产物的ee值仅为52% (Scheme 1).相同条件下, 将配体L1应用于降冰片烯的不对称硅氢化反应中, 所得产物的ee值为53%.
 图式 1
平面手性膦配体L1在烯烃的不对称硅氢化反应中的应用
Scheme1.
Application of planar chiral phosphine L1 in the asymmetric hydrosilylation of alkenes
图式 1
平面手性膦配体L1在烯烃的不对称硅氢化反应中的应用
Scheme1.
Application of planar chiral phosphine L1 in the asymmetric hydrosilylation of alkenes
1983年, Hayashi等[12]报道了首例环状1, 3-二烯烃的不对称硅氢化反应.环状1, 3-二烯烃与硅烷的不对称硅氢化反应以1, 4-顺式加成为主生成手性烯丙基硅烷.烯丙基硅烷通过Fleming-Tamao氧化反应进一步生成手性烯丙醇.他们将一系列二茂铁骨架的单齿膦配体应用于钯催化的环戊二烯及1, 3-环己二烯的不对称硅氢化反应中. 30 ℃下, Cat. 1在环戊二烯与甲基二氯硅烷的反应中, 以87%的产率得到ee值为22%的产物.当底物为1, 3-环己二烯时, 虽然产率高达95%, 产物的ee值仅为2%.随后, 他们[13]将二茂铁骨架的催化剂Cat. 1应用于钯催化的链状1, 3-二烯烃如1-苯基-1, 3-丁二烯(13a)的不对称硅氢化反应中, 以62%的收率得到了两个产物.其中, 1, 4-加成产物和1, 2-加成产物的比例为94:6, ee值分别为63%和30%.
接下来, 他们[14]又合成了含氟长烷基链的配体L2, 并将其与金属钯形成的络合物Cat. 2应用于环戊二烯的不对称硅氢化反应中(Scheme 2).当催化剂的用量为0.016 mol%, 室温下反应90 h, Cat. 2a和Cat. 2b所得产物的ee值分别为57%和55%.而相同条件下, Cat. 1在该反应中没有活性.这可能是含氟侧链的引入使得催化剂溶解性能有了很大提高, 同时大大降低了氮原子的配位能力, 便于底物与金属配位, 从而促进催化反应的进行.然而, 含氟长烷基链的配体L2在链状1, 3-二烯烃的不对称硅氢化反应中, 选择性仍然不理想.
 图式 2
平面手性膦配体L2在环戊二烯的不对称硅氢化反应中的应用
Scheme2.
Application of planar chiral phosphines L2 in the asymmetric hydrosilylation of cyclopentadiene
图式 2
平面手性膦配体L2在环戊二烯的不对称硅氢化反应中的应用
Scheme2.
Application of planar chiral phosphines L2 in the asymmetric hydrosilylation of cyclopentadiene
1998年, Togni等[15]将含有吡唑环的二茂铁骨架的单膦配体L3与金属钯形成的络合物Cat. 3应用于一系列苯乙烯及其衍生物的不对称硅氢化反应中, 虽然催化剂的活性略有提高, 但是仅取得了中等选择性(Scheme 3).同时, 他们发现产物的构型与底物中对位基团的电子效应有关.例如, Cat. 3a催化的富电子的4-氨基苯乙烯得到R构型为主的产物, ee值为64%;而缺电子的4-氯苯乙烯得到S构型的产物, ee值为67%.同时, 他们将Cat. 3应用于Pd(COD)Cl2催化的降冰片烯的不对称硅氢化反应中.研究结果表明, 催化剂的活性和选择性取决于磷及吡唑环上的取代基.增大R2的位阻可能削弱了氮原子的配位能力, 从而使得催化剂的活性增加, 反应能在较低温下进行, 选择性也相应的增加.当R1为氢原子时, 降低磷原子电子云密度, 催化剂的活性和选择性进一步增加.配体L3b的R1为氢原子, 磷原子上的芳基为吸电子的3, 5-二三氟甲基苯基, R2为大位阻基2, 4, 6-三甲基苯基.该配体在该降冰片烯的不对称硅氢化反应中取得了>99.5%的对映选择性.这也是迄今为止, 该底物取得的最高对映选择性.
 图式 3
平面手性膦配体L3在取代苯乙烯及降冰片烯的不对称硅氢化反应中的应用
Scheme3.
Application of planar chiral phosphines L3 in the asymmetric hydrosilylation of substituted styrenes and norbornene
图式 3
平面手性膦配体L3在取代苯乙烯及降冰片烯的不对称硅氢化反应中的应用
Scheme3.
Application of planar chiral phosphines L3 in the asymmetric hydrosilylation of substituted styrenes and norbornene
2001年, Jones等[16]设计合成了一类含有杂环的新型平面手性膦配体L4.采用[Pd(C3H5)Br]2作为催化剂前体, 催化剂用量为0.25 mol%, 并将反应体系降至-40 ℃, 含有呋喃环的配体L4a在苯乙烯及其衍生物的不对称硅氢化反应中取得了87% ee (表 1, Entry 1).将反应温度降至-50 ℃, 其他条件保持不变, 含噻吩环的配体L4b获得了92% ee的产物(表 1, Entry 2). 2006年, Gibson等[17]将相同手性骨架的配体L5用于钯催化的苯乙烯类底物的不对称硅氢化反应中, 仅取了中等的选择性(表 1, Entries 3, 4).
 表 1
平面手性膦配体L4、L5及L6在钯催化的苯乙烯硅氢化反应中的应用a
Table 1.
Application of planar chiral phosphines L4、L5 and L6 in the asymmetric hydrosilylation of styrene
表 1
平面手性膦配体L4、L5及L6在钯催化的苯乙烯硅氢化反应中的应用a
Table 1.
Application of planar chiral phosphines L4、L5 and L6 in the asymmetric hydrosilylation of styrene

Entry L Catalyst/
mol%Condition Yield/%
2ee/%
31b, c, d L4a 0.25 -40 ℃, 48 h >85 87 2b, c, d L4b 0.25 -50 ℃, 48 h >85 92 3e L5a 0.5 r.t., 48 h 99 55 4f L5b 0.5 r.t., 48 h 97 77 5 L6a 1 r.t., 1.6 h 100 86 6 L6b 1 r.t., 8 h 100 25 7 L6c 1 r.t., 1.5 h 100 79 8 L6b 0.1 r.t., 8 h 100 90 9 L6c 0.1 r.t., 3.5 h 100 85 10g L6c 0.1 r.t., 20 s 100 62 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, [L]/[Pd]=2, solvent free unless otherwise stated. b [L]/[Pd]=1.cCH2Cl2 or CDCl3 as solvent.d [Pd(C3H5)Br]2 as precursor. eCH2Cl2 as solvent. fBenzeneas solvent. g[L]/[Pd]=1.2. Johannsen等[18]将二茂铁骨架芳基修饰的配体L6应用于苯乙烯类底物的不对称催化反应中.他们发现增大配体中芳环上的电子云密度, 有利于催化剂活性和选择性的提高.他们采用[Pd(C3H5)Cl]2作为催化剂前体, 配体与金属钯的比例为2, 催化剂的用量为1 mol%, 富电子配体L6a在1.6 h内底物完全转化, 取得了86%的对映选择性(表 1, Entry 5);而在相同条件下, 缺电子配体L6b需要8 h才能完成反应, ee值仅为25%(表 1, Entry 6).保持配体与金属的比例不变, 将催化剂的用量降为0.1 mol%时, 配体L6a所得产物的ee值升高至90% (Entry 8), 配体L6c所得产物的ee值也由79% (表 1, Entry 7) 升高至85%(表 1, Entry 9).此外, 他们以配体L6c为例, 研究了配体与金属的比例对于催化剂的活性和选择性的影响.发现降低配体与金属的比例, 催化剂的选择性下降明显, 但是活性却大大的增加了.如当金属与配体的比例为0.1:0.12时, 底物在20 s内完全转化, 转化频率(TOF)值高达180000 h-1(表 1, Entry 10).
在Chalk-Harrod机理的基础上, 他们以实验结果为依据, 提出了硅氢化反应可能经历了两个不同的催化循环过程(Scheme 4):配体与金属的比例为2时, 反应按慢催化循环方向进行; 当金属与配体的比例小于2时, 反应往快催化循环方向进行.在慢催化循环中, PdP2先与HSiCl3氧化加成, 单膦配体再与苯乙烯发生配位交换, 烯烃插入Pd—H键后, 另一个单膦配体与钯络合物配位, 还原消除后重新生成PdP2.与慢循环不同的是, 当单膦配体比例较小时, 为了维持配位稳定性, 快循环中另一个苯乙烯与钯络合物配位, 再经过还原消除得到二配位的化合物, 最后与HSiCl3氧化加成完成催化循环.快循环中, 第二个烯烃参与配位, 故能很好地解释配体与金属比例降低时, 反应速率反而大大增加.
 图式 4
基于单膦配体用量的两个催化循环
Scheme4.
Two proposed catalytic cycles depending on loadings of phosphine
图式 4
基于单膦配体用量的两个催化循环
Scheme4.
Two proposed catalytic cycles depending on loadings of phosphine
Ohmura等[19]将含氧侧链的二茂铁骨架的手性单膦配体L7应用于1, 3-环己二烯的不对称硅氢化反应中(Scheme 5).为了提高催化剂的的活性及选择性, 他们对配体、硅烷等进行了优化.室温下, 以Pd(Ph-CN)2Cl2为催化剂前体, 采用缺电子基的苯基二氟硅烷与环己二烯反应, 催化剂的用量为1 mol%时, 含氮侧链的配体L1仅取得了9% ee, 而含氧侧链的配体选择性有了大大的提高.例如, 在相同的条件下, L7a取得了54% ee.缺电子配体L7b取得了65% ee.采用[Pd(C3H5)Cl]2作为催化剂前体, 含吸电子基团配体L7c ee值升高至77%.上述研究结果表明, 配体侧链上的取代基影响催化剂的活性及选择性, 这可能与含氧侧链的配体与钯共配位能力较弱有关.
 图式 5
平面手性膦配体L7在环己二烯的不对称硅氢化反应中的应用
Scheme5.
Application of planar chiral phosphines L7 in the asymmetric hydrosilylation of cyclohexadiene
图式 5
平面手性膦配体L7在环己二烯的不对称硅氢化反应中的应用
Scheme5.
Application of planar chiral phosphines L7 in the asymmetric hydrosilylation of cyclohexadiene
2002年Hayashi等[20]将磷上连有两个二茂铁基的含氧侧链配体L8应用于1, 3-癸二烯(13b)的不对称硅氢化反应中(Eq. 1).他们也发现产物的ee值取决于配体的电子效应, 含缺电子基的配体往往能得到更高的ee值.例如, 20 ℃下, 富电子配体L8b仅取得了68% ee, 而缺电子的配体L8c以89%的区域选择性得到了1, 4-加成产物, 产物的ee值为87%, 将反应体系温度降至-5 ℃, 产物的ee值提高至93%.
在此基础上, Hayashi等[21]将含二茂铁结构的手性配体L7和L8应用于钯催化的1-丁烯-3-炔(18a)的不对称硅氢化反应中, 得到了轴手性的丙二烯基硅烷.他们发现产物的光学纯度取决于底物4位取代基的位阻.相同条件下, 位阻小的底物, 催化剂选择性较低(表 2, Entries 1, 2).当底物4位为叔丁基(18b)时, 配体L7a在20 ℃下, 反应9 h, 以81%的产率得到72% ee的产物(表 2, Entry 3).含两个二茂铁基的配体L8a催化活性较低, 但所得产物ee值升为85%(表 2, Entry 4).进一步将反应温度降至0 ℃, ee值上升至90%(表 2, Entry 5). 2006年, Hayashi课题组[22]合成了2位及5位为轴手性的联二萘基取代的新型手性磷杂环戊二烯及其金属茂, 并将其一并应用于18b的不对称硅氢化反应中.磷杂二茂铁配体L9在0 ℃下反应72 h, 得到了92% ee的产物21(表 2, Entry 6).
表 2
平面手性膦配体L7a、L8a及L9在钯催化的1, 3-烯炔的不对称硅氢化反应中的应用a
Table 2.
Application of planar chiral phosphines L7a, L8a and L9 in the asymmetric hydrosilylation of 1, 3-enynes

Entry 18 L Condition Yield/% ee/% of 20 1 18a L7a 20 ℃, 18 h 54 56 2 18a L8a 20 ℃, 64 h 90 77 3 18b L7a 20 ℃, 9 h 81 72 4 18b L8a 20 ℃, 48 h 59 85 5 18b L8a 0 ℃, 160 h 37 90 6b 18b L9 0 ℃, 72 h 82 92 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 1 mol% catalyst, [Ligand]/[Pd]=2.2, solvent free unless otherwise stated. bee of 21. 1.2 轴手性单齿膦配体
轴手性配体在整个不对称领域占据着重要地位, 如具有联二萘骨架的轴手性双膦配体BINAP, 因成功应用于多种类型的不对称催化反应而被称为“明星分子”[23].然而, Hayashi等[24]发现金属钯与BINAP形成的络合物在高达60 ℃时, 仍然不能使烯烃发生不对称硅氢化反应.这可能是双膦配体与钯配位后, 形成了配位稳定的络合物, 占据了钯的两个空轨道, 底物不能与金属有效配位, 故催化反应不能顺利进行.而单膦配体就能够以一个配体与钯配位, 进而催化不对称反应.轴手性的单齿膦配体(图 3)底物普适性好, 在烷基烯烃、苯乙烯、1, 3-二烯烃等底物中均取得了优异的催化效果.
 图3
用于烯烃不对称硅氢化反应的轴手性单齿膦配体
Figure3.
Axially chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes
图3
用于烯烃不对称硅氢化反应的轴手性单齿膦配体
Figure3.
Axially chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes
Hayashi等合成了联二萘骨架的配体L10, 并将其应用于一系列链状末端烯烃的不对称硅氢化反应中, 首次以高区域选择性、高收率及优异的对映选择性生成手性仲醇(表 3).如采用[Pd(C3H5)Cl]2作为催化剂前体, 当催化剂的用量为0.1 mol%时, 40 ℃下反应24 h, 配体L10a (MeO-MOP)在1-辛烯(22a)与三氯硅烷的不对称硅氢化反应中, 取得了83%的产率, 直链产物和支链产物的比例为7:93, 支链产物的对映选择性为95%(表 3, Entry 1).当甲氧基被异丙氧基取代时得到配体L10b, 其对应产物的ee值略有下降至91% (表 3, Entry 2);甲氧基被更大位阻的苄氧取代配体L10c虽然取得了高达95% ee, 可是区域选择性下降明显, 仅为80:20(表 3, Entry 3).将MeO-MOP中的甲氧基变成乙基时所得的配体L10d, 无论是区域选择性还是立体选择性均与MeO-MOP相近, 说明氧原子并不是获取高选择性的必要条件(表 3, Entry 4).
表 3
轴手性单膦配体L10在链状端烯的不对称硅氢化反应中的应用a
Table 3.
Application of axially chiral phosphines L10 in the asymmetric hydrosilylation of acyclic terminated alkenes

Entry 22 L Condition Yield/% 23:24 ee/% of 26 1 22a L10a 40 ℃, 24 h 83 93:7 95 2 22a L10b 40 ℃, 24 h 88 90:10 91 3 22a L10c 40 ℃, 24 h 85 80:20 95 4 22a L10d 40 ℃, 24 h 80 90:10 93 5 22b L10a 40 ℃, 24 h 91 89:11 94 6 22c L10a 40 ℃, 72 h 90 94:6 95 7 22d L10a 40 ℃, 24 h 90 81:19 97 8b 22e L10a 30 ℃, 60 h 81 80:20 92 9 22f L10a 40 ℃, 24 h 100 66:34 96 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 0.1 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. bTHF as solvent. MeO-MOP不仅在链状端烯的不对称硅氢化反应中取得了优异的催化效果, 在环状烯烃中也有较好的应用(Scheme 6). 1992年, Hayashi等[25]将Pd/MeO-MOP催化剂应用于降冰片烯和三氯硅烷的不对称硅氢化反应中, 在催化剂的用量为0.01 mol%时, -20 ℃下反应3 d, 以定量的产率生成外-2-三氯硅基降冰片烷, 经氧化后得到ee值为96%的手性外-降冰片醇.降冰片烯的环上的酯基在硅氢化反应中不受影响.此外, 改变三氯硅氢用量, 降冰片二烯可选择性地生成单硅氢化产物或双硅氢化产物.若三氯硅氢的用量为1 equiv.时, 生成单硅氢化产物, ee值为95%;若三氯硅氢的用量为2.5 equiv.时, 两个双键均发生反应, 所得产物的ee值大于99%.随后, 他们将Pd/MeO-MOP应用范围进一步拓展到呋喃类底物的不对称硅氢化反应中[26]. 2, 3-二氢呋喃在催化剂的用量为0.2 mol%时, 25 ℃下反应12 h, 以86%的产率生成2-三氯硅基四氢呋喃, 与格氏试剂作用后, 得到ee值为82%的手性2-三甲基硅基四氢呋喃. 2, 5-二氢呋喃在催化剂的用量为0.1 mol%时, 40 ℃下反应24 h, 以65%的产率生成3-三氯硅基四氢呋喃, 经氧化后, 以83%的收率得到ee值为95%的手性醇.他们进一步将底物拓展到酯基取代的7-氧杂二环[2, 2, 1]庚烷中, 以100%的区域选择性生成了95% ee的含多个手性中心产物.
 图式 6
MeO-MOP在环状烯烃的不对称硅氢化反应中的应用
Scheme6.
Application of MeO-MOP in the asymmetric hydrosilylation of cyclic alkenes
图式 6
MeO-MOP在环状烯烃的不对称硅氢化反应中的应用
Scheme6.
Application of MeO-MOP in the asymmetric hydrosilylation of cyclic alkenes
配体MeO-MOP虽然在脂肪烯烃的硅氢化反应中取得了优异的催化性能, 但在苯乙烯的不对称硅氢化反应中, 在0 ℃及无溶剂的条件下, 仅取得了14%ee[27].采用苯作为溶剂, 催化剂的选择性升高至71%.然而, 令人遗憾的是, 其他2'位为给电子基的配体(L10d、L10e)或吸电子基的配体(L10f、L10g)取得的ee值均很低, 说明2'位电子效应对于催化剂选择性并不起决定作用.后来, 他们发现配体中2'位的位阻对于选择性有很大影响, 2'位为氢的配体L11a (H-MOP)取得了高达95% ee值. 2001年, Hayashi等[28]合成了结构与H-MOP类似的2位磷原子上为吸电子基[3, 5-(CF2)2C6H3]的配体L11b, 其在钯催化的苯乙烯与三氯硅氢的不对称催化反应中, 取得了优异的催化效果.在0 ℃下, 当催化剂的用量为0.1 mol%时, 底物在1 h内完全转化, 所得产物的ee值为97%.降低温度至-20 ℃, ee值进一步提高至98%. 1995年, 他们[29]通过钯催化的不对称交叉偶联反应合成了2'-位为氢原子的轴手性联芳基骨架的单齿膦配体L12.采用[Pd(C3H5)Cl]2/L12作为催化剂, 0 ℃下反应24 h, 以68%的收率得到了产物, 对映选择性为91%. Bringmann等[30]报道了相同骨架的类似配体L13, 配体. L13的2'为甲基, 在相同条件下仅取得了50% ee.上述结果表明, 2'位取代基在芳基烯烃的不对称硅氢化反应中扮演着重要角色, 尽管具体的原因还不明了.其中, 2'位为氢的配体有着较好的选择性.
1996年, Hayashi等[31]首次将MOP类配体应用于[Pd(C3H5)Cl]2催化的环状1, 3-二烯烃与三氯硅烷的反应中.当底物为环戊二烯时, MeO-MOP在20 ℃下反应14 h, 底物完全转化, 所得产物的ee值为39%(表 4, Entry 1).同样条件下, H-MOP的选择性仅为28%(表 4, Entry 2).与MeO-MOP结构类似的联二菲骨架的配体L14虽然活性不高, 却取得了80%的ee值(表 4, Entry 3).催化活性不高的原因可能是Pd-Ar-MOP催化体系在双烯与三氯氢硅烷加成反应体系中的溶解性较低引起的.后来, 他们[32]将2'位为芳基的配体L15应用于环戊二烯的不对称硅氢化反应中.研究结果表明, 0 ℃下, 钯催化剂的用量为0.25 mol%时, 2'位芳基越富有电子, 位阻越大, 所生成的产物ee值越高.如配体L15a的ee值为69%(表 4, Entry 4), 富电子配体L15b和L15e的ee值分别为76%(表 4, Entry 5) 和79%(表 4, Entry 8), 而既富有电子又具有较大位阻的配体L15f, ee值高达88%(表 4, Entry 9).进一步降低反应温度至-20 ℃, ee值上升至90%(表 4, Entry 10).与之相对应的是缺电子基的配体L15c在相同条件下(0 ℃)仅取得了47% ee(表 4, Entry 6).而当底物为环己二烯时, 0 ℃下, 配体L15f 仅取得79% ee(表 4, Entry 13).进一步降低反应温度不能生成目标产物, 这可能是钯催化剂低温下不能溶解于反应体系所致.为了提高此催化体系的溶解性, 他们[33]在配体L15f的6位和6'位上引入长链烷基, 得到配体L16.配体L16能很好的溶于反应体系, 将其应用于钯催化的环状1, 3-二烯烃与三氯硅烷的加成反应中.当底物为环戊二烯, 钯催化剂用量为0.25 mol%, 配体为0.5 mol%时, -30 ℃下, 配体L16取得了75%的产率以及91%的ee值(表 4, Entry 11). -10 ℃下, 配体L16在1, 3-环己二烯的不对称催化中, 取得了70%的产率以及83%的ee值(表 4, Entry 12).除此之外, 轴手性的单齿膦配体还能催化链状l, 3-二烯烃的硅氢加成反应, 然而仅取得了中等对映选择性[34].
表 4
轴手性膦配体用于的环状1, 3-二烯烃的不对称硅氢化反应a
Table 4.
Axially phosphines used in the asymmetric hydrosilylation of cyclic 1, 3-dienes

Entry 7/8 L Condition Yield/% of
9/10ee/% of
11/121b 7 L10a 20 ℃, 14 h 100 39 2b 7 L11a 20 ℃, 3 h 91 28 3b 7 L14 20 ℃, 120 h 99 80 4 7 L15a 0 ℃, 24 h 84 69 5 7 L15b 0 ℃, 24 h 84 76 6 7 L15c 0 ℃, 24 h 83 47 7 7 L15d 0 ℃, 24 h 85 76 8 7 L15e 0 ℃, 24 h 83 79 9 7 L15f 0 ℃, 24 h 79 88 10 7 L15f -20 ℃, 72 h 89 90 11 7 L16 -30 ℃, 168 h 75 91 12 8 L16 -10 ℃, 168 h 70 83 13 8 L15f 0 ℃, 72 h 75 79 aAll reactions were conducted with [Pd (C3H5)Cl]2as precursor, 0.25 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. b0.1 mol% catalyst. 2004年, Pregosin等[35]合成并表征了配体L17.与典型的MOP类配体相比, 这类配体结构的显著特点是联二萘骨架的2位磷原子上连有大位阻的芳基.他们将这些配体与Hayashi之前报道的2位为二苯基膦的配体一并应用于苯乙烯的不对称硅氢化反应中.在5 ℃下, 采用[Pd(C3H5)Cl]2作为催化剂前体, 三氯硅氢作为硅烷, 当催化剂的用量为0.096 mol%时, 2'位为氢的配体L11a (H-MOP)取得了92% ee(表 5, Entry 1), 而在相同条件下, 较大位阻的配体L15a也取得了88% ee(表 5, Entry 2).当2'位为甲氧基的配体L10a (MeO-MOP)的ee值仅为7%(表 5, Entry 3), 而与之相对应的大位阻配体L17b却取得了57% ee(表 5, Entry 4);当2'位为氰基时, 也是大位阻配体的ee值远远高于相应的二苯基膦配体(表 5, Entries 5, vs. 6).他们的研究结果表明, 2位的位阻对于催化剂的选择性有着显著的影响, 大位阻基团的引入可能有利于获取高对映选择性的产物.虽然位阻效应导致催化剂选择性差异的具体原因还不清楚, 但是, 他们认为, 大位阻基团使得磷-碳键的旋转受阻, 有利于创造刚性的“手性口袋”, 从而促进催化剂选择性的提高[36].
表 5
轴手性膦配体L17~L22在苯乙烯的不对称硅氢化反应中的应用a
Table 5.
Application of axially chiral phosphines L17~L22 in the asymmetric hydrosilylation of styrene

Entry L Condition Yield/% of 2 ee/% of 3 1 L11a 0 ℃, 20 h 100 92 2 L17a 5 ℃, 16 h 100 88 3 L10a 5 ℃, 16 h 100 7 4 L17b 0 ℃, 43 h 100 57 5 L10g 5 ℃, 2 h 100 42 6 L17c 5 ℃, 16 h 100 81 7 L18 r.t., 70 h 100 18 8b L18 r.t., 70 h 100 72 9 L19 0 ℃, 10 h 78 95 10 L20 r.t., 24 h >99 80 11b L21 r.t., 18 h 92 91 12c L22a r.t., 3 h 63 54 13c L22b r.t., 3 h 90 54 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 0.1 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. bBenzeneas solvent. c 2.5 mol% catalyst. 此外, Gladiali等[37]将双膦配体BINAP部分氧化得到单齿膦配体L18 (BINAPO), 并将其应用于苯乙烯的不对称硅氢化反应中.室温下, 当配体与金属的比例为2时, 底物与三氯硅氢反应70 h后, 底物完全转化, 仅取得了18% ee(表 5, Entry 7).当采用苯作为溶剂时, 催化剂的对映选择性升高至72%(表 5, Entry 8).受此启发, 2004年Kurita等[38]报道了单膦配体L19 (BINAPSb)的合成及性能研究.他们将其应用于苯乙烯的不对称硅氢化反应中, 0 ℃下反应10 h时, 以78%的收率、95%的ee得到S-苯乙醇(表 5, Entry 9).而双铋配体在相同条件下反应24 h, 仅取得了12% ee.这一结果说明, 配体中单个磷原子的存在是该反应中获得较好催化效果的必要条件.
2011年, Higham等[39]采用联二萘骨架的伯膦和二氯乙烷作为原料, 在甲基锂的作用下, 合成了稳定的联二萘骨架的手性磷杂环丙烷L20.将2 equiv.配体与顺式[Pd(COD)Cl]2反应, 通过高分辨质谱、核磁共振及X射线单晶衍射等研究了手性磷杂环丙烷与后过渡金属的配位行为, 了解到具有对热及空气稳定的手性磷杂环丙烷与后过渡金属配位时稳定性并没有下降.他们将这种新型的稳定的手性磷杂环丙烷配体用于[Pd(C3H5)Cl]2催化的苯乙烯的不对称硅氢化反应中, 在反应条件未经优化的前提下, 取得了80% ee(表 5, Entry 10).
2012年, Lemaire等[40]从手性BINOL出发经过5步反应得到了兼具P手性和轴手性的配体L21.室温下, 采用[Pd(C3H5)Cl]2作为催化剂前体, 当催化剂的用量为0.04 mol%时, 配体取得了91%的对映选择性(表 5, Entry 11).同时, 他们发现仅改变配体中磷的构型, 并不影响产物的构型; 而改变轴手性的构型, 产物的构型发生变化.这说明磷原子的构型在不对称诱导中不起决定性的作用, 配体本身的位阻/电子效应可能更大程度上影响催化性能.最近, Panossian等[41]合成了轴手性的“Buchwald型”联苯基单齿膦配体L22, 并将其应用于苯乙烯的不对称硅氢化反应中, 虽然催化剂在该反应中活性较好, 然而仅取得了54% ee(表 5, Entries 12, 13).
1.3 其他手性单齿膦配体
1972年, Kumada等[6, 42]将萜衍生薄荷基二苯基膦L23和新薄荷基二苯基膦L24(图 4)应用于苯乙烯的不对称硅氢化反应中.室温下, 当采用Pd(PhCN)2Cl2作为催化剂前体, 室温下反应5 h, 配体L23取得了34% ee, 配体L24取得了22% ee.而磷手性的配体L25需要高达120 ℃的反应温度, 仅得到了2% ee.这些配体活性差异可能在于配体L25位阻较小, 使得有2 equiv.的配体与金属配位, 底物插入时需要更大能量使得1 equiv.配体配位解离.手性三烷基膦配体L26[43]应用于环戊二烯的硅氢化反应中, 当配体与金属的比例为1时, 25~30 ℃下, 取得了70%的产率以及54% ee; 而配体与金属比例为2时, 在高达70 ℃温度下, 仅取得了26%的产率以及44% ee.第二当量的富电子三烷基膦配体在该反应中很明显是不利的. Achiwa等[44]将S-缬氨醇衍生的手性β-胺基烷基磷配体L27在环戊二烯的不对称硅氢化反应中选择性非常低.在相同条件下, 配体L28a和L28c取得的对映选择性分别为61%和72%.而配体L28b仅取得了7% ee, 说明β-胺基上的氢原子对于选择性是十分重要的.
 图4
用于烯烃不对称硅氢化反应的其他手性单齿膦配体
Figure4.
Structures of other chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes
图4
用于烯烃不对称硅氢化反应的其他手性单齿膦配体
Figure4.
Structures of other chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes
近年来, 人工合成的螺旋聚合物的应用为不对称催化中构筑手性环境提供了新方法. 2010年, Suginome等[45]采用手性有机钯引发剂, 通过不对称活性聚合反应合成了结构新颖的含二苯基膦单元的聚喹喔啉结构的手性聚合物配体L29.他们将螺旋手性聚合物作为配体, 应用于钯催化的苯乙烯的不对称硅氢化反应中.其中, 右手螺旋的聚合物配体得到了光学纯度为96%的苯乙醇.值得一提的是, 配体与金属形成的络合物不溶于反应体系中, 便于催化剂的回收与再利用.催化剂回收9次后, 其活性和选择性基本保持不变.另外, 溶剂可控制聚合物骨架的构型, 因此可以通过一种手性催化剂可控生成两种不同构型的产物.
2 手性单齿亚磷酰胺酯配体在钯催化的烯烃的不对称硅氢化反应中的应用
亚磷酰胺酯配体结构可分为亚磷酰基和胺基两部分.早在1994年Feringa等[46]首次报道了联二萘骨架的单齿手性亚磷酰胺酯配体的合成, 在随后的几年, 将其成功地运用于1, 4-共轭加成[47]、不对称氢化反应[48]以及烯丙基取代反应[49]中, 取得了高活性及高对映选择性.相对而言, 亚磷酰胺配体在烯烃硅氢化反应中的报道较少.目前.亚磷酰胺酯配体(图 5)主要应用于苯乙烯类底物的不对称硅氢化反应中.
 图5
用于烯烃不对称硅氢化反应的单齿亚磷酰胺酯配体
Figure5.
Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of alkenes
图5
用于烯烃不对称硅氢化反应的单齿亚磷酰胺酯配体
Figure5.
Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of alkenes
2002年, Johannsen等[50]首次将Feringa合成的用于铜-催化的烯酮与二烷基锌的不对称加成反应中的手性亚磷酰胺酯配体应用于苯乙烯的不对称硅氢化反应中.研究结果表明, 催化剂的选择性取决于配体中胺基的结构.如采用非手性胺制备的配体L30a (MonoPhos)仅取了55% ee (表 6, Entry 1).而二异丙基胺所得的配体L30b, 仅取得了20% ee (表 6, Entry 2).同时, 他们将由S-BINOL与手性胺制备的配体L31, 用于不对称硅氢化反应中, 取得了高达99%的对映选择性(表 6, Entry 3), 这是迄今为止钯催化苯乙烯取得的最高的对映选择性.而手性不匹配的配体L32, 虽然活性与配体L31相当, 然而仅取得了60%ee(表 6, Entry 4).
表 6
手性亚磷酰胺酯配体用于钯催化的苯乙烯硅氢化反应a
Table 6.
Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of styrene

Entry L Catalyst/mol% Condition Yield/% of 2 ee/% of3 1b L30a 1 r.t., 24 h 100 55 2b L30b 1 r.t., 24 h 100 20 3 L31 0.25 20 ℃, 16 h 87 99 4 L32 1 r.t., 24 h 100 60 5 L33 0.25 15~20 ℃, 2 h 99 97 6 L34 0.125 r.t., 36 h 94 90 7 L35 0.25 0 ℃, 8 h 30 5 8b L36a 0.25 0 ℃, 6 h >95 85 9b L36a 0.25 -20 ℃, 8 h >95 92 10b L36b 0.25 -20 ℃, 16 h >95 96 11b L36c 0.25 -20 ℃, 8 h >95 96 12b L36a 0.1 0 ℃, 20 min >95 92 13b L36a 0.02 0 ℃, 12 h >95 90 14b L36a 0.01 0 ℃, 72 h >95 81 15 L37a 0.25 r.t., 16 h >99 82.5 16 L37b 0.25 r.t., 16 h >99 85 17 L37c 0.25 r.t., 16 h >99 90 18 L38 0.25 -20 ℃, 72 h 99 90 aAll reactions were conducted with [Pd (C3H5)Cl]2 as precursor, [L]/[Pd]=2, solvent free unless otherwise stated.b [L]/[Pd]=1. 2004年, 周其林课题组[51]从新颖的螺二氢茚骨架出发合成了一系列亚磷酰胺酯配体.与Johannsen的研究结果类似, 他们也发现催化剂的活性与选择性取决于配体中氮原子上的基团.当配体中胺基部分为二甲氨基、二异丙基胺基、吡咯烷基时, 所对应的催化剂活性不高, 且选择性仅为中等.采用手性匹配的胺与酚制得的配体L33, 其对应的催化剂在苯乙烯的不对称硅氢化反应中不仅活性高, 且取得了高达97%ee(表 6, Entry 5).当底物为2-氯苯乙烯时, 所得产物的ee值为99.1%.
高选择性配体L31和配体L33均由结构对称的且手性匹配的仲胺制备而来.李新生等[52]报道了由结构不对称的手性仲胺合成的联二萘骨架的亚磷酰胺酯配体L34, 在苯乙烯类底物的不对称硅氢化反应中取得了90% ee (表 6, Entry 6).这说明, 高对映选择性的手性亚磷酰胺酯配体不一定需要结构对称的手性胺作为原料.
2008年, 我们组[53]报道了树状结构手性亚磷酰胺酯配体, 在铑催化的官能化烯烃如α-脱氢氨基酸酯及芳基烯酰胺的不对称氢化反应中, 取得了优异的催化性能, 体现了十分明显“树状分子正效应”.然而, 我们[54]将树状结构配体L35应用于钯催化的苯乙烯的不对称硅氢化反应中, 仅取得了5% ee (表 6, Entry 7).这说明增大胺基的位阻可能不利于实现高效的不对称诱导.于是, 我们从联二萘骨架的3, 3'取代的手性联二萘酚出发, 采用价廉易得的非手性的二苄胺作为原料, 合成了一类新型的亚磷酰胺酯配体L36.通过对配体的结构与催化性能的研究, 发现3, 3'为芳基的配体所得产物的选择性较高(表 6, Entries 8~10).其中, 3, 3'位为4-甲氧基苯基的配体L36c, 取得了96% ee(表 6, Entry 11).不仅如此, 该类配体成功地应用于一系列苯乙烯类底物的催化不对称硅氢化反应中, 取得了高活性(TOF值高达3000, 表 6, Entry 12)、高产出率(TON值高达10000, 表 6, Entry 14).这一结果可与由手性胺生成的亚磷酰胺酯配体的催化体系相媲美.
随后, Beller等[55]报道了由3, 3'位取代的部分氢化的联二萘酚和非手性胺制备的手性亚磷酰胺酯配体L37.他们详细研究了金属前体、底物与硅烷的比例、配体与金属的比例、配体的结构及偶合常数J (P, Se)与对映选择性之间的关系.在室温、无溶剂条件下, 该类配体在苯乙烯类底物的不对称氢化反应中, 有着很好的催化活性、选择性及底物广谱性.最近, Han等[56]也合成了一系列联二萘3, 3'位取代的亚磷酰胺酯配体, 与我们的研究结果相似, 由非手性胺合成的3, 3'位芳基取代配体L38在苯乙烯的不对称硅氢化反应中, 取得了高达90%ee (表 6, Entry 18).这些结果都说明高效的亚磷酰胺酯配体不一定需要手性胺作为原料.
手性亚磷酰胺酯配体在苯乙烯的硅氢化反应中取得了优异的催化效果, 然而在其他烯烃的不对称硅氢化反应中却鲜有报道. 2013年, Hayashi等[57]将由手性胺制备的配体L31用于1, 3-环己二烯的不对称硅氢化反应中.虽然该配体在苯乙烯的硅氢化反应中取得了99% ee, 然而在该反应中的ee值仅为57%(表 7, Entry 1).令人意外的是, 采用非手性胺合成的配体L39在相同条件下取得了87% ee(表 7, Entry 2).这也是迄今为止该底物在钯催化的不对称硅氢化反应中取得的最高对映选择性.受此启发, Han等[58]也将非手性胺制得的配体L38用于1, 3-环己二烯的不对称硅氢化反应中, 取得了65% ee(表 7, Entry 4).与Hayashi的结果类似, 手性胺对应的配体L40选择性较差(表 7, Entry 6, 33% ee).
表 7
手性亚磷酰胺酯配体用于钯催化的1, 3-环己二烯的硅氢化反应a
Table 7.
Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of 1, 3-cychlohexadiene

Entry L Condition Yield/% of 10a ee/% of 12a 1 L31 20 ℃, 22 h 93 57 2 L39 20 ℃, 20 h 82 87 3 L39 -10 ℃, 84 h 97 87 4 L38 20 ℃, 20 h 90 65 5 L38 -10 ℃, 72 h 61 72 6 L40 20 ℃, 20 h 99 33 a All reactions were conducted with [Pd(C3H5)Cl]2 as precursor, [Ligand]/[Pd]=2, solvent free, 1 mol% catalyst. 3 手性单齿亚膦酸酯配体在钯催化的烯烃的不对称硅氢化反应中的应用
2012年, Higham等[59]报道了具有两个联二萘单元的新型手性单齿亚膦酸酯配体.该配体的合成是以联二萘骨架的伯膦作为原料与手性联二萘酚(BINOL)反应制得.由于配体中含有两个手性单元, 他们制备了四种不同构型的配体, 通过核磁以及X射线单晶衍射对钯与配体的配位行为进行了详细研究.研究结果表明, 配体中除了磷原子与金属钯配位外, 萘环上的碳原子也与金属作用占据剩下的一个配位空轨道.同时, 他们将4种不同构型的配体应用于[Pd(C3H5)Cl]2催化的苯乙烯与三氯硅氢的不对称硅氢化反应中.其中, 由R构型的伯膦与S构型的BINOL所得的配体L41(图 6)取得了最高对映选择性(80% ee).最近, 他们组[60]报道了类似结构的配体L42, 并将其应用于苯乙烯及其衍生物的不对称硅氢化反应中.当催化剂前体[Pd(C3H5)Cl]2的用量为0.125 mol%, 配体与金属的比例为2时, 室温下, 配体L42a取得了79% ee, 配体L42b取得了80% ee.将反应温度降为0 ℃, 72 h后, 配体L42b取得了95% ee.
 图6
用于烯烃不对称硅氢化反应的单齿亚膦酸酯配体
Figure6.
Chiral monodentate phophonites used in the asymmetric hydrosilylation of alkenes
图6
用于烯烃不对称硅氢化反应的单齿亚膦酸酯配体
Figure6.
Chiral monodentate phophonites used in the asymmetric hydrosilylation of alkenes
4 双膦配体稳定的纳米钯催化剂在不对称硅氢化反应中的应用
均相的BINAP-钯络合物不能催化烯烃的不对称硅氢化反应. 2003年, Fujihara等[61]首次合成了手性双膦配体BINAP稳定的直径小于2 nm的钯纳米颗粒, 并将其应用于苯乙烯的不对称硅氢化反应中.研究结果表明, 室温下反应5 h, BINAP-钯纳米颗粒使得底物完全转化, 以81%的收率、75% ee得到S-苯乙醇.当反应温度降至0 ℃时, 催化剂活性得以保持, 对映选择性上升至95%.均相的BINAP-钯络合物在烯烃的不对称硅氢化反应中没有活性, 这充分说明BINAP-钯纳米颗粒中特殊的纳米结构起到了诱导不对称催化反应的结果.
5 总结与展望
综上所述, 经过三十多年的发展, 随着许多优秀手性单膦配体的合成与应用, 钯催化的烷基烯烃、苯乙烯、1, 3-二烯烃等的不对称硅氢化反应取得了较大的进展, 在整个不对称催化基础研究领域占据非常重要的地位.尽管烯烃的不对称硅氢化已经发展得比较成熟, 但还有很大的提升空间.首先, 开发价廉易得、结构简单、新颖、可操作性强(不需无水无氧)的, 具有高活性、高区域选择性、高对映选择性的手性单膦配体仍然是今后研究的重要内容.同时, 深入研究烯烃的不对称硅氢化反应机理, 为设计高效催化体系提供理论依据及指导, 也是将来需要进一步关注的研究热点.再次, 发展负载型手性单膦配体[62], 借鉴廉价金属[63, 64]及非金属[65]催化体系的成功经验, 为解决钯的价格昂贵和回收困难导致难以实现大规模工业化应用的问题.另外, 相对于不对称氢化反应, 单膦配体在烯烃的硅氢化反应中底物适用范围还较窄, 可以尝试将应用面拓展到更多类别更困难的底物.我们相信, 随着科研工作者不断深入研究不对称硅催化反应, 未来钯-单膦催化剂在基础研究及工业应用方面都将有着更加广阔的前景.
-
-
[1]
(a) Kiss, G. Chem. Rev. 2001, 101, 3435.
(b) Liebscher, Y. J. Chem. Rev. 2007, 107, 133.
(c) Negishi, E. I.; Anastasia, L. Chem. Rev. 2003, 103, 1979.
(d) Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564. -
[2]
(a) Lyons, T. W. ; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(b) Wu, X. F. ; Neumann, H. ; Beller, M. Chem. Rev. 2013, 113, 1.
(c) Cacchi, S. ; Fabrizi, G. Chem. Rev. 2005, 105, 2873.
(d) Li, Z. ; Duan, W. L. Chin. J. Org. Chem. 2016, 36, 1805 (in Chinese).
(李振, 段伟良, 有机化学, 2016, 36, 1805. )
(e) Li, J. X. ; Zhang, Z. M. ; Li, C. S. ; Luo, W. ; Yang, S. R. Chin. J. Org. Chem. 2015, 35, 2199 (in Chinese).
(李建晓, 张振明, 李春生, 罗维, 杨少容, 有机化学, 2015, 35, 2199. ) -
[3]
(a) Zimmer, R.; Dinesh, C. U.; Nandanan, E.; Khan, F. A. Chem. Rev. 2000, 100, 3067.
(b) Tietze, L. F.; Ila, H.; Bell, H. P. Chem. Rev. 2004, 104, 3453. -
[4]
McDonald, R. I.; Liu, G. S.; Stahl, S. S. Chem. Rev. 2011, 111, 2981. doi: 10.1021/cr100371y
-
[5]
(a) Brunner, H.; Becker, R.; Riepl, G. Organometallics 1984, 3, 1354.
(b) Nishiyama, H.; Yamaguchi, S.; Kondo, M.; Itoh, K. J. Org. Chem. 1992, 57, 4306. -
[6]
Kiso, Y.; Yamamoto, K.; Tamao, K.; Kumada, M. J. Am. Chem. Soc. 1972, 94, 4373. doi: 10.1021/ja00767a074
-
[7]
(a) Teichert, J. F. ; Feringa, B. L. Angew. Chem. , Int. Ed. 2010, 49, 2486.
(b) Zhang, Z. ; Xie, F. ; Yang, B. ; Yu, H. ; Zhang, W. Chin. J. Org. Chem. 2011, 31, 429 (in Chinese).
(张振锋, 谢芳, 杨波, 余焓, 张万斌, 有机化学, 2011, 31, 429. )
(c) Minnaard, A. J. ; Feringa, B. L. ; Lefort, L. ; de Vries, J. G. Acc. Chem. Res. 2007, 40, 1267.
(d) de Vries, A. H. M. ; de Vries, J. G. Platinum Metals Rev. 2006, 50, 54. -
[8]
(a) Feng, X. Q. ; Duan, H. F. Chin. J. Org. Chem. 2015, 35, 259 (in Chinese).
(冯向青, 杜海峰, 有机化学, 2015, 35, 259. )
(b) Rong, J. ; Ni, C. F. ; Wang, Y. Z. ; Kuang, C. W. ; Gu, Y. C. ; Hu, J. B. Acta Chim. Sinica 2017, 75, 105 (in Chinese).
(荣健, 倪传法, 王云泽, 匡翠文, 顾玉诚, 胡金波, 化学学报, 2017, 75, 105. )
(c) Park, H. S. ; Kim, M. Y. ; Ahn, H. J. ; Han, J. W. Bull. Korean Chem. Soc. 2016, 37, 795.
(d) Ogasawara, M. ; Area, S. ; Watanabe, S. ; Nakajima, K. ; Takahashi, T. ACS Catal. 2016, 6, 1308. -
[9]
(a) Gibson, S. E.; Rudd, M. Adv. Synth. Catal. 2007, 349, 781.
(b) Han, J. W.; Hayashi, T. Tetrahedron: Asymmetry 2010, 21, 2193.
(c) Han, J. W.; Hayashi, T. Tetrahedron: Asymmetry 2014, 25, 479. -
[10]
刘振德, 何煦昌, 化学进展, 2006, 18, 1489. doi: 10.3321/j.issn:1005-281X.2006.11.011Liu, Z.; He, X. Proc. Chem. 2006, 18, 1489(in Chinese). doi: 10.3321/j.issn:1005-281X.2006.11.011
-
[11]
Hayashi, T.; Tamao, K.; Katsuro, Y.; Nakae, I.; Kumada, M. Tetrahedron Lett. 1980, 21, 1871. doi: 10.1016/S0040-4039(00)92802-8
-
[12]
Uozumi, Y.; Kitayama, K.; Hayashi, T. Tetrahedron:Asymmetry 1993, 4, 2419. doi: 10.1016/S0957-4166(00)82214-4
-
[13]
Hayashi, T.; Kabeta, K. Tetrahedron Lett. 1985, 26, 3023. doi: 10.1016/S0040-4039(00)98608-8
-
[14]
Hayashi, T.; Matsumoto, Y.; Morikawa, I.; Ito, Y. Tetrahedron:Asymmetry 1990, 1, 151. doi: 10.1016/S0957-4166(00)82367-8
-
[15]
Pioda, G.; Togni, A. Tetrahedron:Asymmetry 1998, 9, 3903. doi: 10.1016/S0957-4166(98)00409-1
-
[16]
Weber, I.; Jones, G. B. Tetrahedron Lett. 2001, 42, 6983. doi: 10.1016/S0040-4039(01)01471-X
-
[17]
Gibson, S. E.; Rendell, J. T.; Rudd, M. Synthesis 2006, 3631.
-
[18]
(a) Pedersen, H. L.; Johannsen, M. Chem. Commun. 1999, 2517.
(b) Pedersen, H. L.; Johannsen, M. J. Org. Chem. 2002, 67, 7982. -
[19]
Ohmura, H.; Matsuhash, H.; Tanaka, M.; Kuroboshi, M.; Hiyama, T.; Hatanaka, Y.; Goda, K. J. Organomet. Chem. 1995, 499, 167. doi: 10.1016/0022-328X(95)00311-D
-
[20]
Han, J. W.; Tokunaga, N.; Hayashi, T. Helv. Chim. Acta 2002, 85, 3848. doi: 10.1002/1522-2675(200211)85:11<3848::AID-HLCA3848>3.0.CO;2-V
-
[21]
Han, J. W.; Tokunaga, N.; Hayashi, T. J. Am. Chem. Soc. 2001, 123, 12915. doi: 10.1021/ja017138h
-
[22]
Ogasawara, M.; Ito, A.; Yoshida, K.; Hayashi, T. Organometallics 2006, 25, 2715. doi: 10.1021/om060138b
-
[23]
(a) Noyori, R. ; Takaya, H. Acc. Chem. Res. 1990, 23, 345.
(b) Berthod, M. ; Mignani, G. ; Woodward, G. ; Lemaire, M. Chem. Rev. 2005, 105, 1801.
(c) Gao, A. ; Ye, Q. ; Yu, J. ; Liu, W. Chin. J. Org. Chem. 2017, 37, 47 (in Chinese).
(高安丽, 叶青松, 余娟, 刘伟平, 有机化学, 2017, 37, 47. ) -
[24]
Uozumi, Y.; Hayashi, T. J. Am. Chem. Soc. 1991, 113, 9887. doi: 10.1021/ja00026a044
-
[25]
Uozumi, Y.; Lee, S. Y.; Hayashi, T. Tetrahedron Lett. 1992, 33, 7185. doi: 10.1016/S0040-4039(00)60868-7
-
[26]
Uozumi, Y.; Hayashi, T. Tetrahedron Lett. 1993, 34, 2335. doi: 10.1016/S0040-4039(00)77607-6
-
[27]
Kitayama, K.; Uozumi, Y.; Hayashi, T. J. Chem. Soc., Chem. Commun. 1995, 1533.
-
[28]
Hayashi, T.; Hirate, S.; Kitayama, K.; Tsuji, H.; Torii, A.; Uo-zumi, Y. J. Org. Chem. 2001, 66, 1441. doi: 10.1021/jo001614p
-
[29]
Hayashi, T.; Niizuma, S.; Kamikawa, T.; Suzuki, N.; Uozumi, Y. J. Am. Chem. Soc. 1995, 117, 9101. doi: 10.1021/ja00140a041
-
[30]
Bringmann, G.; Wuzik, A.; Breuning, M.; Henschel, P.; Peters, K.; Peters, E. M. Tetrahedron:Asymmetry 1999, 10, 3025. doi: 10.1016/S0957-4166(99)00299-2
-
[31]
Kitayama, K.; Tsuji, H.; Uozumi, Y.; Hayashi, T. Tetrahedron Lett. 1996, 37, 4169. doi: 10.1016/0040-4039(96)00786-1
-
[32]
Hayashi, T.; Han, J. W.; Takeda, A.; Tang, J.; Nohmi, K.; Mukaide, K.; Tsuji, H.; Uozumi, Y. Adv. Synth. Catal. 2001, 343, 279. doi: 10.1002/(ISSN)1615-4169
-
[33]
Han, J. W.; Hayashi, T. Chem. Lett. 2001, 976.
-
[34]
Han, J. W.; Hayashi, T. Tetrahedron:Asymmetry 2002, 13, 325. doi: 10.1016/S0957-4166(02)00094-0
-
[35]
Dotta, P.; Kumar, P. G. A.; Pregosin, P. S.; Albinati, A.; Rizzato, S. Organometallics 2004, 23, 2295. doi: 10.1021/om034381b
-
[36]
Tschoerner, M.; Pregosin, P.; Albinati, A. Organometallics 1999, 18, 670. doi: 10.1021/om980783l
-
[37]
Gladiali, S.; Pulacchini, S.; Fabbri, D.; Manassero, M.; Sansoni, M. Tetrahedron:Asymmetry 1998, 9, 391. doi: 10.1016/S0957-4166(98)00009-3
-
[38]
Yasuike, S.; Kawara, S.; Okajima, S.; Seki, H.; Yamaguchi, K.; Kurita, J. Tetrahedron Lett. 2004, 45, 9135. doi: 10.1016/j.tetlet.2004.10.020
-
[39]
Ficks, A.; Martinez-Botella, I.; Stewart, B.; Harrington, R. W.; Clegg, W.; Higham, L. J. Chem. Commun. 2011, 47, 8274. doi: 10.1039/c1cc12440d
-
[40]
Duclos, M. C.; Singjunla, Y.; Petit, C.; Favre-Réguillon, A.; Jeanneau, E.; Popowycz, F.; Métay, E.; Lemaire, M. Tetrahedron Lett. 2012, 53, 5984. doi: 10.1016/j.tetlet.2012.07.136
-
[41]
Fer, M. J.; Cinqualbre, J.; Bortoluzzi, J.; Chesse, M.; Leroux, F. R.; Panossian, A. Eur. J. Org. Chem. 2016, 26, 4545.
-
[42]
Kiso, Y.; Yamamoto, K.; Tamao, K.; Kumada, M. J. Organomet. Chem. 1981, 210, 9. doi: 10.1016/S0022-328X(00)86640-1
-
[43]
(a) Marinetti, A. Tetrahedron Lett. 1994, 35, 5861.
(b) Marinetti, A.; Ricard, L. Organometallics 1994, 13, 3956. -
[44]
(a) Okada, T.; Morimoto, T.; Achiwa, K. Chem. Lett. 1990, 999.
(b) Sakuraba, S.; Okada, T.; Morimoto, T.; Achiwa, K. Chem. Pharm. Bull. 1995, 43, 927. -
[45]
Yamomoto, T.; Yamada, T.; Nagata, Y.; Suginome, M. J. Am. Chem. Soc. 2010, 132, 7899. doi: 10.1021/ja102428q
-
[46]
Hulst, R.; de Vries, N. K.; Feringa, B. L. Tetrahedron:Asymmetry 1994, 5, 699. doi: 10.1016/0957-4166(94)80032-4
-
[47]
(a) deVries, A. H. M.; Meetsma, A.; Feringa, B. L. Angew. Chem. Int. Ed. 1996, 35, 2375.
(b) Feringa, B. L. Acc. Chem. Res. 2000, 33, 346.
(c) López, F.; Minnaard, A. J.; Feringa, B. L. Acc. Chem. Res. 2007, 40, 179. -
[48]
(a) Van den Berg, M.; Minnaard, A. J.; Schudd, E. P.; van Esch, J.; de Vries, A. H. M.; de Vries, J. G. J. Am. Chem. Soc. 2000, 122, 11539.
(b) Hu, A. G.; Fu, Y.; Xie, J. H.; Zhou, H.; Wang, L. X.; Zhou, Q. L. Angew. Chem., Int. Ed. 2002, 41, 2348.
(c) Hou, G. H.; Xie, J. H.; Yan, P. C.; Zhou, Q. L. J. Am. Chem. Soc. 2009, 131, 1366.
(d) Liu, Y.; Ding, K. L. J. Am. Chem. Soc. 2005, 127, 10488. -
[49]
(a) Lipowsky, G.; Miller, N.; Helmchen, G. Angew. Chem., Int. Ed. 2004, 43, 4595.
(b) Ohmura, T.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 15164.
(c) Kiener, C. A.; Shu, C.; Incarvito, C.; Hartwig, J. F. J. Am. Chem. Soc. 2003, 125, 14272. -
[50]
Jensen, J. F.; Svendsen, B. Y.; la Cour, T. V.; Pedersen, H. L.; Johannsen, M. J. Am. Chem. Soc. 2002, 124, 4558. doi: 10.1021/ja025617q
-
[51]
Guo, X. X.; Xie, J. H.; Hou, G. H.; Shi, W. J.; Wang, L. X.; Zhou, Q. L. Tetrahedron:Asymmetry 2004, 15, 2231. doi: 10.1016/j.tetasy.2004.05.038
-
[52]
Li, X. S.; Song, J. A.; Xu, D. S.; Kong, L. C. Synthesis 2008, 925.
-
[53]
Zhang, F.; Li, Y.; Li, Z. W.; He, Y. M.; Zhu, S. F.; Fan, Q. H.; Zhou, Q. L. Chem. Commun. 2008, 6048.
-
[54]
Zhang, F.; Fan, Q. H. Org. Biomol. Chem. 2009, 7, 4470. doi: 10.1039/b909334f
-
[55]
Junge, K.; Wendt, B.; Enthaler, S.; Beller, M. ChemCatChem 2010, 2, 453. doi: 10.1002/cctc.v2:4
-
[56]
Park, H. S.; Namgung, S.; Shin, H. M.; Ahn, H. J.; Han, J. W. Bull. Korean Chem. Soc. 2014, 35, 2243. doi: 10.5012/bkcs.2014.35.8.2243
-
[57]
Park, H. S.; Han, J. W.; Shintani, R.; Hayashi, T. Tetrahedron:Asymmetry 2013, 24, 418. doi: 10.1016/j.tetasy.2013.02.002
-
[58]
Park, H. S.; Shin, H. M.; Namgung, S.; Han, J. W. Bull. Korean Chem. Soc. 2014, 35, 2613. doi: 10.5012/bkcs.2014.35.9.2613
-
[59]
Ficks, A.; Hiney, R. M.; Harrington, R. W.; Gilheany, D. G.; Higham, L. J. Dalton Trans. 2012, 41, 3515. doi: 10.1039/c2dt12214f
-
[60]
Fleming, J. T.; Ficks, A.; Waddell, P. G.; Paul, G.; Harrington, R. W.; Higham, L. J. Dalton Trans. 2016, 45, 1886. doi: 10.1039/C5DT04475H
-
[61]
Tamura, M.; Fujihara, H. J. Am. Chem. Soc. 2003, 125, 15742. doi: 10.1021/ja0369055
-
[62]
(a) Zhong, M. M.; Zhang, X. M.; Zhao, Y. P.; Li, C.; Yang, Q. H. Green Chem. 2015, 17, 1702.
(b) Shi, L.; Wang, X. W.; Sandoval, C. A.; Li, M. X.; Qi, Q. Y.; Li, Z. T.; Ding, K. L. Angew. Chem., Int. Ed. 2006, 45, 4108. -
[63]
(a) Xu, D. P. ; Xiao, W. J. ; Peng, J. J. ; Li, J. Y. ; Bai, Y. Chin. J. Org. Chem. 2014, 34, 2195 (in Chinese).
(徐大鹏, 肖文军, 彭家建, 厉嘉云, 白赢, 有机化学, 2014, 34, 2195. )
(b) Chen, L. Z. ; Peng, J. J. ; Li, J. Y. ; Bai, Y. ; Qiu, H. Y. ; Lai, G. Q. Chin. J. Org. Chem. 2008, 28, 761 (in Chinese).
(陈玲珍, 彭家建, 厉嘉云, 白赢, 邱化玉, 来国桥, 有机化学, 2008, 28, 761. )
(c) Liu, S. ; Peng, J. J. ; Li, J. Y. ; Bai, Y. ; Xiao, W. J. ; Lai, G. Q. Chin. J. Org. Chem. 2012, 32, 1827 (in Chinese).
(刘帅, 彭家建, 厉嘉云, 白赢, 肖文军, 来国桥, 有机化学, 2012, 32, 1827. ) -
[64]
(a) Liu, Y. Y. ; Zhang, W. B. Chin. J. Org. Chem. 2016, 36, 2249 (in Chinese).
(刘媛媛, 张万斌, 有机化学, 2016, 36, 2249. )
(b) Li, T. T. ; Yu, P. ; Lin, J. S. ; Zhi, Y. G. ; Liu, X. Y. Chin. J. Chem. 2016, 34, 490.
(c) Yang, M. B. ; Wang, W. L. ; Liu, Y. ; Feng, L. J. ; Ju, X. X. Chin. J. Chem. 2014, 32, 833.
(d) Gribble, M. W. ; Pirnot, M. T. ; Bandar, J. S. ; Liu, R. Y. ; Buchwald, S. L. J. Am. Chem. Soc. 2017, 139, 2192. -
[65]
Yang, L.; Lu, W.; Zhou, W.; Zhang, F. Green Chem. 2016, 18, 2941 doi: 10.1039/C6GC00362A
-
[1]
-

图 2 用于烯烃不对称硅氢化反应的平面手性膦配体
Figure 2 Planar chiral phosphines used in the asymmetric hydrosilylation of alkenes

图式 1 平面手性膦配体L1在烯烃的不对称硅氢化反应中的应用
Scheme 1 Application of planar chiral phosphine L1 in the asymmetric hydrosilylation of alkenes

图式 2 平面手性膦配体L2在环戊二烯的不对称硅氢化反应中的应用
Scheme 2 Application of planar chiral phosphines L2 in the asymmetric hydrosilylation of cyclopentadiene

图式 3 平面手性膦配体L3在取代苯乙烯及降冰片烯的不对称硅氢化反应中的应用
Scheme 3 Application of planar chiral phosphines L3 in the asymmetric hydrosilylation of substituted styrenes and norbornene

图式 4 基于单膦配体用量的两个催化循环
Scheme 4 Two proposed catalytic cycles depending on loadings of phosphine

图式 5 平面手性膦配体L7在环己二烯的不对称硅氢化反应中的应用
Scheme 5 Application of planar chiral phosphines L7 in the asymmetric hydrosilylation of cyclohexadiene

图 3 用于烯烃不对称硅氢化反应的轴手性单齿膦配体
Figure 3 Axially chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes

图式 6 MeO-MOP在环状烯烃的不对称硅氢化反应中的应用
Scheme 6 Application of MeO-MOP in the asymmetric hydrosilylation of cyclic alkenes

图 4 用于烯烃不对称硅氢化反应的其他手性单齿膦配体
Figure 4 Structures of other chiral monodentate phosphines used in the asymmetric hydrosilylation of alkenes

图 5 用于烯烃不对称硅氢化反应的单齿亚磷酰胺酯配体
Figure 5 Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of alkenes

图 6 用于烯烃不对称硅氢化反应的单齿亚膦酸酯配体
Figure 6 Chiral monodentate phophonites used in the asymmetric hydrosilylation of alkenes
表 1 平面手性膦配体L4、L5及L6在钯催化的苯乙烯硅氢化反应中的应用a
Table 1. Application of planar chiral phosphines L4、L5 and L6 in the asymmetric hydrosilylation of styrene
Entry L Catalyst/
mol%Condition Yield/%
2ee/%
31b, c, d L4a 0.25 -40 ℃, 48 h >85 87 2b, c, d L4b 0.25 -50 ℃, 48 h >85 92 3e L5a 0.5 r.t., 48 h 99 55 4f L5b 0.5 r.t., 48 h 97 77 5 L6a 1 r.t., 1.6 h 100 86 6 L6b 1 r.t., 8 h 100 25 7 L6c 1 r.t., 1.5 h 100 79 8 L6b 0.1 r.t., 8 h 100 90 9 L6c 0.1 r.t., 3.5 h 100 85 10g L6c 0.1 r.t., 20 s 100 62 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, [L]/[Pd]=2, solvent free unless otherwise stated. b [L]/[Pd]=1.cCH2Cl2 or CDCl3 as solvent.d [Pd(C3H5)Br]2 as precursor. eCH2Cl2 as solvent. fBenzeneas solvent. g[L]/[Pd]=1.2.  下载: 导出CSV
下载: 导出CSV
表 2 平面手性膦配体L7a、L8a及L9在钯催化的1, 3-烯炔的不对称硅氢化反应中的应用a
Table 2. Application of planar chiral phosphines L7a, L8a and L9 in the asymmetric hydrosilylation of 1, 3-enynes
Entry 18 L Condition Yield/% ee/% of 20 1 18a L7a 20 ℃, 18 h 54 56 2 18a L8a 20 ℃, 64 h 90 77 3 18b L7a 20 ℃, 9 h 81 72 4 18b L8a 20 ℃, 48 h 59 85 5 18b L8a 0 ℃, 160 h 37 90 6b 18b L9 0 ℃, 72 h 82 92 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 1 mol% catalyst, [Ligand]/[Pd]=2.2, solvent free unless otherwise stated. bee of 21.
下载: 导出CSV
表 3 轴手性单膦配体L10在链状端烯的不对称硅氢化反应中的应用a
Table 3. Application of axially chiral phosphines L10 in the asymmetric hydrosilylation of acyclic terminated alkenes
Entry 22 L Condition Yield/% 23:24 ee/% of 26 1 22a L10a 40 ℃, 24 h 83 93:7 95 2 22a L10b 40 ℃, 24 h 88 90:10 91 3 22a L10c 40 ℃, 24 h 85 80:20 95 4 22a L10d 40 ℃, 24 h 80 90:10 93 5 22b L10a 40 ℃, 24 h 91 89:11 94 6 22c L10a 40 ℃, 72 h 90 94:6 95 7 22d L10a 40 ℃, 24 h 90 81:19 97 8b 22e L10a 30 ℃, 60 h 81 80:20 92 9 22f L10a 40 ℃, 24 h 100 66:34 96 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 0.1 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. bTHF as solvent.
下载: 导出CSV
表 4 轴手性膦配体用于的环状1, 3-二烯烃的不对称硅氢化反应a
Table 4. Axially phosphines used in the asymmetric hydrosilylation of cyclic 1, 3-dienes
Entry 7/8 L Condition Yield/% of
9/10ee/% of
11/121b 7 L10a 20 ℃, 14 h 100 39 2b 7 L11a 20 ℃, 3 h 91 28 3b 7 L14 20 ℃, 120 h 99 80 4 7 L15a 0 ℃, 24 h 84 69 5 7 L15b 0 ℃, 24 h 84 76 6 7 L15c 0 ℃, 24 h 83 47 7 7 L15d 0 ℃, 24 h 85 76 8 7 L15e 0 ℃, 24 h 83 79 9 7 L15f 0 ℃, 24 h 79 88 10 7 L15f -20 ℃, 72 h 89 90 11 7 L16 -30 ℃, 168 h 75 91 12 8 L16 -10 ℃, 168 h 70 83 13 8 L15f 0 ℃, 72 h 75 79 aAll reactions were conducted with [Pd (C3H5)Cl]2as precursor, 0.25 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. b0.1 mol% catalyst.
下载: 导出CSV
表 5 轴手性膦配体L17~L22在苯乙烯的不对称硅氢化反应中的应用a
Table 5. Application of axially chiral phosphines L17~L22 in the asymmetric hydrosilylation of styrene
Entry L Condition Yield/% of 2 ee/% of 3 1 L11a 0 ℃, 20 h 100 92 2 L17a 5 ℃, 16 h 100 88 3 L10a 5 ℃, 16 h 100 7 4 L17b 0 ℃, 43 h 100 57 5 L10g 5 ℃, 2 h 100 42 6 L17c 5 ℃, 16 h 100 81 7 L18 r.t., 70 h 100 18 8b L18 r.t., 70 h 100 72 9 L19 0 ℃, 10 h 78 95 10 L20 r.t., 24 h >99 80 11b L21 r.t., 18 h 92 91 12c L22a r.t., 3 h 63 54 13c L22b r.t., 3 h 90 54 aAll reactions were conducted with [Pd(C3H5)Cl]2 as precursor, 0.1 mol% catalyst, [L]/[Pd]=2, solvent free unless otherwise stated. bBenzeneas solvent. c 2.5 mol% catalyst.
下载: 导出CSV
表 6 手性亚磷酰胺酯配体用于钯催化的苯乙烯硅氢化反应a
Table 6. Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of styrene
Entry L Catalyst/mol% Condition Yield/% of 2 ee/% of3 1b L30a 1 r.t., 24 h 100 55 2b L30b 1 r.t., 24 h 100 20 3 L31 0.25 20 ℃, 16 h 87 99 4 L32 1 r.t., 24 h 100 60 5 L33 0.25 15~20 ℃, 2 h 99 97 6 L34 0.125 r.t., 36 h 94 90 7 L35 0.25 0 ℃, 8 h 30 5 8b L36a 0.25 0 ℃, 6 h >95 85 9b L36a 0.25 -20 ℃, 8 h >95 92 10b L36b 0.25 -20 ℃, 16 h >95 96 11b L36c 0.25 -20 ℃, 8 h >95 96 12b L36a 0.1 0 ℃, 20 min >95 92 13b L36a 0.02 0 ℃, 12 h >95 90 14b L36a 0.01 0 ℃, 72 h >95 81 15 L37a 0.25 r.t., 16 h >99 82.5 16 L37b 0.25 r.t., 16 h >99 85 17 L37c 0.25 r.t., 16 h >99 90 18 L38 0.25 -20 ℃, 72 h 99 90 aAll reactions were conducted with [Pd (C3H5)Cl]2 as precursor, [L]/[Pd]=2, solvent free unless otherwise stated.b [L]/[Pd]=1.
下载: 导出CSV
表 7 手性亚磷酰胺酯配体用于钯催化的1, 3-环己二烯的硅氢化反应a
Table 7. Chiral monodentate phosphoramidites used in the asymmetric hydrosilylation of 1, 3-cychlohexadiene
Entry L Condition Yield/% of 10a ee/% of 12a 1 L31 20 ℃, 22 h 93 57 2 L39 20 ℃, 20 h 82 87 3 L39 -10 ℃, 84 h 97 87 4 L38 20 ℃, 20 h 90 65 5 L38 -10 ℃, 72 h 61 72 6 L40 20 ℃, 20 h 99 33 a All reactions were conducted with [Pd(C3H5)Cl]2 as precursor, [Ligand]/[Pd]=2, solvent free, 1 mol% catalyst.
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 8
- 文章访问数: 2458
- HTML全文浏览量: 449
 下载:
下载:
