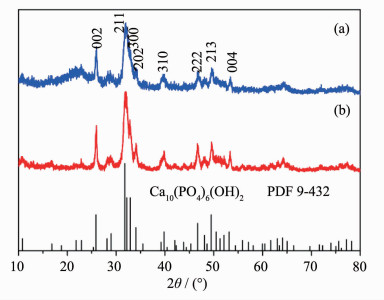
 Figure 4.
XRD patterns of HAP prepared by direct precipitation method with PEG (a) and PVP (b) as surfactants, and the standard data for bulk Ca10(PO4)6(OH)2 as a reference
Figure 4.
XRD patterns of HAP prepared by direct precipitation method with PEG (a) and PVP (b) as surfactants, and the standard data for bulk Ca10(PO4)6(OH)2 as a reference
Citation:
XIAO Xin-Li, KONG De-Yan, ZHANG Ju-Sheng. Morphologically Controlled Synthesis of Hydroxyapatite at Mild Conditions[J]. Chinese Journal of Inorganic Chemistry,
2016, 32(2): 289-296.
doi:
10.11862/CJIC.2016.038

温和条件下制备形貌可控的羟基磷灰石
摘要:
在温和条件下采用简单的实验方法成功制备出不同结构和形貌的羟基磷灰石 (HAP) 纳米粒子.采用沉淀-水解两步法, 以PEG和PVP为表面活性剂, 在沉淀步骤制备出前驱体CaHPO4粒子, 然后在100 ℃、常压条件下水解获得高结晶度的HAP纳米棒.采用PEG制备的纳米棒形貌均匀、纵横比高; 采用PVP制备的纳米棒尺寸范围较宽且尺寸较小.直接采用一步沉淀法也能够成功制备出HAP纳米粒子.采用PEG制备出麦穗状的纳米粒子; 采用PVP制备出尺寸小的纳米棒和纳米粒子混合物.HAP纳米晶的结构和形貌因制备条件不同发生很大变化, 故而控制其合成, 有望用于生物医药领域.
English
Morphologically Controlled Synthesis of Hydroxyapatite at Mild Conditions
Abstract:
Simple methods are introduced to obtain hydroxyapatite (HAP, Ca10(PO4)6(OH)2) nanocrystals with different structures and morphologies at mild conditions. The precipitation-hydrolysis two-step method can produce CaHPO4 precursor by assistance of polyethylene glycol (PEG) and polyvinylpyrrolidone (PVP) during the precipitation step, and the hydrolysis of CaHPO4 at 100 ℃ under atmospheric pressure leads to the formation of highly crystalline rod-like HAP nanocrystals. The HAP nanorods prepared with PEG possess uniform morphology with high aspect ratio while those prepared with PVP have smaller size with broader size distribution. One-step precipitation method can produce HAP directly, and the nanocrystals prepared with PEG are like wheat ears, while those prepared with PVP were composed of small nanoparticles and nanorods. The morphology of HAP samples was influenced by the organic additives, because the surfactants stabilized and confined the formation and growth of HAP nanocrystals. The morphology and structure of HAP nanoparticles changed greatly via the difference synthetic methods. Therefore, the controlling synthesis of HAP can be achieved by the change of synthetic conditions. The obtained HAP nanoparticles will be applicable for the future biomedical applications.
-
0 Introduction
As the main mineral constituent of natural bone, the bioactive hydroxyapatite (HAP, Ca10(PO4)6(OH)2) has attracted much attention due to its extensive applications in various fields such as tooth implants, bone filling and absorbents[1-5]. HAP is used as powders in most cases, and its usefulness is influenced greatly by the powder properties such as structure, composition, crystallite size, and morphology. For their applications in many fields including separation processes, powder carriers for removing heavy metal ions or drugs, HAP nanoparticles with high surface area are more desired[6-9]. Consequently, the HAP crystals with specific morphological and structural properties have attracted much attention. It is observed that the primary HAP particles in bone are in the form of nanoscopic plate-like crystals, while those in tooth enamel are highly elongated nanofilaments[10]. Meanwhile, it is well documented that fibrous and needle-like morphologies with high specific surface area are advantageous for adsorption and ion exchange. Moreover, the size and shape of the HAP crystallite influence the toxicity to cells, osseointegration, resorption of orthopedic and other intended devices[6, 11]. Therefore, significant progress has been made for example in controlling crystal morphology.
Wet chemical methods such as hydrothermal[12], homogeneous precipitation[13], and electrochemical method[14] have been employed to synthesize nanosacle HAP with different morphologies, and the obtained HAP nanocrystals possess various sizes and morphologies, depending on the reactants used and reaction conditions[15-17]. For example, rod-like HAP nanoparticles were synthesized through a mild and biomimetic pathway[18]; The nano-sized HAP powders were obtained using the hydrolysis of dicalcium phosphate (DCP) and CaCO3 at the mild conditions, and the crystallinity of HAP is improved with increasing annealing temperature[19-22]. HAP nanorods were also synthesized using simple hydrothermal procedure, sol-gel route and co-precipitation method[23-26]. However, it is still a big challenge to develop simple and reliable synthetic methods at mild conditions.
In this paper, we developed simple ways to synthesize different HAP nanorods at 100 ℃ at atmospheric pressure and the influence of organic surfactants and synthetic routes on the morphology and structure of HAP nanoparticles were studied. The preparation techniques will benefit the science of wet-chemical materials processing and the obtained HAP nanoparticles will be applicable for the future biomedical applications.
1 Experimental
1.1 Materials
Ca (NO3)2 ·4H2O (AR, Tianjin Bodi Chemical Co., Ltd.), (NH4)2HPO4 (AR, Shantou Xilong Chemical Factory), polyethylene glycol (PEG) (AR, molecular weight 10 000, Sinopharm Chemical Reagent Co., Ltd.), polyvinylpyrrolidone K30 (PVP) (AR, Sinopharm Chemical Reagent Co., Ltd.), ammonia (NH3·H2O) (AR, Xilong Chemical Industry Incorporated Co., Ltd) were used as starting materials; Distilled water and ethanol (AR, Tianjin Kemiou Chemical Reagent Co., Ltd) were used as the solvent.
1.2 Synthesis of HAP nanorod by the two-step method
1.3 Syntheses of HAP nanocrystals by the one-step method
To compare the influence of preparation routes on the nanocrystals, HAP samples were prepared by the one step precipitation technique. Typically, PEG or PVP was dissolved in Ca (NO3)2·4H2O solution, then (NH4)2HPO4 solution was added and stirred. At the same time, the ammonia was added dropwise to this suspension to adjust a specific value of pH at 10, then the suspensions were kept at 100 ℃ in a temperature-controlled electromantle for 24 h. HAP nanoparticles were obtained by the centrifugation, redispersion and drying procedures as the same as the above part.
1.4 Characterization
X-ray diffraction (XRD) was carried out on a Rigaku-Dmax rB diffractometer with Cu Kα radiation (λ=0.154 05 nm). The XRD accelerating voltage and emission current were 40 kV and 50 mA, respectively. TEM images were obtained using a JEOL H7650 transmission electron microscope operating at 100 kV. Samples for TEM were prepared by depositing a drop of ethanol suspension of the powder sample onto a copper grid and dried in air. Fourier transform infrared (FTIR) spectra were measured with Nicolet 360 infrared spectrophotometer with the KBr pellet technique. All the measurements were performed at room temperature.
1.2.1 Syntheses of HAP precursor samples
The precursor samples were performed at the first precipitation step. Typically, 4.725 g PEG or 0.251 g PVP was dissolved in 20 mL distilled water in a beaker, then 11.812 g Ca (NO3)2·4H2O was added and stirred at the moderate speed to form the solution A. At the same time, 2.642 g (NH4)2HPO4 was dissolved in 15 mL distilled water in another beaker to form the solution B. Then, the solution B was added dropwise to the solution A to give a white precipitate and the mixture was kept stirring at the moderate speed for 2 hours. The solid sample was obtained by centrifugation for 5 minutes at a speed of 3 000 r· min-1, then the settled particles were dispersed in ethanol and centrifuged four times. Finally, the solid sample was dried at 80 ℃ in air.
1.2.2 Syntheses of HAP nanorods
The HAP samples were obtained at the second hydrolysis step. 0.3 g of the above precursor was added into the 250 mL round-bottomed flask containing 50 mL of distilled water, and the slurry was stirred at the moderate speed at 100 ℃ for 14 h in a temperature-controlled electromantle for the hydrolysis of the precursor samples. The pH value of the suspension was adjusted and kept at 10.0 by the addition of ammonium hydroxide dropwise during the hydrolysis reaction. The samples were obtained by centrifugation for 5 minutes at a speed of 3 500 r·min-1, and then redispersed in ethanol and repeated the centrifugation and redispersion three times, Finally, HAP nanorods were obtained by drying at 80 ℃ in air.
2 Results and discussion
2.1 HAP nanocrystals obtained by the precipi-tation-hydrolysis two-step method
2.2 HAP nanocrystals obtained with one-step precipitation method
The chemical reaction for the formation of HAP samples by the precipitation method can be simplified as Eq.3:
10Ca (NO3)2·4H2O+6(NH4)2HPO4+8NH3·H2O→Ca10(PO4)6(OH)2+20NH4NO3+46H2O (3)
The structures of HAP nanoparticles produced by direct precipitation method are characterized by XRD, as shown in Fig. 4. The crystal cell parameters of the HAP samples calculated according to their XRD data are a=0.943 8 nm, c=0.686 7 nm (Fig. 4a); and a=0.940 7 nm, c=0.686 6 nm (Fig. 4b), respectively. These results are also in agreement with the reported values of bulk HAP crystal with space group P63/m[17]. The obvious broadening of diffraction peaks implies that the size of HAP nanoparticles prepared by the direct precipitation method is much smaller than those prepared by the hydrolysis process. The crystallinity of HAP is different from each other, as the crystallinity of HAP in Fig. 4b is higher than that in Fig. 4a. The results indicated that the structure and crystallinity of HAP samples are influenced by the surfactant additives.
Figure 4.
XRD patterns of HAP prepared by direct precipitation method with PEG (a) and PVP (b) as surfactants, and the standard data for bulk Ca10(PO4)6(OH)2 as a reference
The HAP sample prepared with the additive of PEG consists of the nanoparticles with the shape of wheat ears, exhibiting narrow size distribution, as shown in Fig. 5a. The more detailed image of the nanoparticle is shown in Fig. 5b, and it is observed that the anisotropy crystal with the long axis of 237 nm and the short axis of 49 nm consists of small nanocrystals. The HAP sample obtained with the additive of PVP in Fig. 5c consists of small nanoparticles and small nanorods. The SAED pattern shown in Fig. 5d revealed the polycrystalline nature of the HAP sample in Fig. 5c, and the diffraction rings indicated that the nanoparticles are different from the above mentioned nanocrystals in growth. The apparent changes in size and morphology of HAP nanoparticles indicate that the crystal growth of HAP samples were greatly influenced by the preparation methods and organic additives.
 Figure 5.
TEM micrographs of HAP samples prepared by the direct precipitation synthetic route: the sample prepared with PEG (a), a single particle (b), the sample prepared with PVP (c), and the corresponding SAED (d)
Figure 5.
TEM micrographs of HAP samples prepared by the direct precipitation synthetic route: the sample prepared with PEG (a), a single particle (b), the sample prepared with PVP (c), and the corresponding SAED (d)
2.3 The effect of the organic additives
For the HAP nanorods prepared by the two-step method, the CaHPO4 precursors formed at the first precipitation step[7, 24]. The surfactant additives affected the structure of monetite precursors, and the different surfactant affected the monetite crystals in the different reactions. PEG absorbed on the surfaces of monetite nuclei via the hydrogen bonding reaction, which made the nuclei stable at the initial stage, but confine the formation of large monetite particles during the growth process, and the final shape and size of monetite particles could be well controlled[14, 19]. PVP influenced the structure and crystallinity of monetite, because of hydrogen bonding with HPO42- ions and sterochemical interaction. Overall, the organic additives used as the surfactants acted as active sites for nucleation and then stabilized and confined the formation of large monetite crystals, and finally controlled the monetite particles.
During the hydrolysis process at a basic environment of pH=10, the CaHPO4 crystals would be dissolved, and the HPO42- transformed into PO43-. At such a basic environment with rather high OH- concentration, the selective absorption of OH- ions on different planes of HAP possibly affected the movement of Ca2+ and PO43- in the formed HAP nuclei, which would result in the preferred growth in one particular direction, and then the nanorods with high aspect ratio are obtained. Some HAP nuclei are less stable than the nuclei with the organic additives, and they would be dissolved or absorbed by other nuclei to form a more stable crystal through Ostwald ripening. Due to the relatively high temperature, more and more organic additives were detached from the HAP crystals with the extension of reaction time, and then exerted weaker influence on the morphology of the crystal. Finally the organic additives were detached from the HAP crystals[24].
To study the effect of surfactant additives, the surface properties of HAP nanocrystals were characterized by FTIR spectra in Fig. 6. The broad absorption band at 3 590 cm-1 is due to the stretching vibration (νs) of the structural hydroxyl anions. The broad weak peak at 3 460 cm-1 is assigned to the physisorbed water. The characteristic bands of PO43- appear at 1 108, 1 034, 950, 610, 570, and 462 cm-1. The absorption peaks at 1 108 and 1 034 cm-1 are both assigned to the triply degenerated asymmetric stretching mode of P-O bond (ν3) of PO43- tetrahedron. The weak peak at 950 cm-1 corresponds to the symmetric stretching mode of the P-O bond (ν1). The peaks in the low wavenumber region at 610 and 570 cm-1 are assigned to the triply degenerated bending mode of the O-P-O bond (ν4) and 462 cm-1 is attributed to the bending vibrations mode (ν2) of the O-P-O in PO43- group. The peaks observed at 884 cm-1 reflect the carbonate groups (CO32-) into the crystal structure of HAP (B-type of the carbonate containing HAP). The presence of CO32-ions in HAP lattice may be due to CO2 from atmosphere via the hydrolysis reaction, which is a common phenomenon in HAP structures[1, 5, 19]. The results in Fig. 6a and 6b indicates that the surfactant additives detached from the HAP nanocrystals when the HAP nanorods had been completely developed via a two-step method. The surface properties of HAP samples prepared by the one-step precipitation method are different from those prepared by the two-step method. In Fig. 6c, the absorption bands at 1 393 cm-1 is assigned to the scissoring vibrations of the CH2 groups in PEG. The broad band around 1 630 cm-1 is due to H2O absorbed in the HAP nanoparticles. The result indicates that a great number of PEG molecules absorbed on the surfaces of HAP nanoparticles prepared by the one-step method, and lots of H2O molecules absorbed on the surface of PEG additive. Seen from Fig. 6d, the peak at 1 623 cm-1 is due to the amide Ⅰ band of PVP[23-24], which demonstrated that PVP is absorbed on the surface of HAP nanoparticles prepared by the one-step method. These results indicate that the surfactants detached from HAP nanorods prepared by a two-step precipitation-hydrolysis method, and the surfactants absorbed on HAP nanoparticles prepared with a one-step precipitation method. The spectra of FTIR confirmed the formation mechanisms of HAP nanorods[25-26].
 Figure 6.
FTIR spectra of the as-prepared HAP nanocrystals via the two-step method with PEG (a) or PVP (b), via the one-step method with PEG (c) or PVP (d)
Figure 6.
FTIR spectra of the as-prepared HAP nanocrystals via the two-step method with PEG (a) or PVP (b), via the one-step method with PEG (c) or PVP (d)
2.4 Effect of the addition order of OH- groups
The addition order of ammonia is a key factor in modifying the morphology of HAP particles. During the two-step precipitation-hydrolysis method, ammonia was added into the precursor CaHPO4 suspension at the second step, and CaHPO4 was totally dissolved under a high pH value. The organic additives were detached from the crystals with the extension of reaction time. Meanwhile, OH- templates the nucleation process where free Ca2+ and PO43- react with OH- in uniaxial direction. Accordingly, many tiny seeds of HAP were formed on the CaHPO4 surface, the seeds grew up into an oriented array of nanorods. Finally, the randomly arranged nanorods evolved from the seeds with the dissolution of the organic additives and CaHPO4[19, 21].
During the one-step precipitation process at a high pH value, the HPO42- anions become PO43- anions, and then the PO43- and OH- anions reacted with Ca2+ cations to form HAP nuclei. The organic additives used as the surfactants acted as active sites for nucleation and stabilized the nuclei and then confined the growth of large HAP crystals, and finally controlled the shape of the HAP nanoparticles[8].
2.1.2 Structural and morphological properties of HAP samples
The chemical reaction for the formation of HAP by the hydrolysis process can be simplified as Eq.2:
10CaHPO4+12NH3·H2O →Ca10(PO4)6(OH)2+4(NH4)3PO4+10H2O (2)
The structures of HAP samples produced by the hydrolysis of CaHPO4 are characterized by XRD, as shown in Fig. 2. It can be observed that the as-synthesized HAP powders are of a single phase and all the peaks are in good agreement with hexagonal crystal structure of bulk HAP phase (PDF Card No.9-432). The results of XRD indicate that the HAP samples prepared by the two-step process are all highly crystalline. The calculated crystal cell parameters of the HAP samples according to their XRD data are a=0.942 6 nm, c=0.688 7 nm in Fig. 2a; a=0.942 2 nm, c=0.687 8 nm in Fig. 2b, respectively. These results are in good agreement with the reported values of bulk HAP crystal (a=0.941 8 nm, c=0.688 4 nm) with space group P63/m[17]. The strong diffractions of (002), (211), (300) planes further demonstrated that the HAP samples are of the same structure with the preference. Compared with the intensity ratios of the standard data, it can be inferred that the preferences of nanocrystal growth in the different samples are all (002) planes. The crystallinity of HAP is different from each other, as the crystallinity of HAP in Fig. 2b is higher than that in Fig. 2a. The results indicated that the structure and crystallinity of HAP samples are influenced by the monetite precursors.
 Figure 2.
XRD patterns of the HAP samples prepared by hydrolysis with PEG (a) and PVP (b) as additives, and the standard data of Ca10(PO4)6(OH)2 as reference
Figure 2.
XRD patterns of the HAP samples prepared by hydrolysis with PEG (a) and PVP (b) as additives, and the standard data of Ca10(PO4)6(OH)2 as reference
TEM micrographs of the HAP samples prepared with different additives by the two-step synthetic route are shown in Fig. 3. The HAP sample prepared with the additive of PEG in Fig. 3a consists of the aggregated nanorods, exhibiting high aspect ratio and narrow size distribution. These nanorods prepared with the additive of PVP in Fig. 3d are smaller in length and possess broader size distribution. Significant change in size and morphology of the particles is mainly caused by the different organic additives. The aspect ratio of the HAP nanorods in Fig. 3a is higher than that in Fig. 3d which indicates that HAP nanorods prepared with the additive of PEG have larger surface area, yielding an increased adsorption on the surface as the aspect ratio determines the absorbability of the HAP particles in general. The result shows that the growth of one-dimensional HAP nanocrystals was affected by the organic additives, and this attributes to the different chemical or physical interaction between the organic additives and the nanocrystals in the growth process. The SAED patterns revealed the single-crystal nature of the HAP nanorods in Fig. 3b and 3e, and the diffraction spots indicated that the nanorods grew preferentially along the (002) direction, in agreement with the XRD results. These results indicated that HAP nanorods were obtained by the two-step method, and the surfactant additives greatly influenced the morphology of HAP nanorods, but the additives hardly affected the structure of HAP nanorods.
 Figure 3.
TEM micrographs of HAP samples prepared with different additives by the precipitation-hydrolysis synthetic routes: a bundle of HAP nanorods with PEG (a), one single nanorod (b), and the SAED (c) corresponding to (b), a bundle of HAP nanorods with PVP (d), one single nanorod (e), and the SAED (f) corresponding to (e)
Figure 3.
TEM micrographs of HAP samples prepared with different additives by the precipitation-hydrolysis synthetic routes: a bundle of HAP nanorods with PEG (a), one single nanorod (b), and the SAED (c) corresponding to (b), a bundle of HAP nanorods with PVP (d), one single nanorod (e), and the SAED (f) corresponding to (e)
2.1.1 Structural properties of CaHPO4 precursors
The chemical reaction for the formation of monetite can be simplified as Eq.1:
Ca (NO3)2·4H2O+(NH4)2HPO4 →CaHPO4+2NH4NO3+8H2O (1)
The structures of the precursors are characterized by XRD. Fig. 1 shows XRD patterns of the precursor samples prepared with different additives: PEG (a), PVP (b) and the standard data for bulk materials as reference. The results of XRD indicate that all the peaks of the precursor samples are in good agreement with triclinic crystal structure of bulk monetite phase CaHPO4 (PDF Card No. 71-1759), which is known to be a stable phase under acidic conditions[9]. The crystal cell parameters of the precursors calculated according to their XRD data are a=0.691 0 nm, b=0.662 5 nm, c=0.694 7 nm (Fig. 1a); and a=0.690 6 nm, b=0.660 4 nm, c=0.695 3 nm (Fig. 1b), respectively. These results are in agreement with the reported values for bulk monetite phase CaHPO4 (a=0.691 6 nm, b=0.661 9 nm, c=0.694 6 nm). Meanwhile, the structures of samples prepared with the different organic addictives are subtle different from that of bulk CaHPO4. The particularly strong diffractions of (200) plane in the products indicate that the as-prepared monetite might be of structure with (200) preference. The crystallinity of monetite phase is different from each other, as the crystallinity of monetite in Fig. 1b is higher than that in Fig. 1a. The relative intensity of the diffraction peaks of (100), (200), (003) planes of the monetite samples in Fig. 1(a) is 17:100:25.5 and that in Fig. 1(b) is 21:100:21, which differ from 15:100:15.5 in the bulk crystal. The results indicated that the structures of monetite samples prepared with the different surfactants are different, and the crystal growth of monetite is influenced by the surfactant additives.
 Figure 1.
XRD patterns of the precursor samples prepared with different additives PEG (a) and PVP (b), and the standard data for bulk monetite phase CaHPO4 works as the reference
Figure 1.
XRD patterns of the precursor samples prepared with different additives PEG (a) and PVP (b), and the standard data for bulk monetite phase CaHPO4 works as the reference
3 Conclusions
In summary, nanocrystalline HAP particles with different structures and morphologies have been successfully obtained by controlling the synthesis routes under mild conditions. The rod-like HAP nanocrystals were prepared via the precipitation-hydrolysis two-step route. The organic additives acted as active sites for nucleation and then stabilized and confined the formation of CaHPO4 precursors during the precipitation step. OH- templates the nucleation and crystal growth of HAP during the hydrolysis step. HAP nanorods prepared with PEG possess uniform morphology and high aspect ratio, while those prepared with PVP possess smaller size and broader size distribution. One-step precipitation process can produce HAP directly, and HAP prepared with PEG are in the shape of wheat ears, while those prepared with PVP consist of small nanoparticles and small nanorods. This research has offered a convenient way to obtain HAP nanocrystals with desired structures and morphologies, which is of great value for their applications.
-
-
[1]
Zhang C M, Cheng Z Y, Yang P P, et al. Langmuir, 2009, 25(23):13591-13598 doi: 10.1021/la9019684
-
[2]
Venkatesan J, Kim S K. J. Biomed. Nanotechnol., 2014, 10 (10):3124-3140 doi: 10.1166/jbn.2014.1893
-
[3]
Kong D Y, Xiao X L, Qiu X Y. et al. Funct. Mater. Lett., 2015, 8(6):1550075 doi: 10.1142/S1793604715500757
-
[4]
Qi C, Zhu Y J, Lu B Q, et al. J. Mater. Chem., 2012, 22(42): 22642-22650 doi: 10.1039/c2jm35280j
-
[5]
Kramer E, Podurgiel J, Wei M. Mater. Lett., 2014, 131:145-147 doi: 10.1016/j.matlet.2014.05.105
-
[6]
Prakash K H, Kumar R, Ooi C P, et al. Langmuir, 2006, 22 (26):11002-11008 doi: 10.1021/la0621665
-
[7]
Kong D Y, Xiao X L, Yang Y L. IET Micro Nano Lett., 2015, 10(6):302-306 doi: 10.1049/mnl.2014.0683
-
[8]
Sadasivan S, Khushalani D, Mann S. Chem. Mater., 2005, 17(10):2765-2770 doi: 10.1021/cm047926g
-
[9]
Mohandes F, Salavati-Niasan M. Chem. Eng. J., 2014, 252: 173-184
-
[10]
Uota M, Arakawa H, Kitamura N, et al. Langmuir, 2005, 21 (10):4724-4728 doi: 10.1021/la050029m
-
[11]
Cai Z Y, Zhang T, Di L Z, et al. Biomed. Microdevices, 2011, 13(4):623-631 doi: 10.1007/s10544-011-9532-8
-
[12]
杜博, 何福明, 王小祥.无机化学学报, 2013, 29 (5):973-978DU Bo, HE FuMing, WANG XiaoXiang. Chinese J. Inorg. Chem., 2013, 29 (5):973-978
-
[13]
Kwon S H, Jun Y K, Hong S H, et al. J. Eur. Ceram. Soc., 2003, 23(7):1039-1045 doi: 10.1016/S0955-2219(02)00263-7
-
[14]
Yang Y S, Wu Q Z, Wang M, et al. Cryst. Growth Des., 2014, 14(9):4864-4871 doi: 10.1021/cg501063j
-
[15]
Michael P, Shanthi S L, Ashoka M, et al. Mater. Lett., 2009, 63:21232125
-
[16]
Yao X, Yao H W, Li G Y, et al. J. Mater. Sci., 2010, 45(7): 1930-1936 doi: 10.1007/s10853-009-4182-4
-
[17]
Zhang Y J, Lu J J, Cryst. Growth Des., 2008, 8(7):2101-2107 doi: 10.1021/cg060880e
-
[18]
Shih W J, Chen Y F, Wang M C, et al. J. Cryst. Growth, 2004, 270(1/2):211-218
-
[19]
Zou Z Y, Liu X G, Chen L, et al. J. Mater. Chem., 2012, 22 (42):22637-22641 doi: 10.1039/c2jm35430f
-
[20]
Cai Z Y, Yang D A, Zhang N, et al. Acta Biomater., 2009, 5:628-635 doi: 10.1016/j.actbio.2008.09.016
-
[21]
殷馨, 刘慧慧, 孔爱国, 等.无机化学学报, 2014, 30(6):1287-1292YIN Xin, LIU Hui-Hui, KONG Ai-Guo, et al. Chinese J. Inorg. Chem., 2014, 30(6):1287-1292
-
[22]
Sadat-Shojai M, Ataia M, Nodehi A, et al. Dent. Mater., 2010, 26(5):471-482 doi: 10.1016/j.dental.2010.01.005
-
[23]
Padmanabhan S K, Balakrishnan A, Chu M C, et al. Particuology, 2009, 7(6):466-470 doi: 10.1016/j.partic.2009.06.008
-
[24]
Kong D Y, Xiao X L, Qiu X Y, et al. Chin. J. Chem., 2015, 33(9):1024-1030 doi: 10.1002/cjoc.201500343
-
[25]
Kuriakose T A, Kalkura S N, Palanichamy M, et al. J. Cryst. Growth, 2004, 263(1/2/3/4):517-523
-
[26]
Kumar R, Prakash K H, Cheang P, et al. Langmuir, 2004, 20 (13):5196-5200 doi: 10.1021/la049304f
-
[1]
-

Figure 1 XRD patterns of the precursor samples prepared with different additives PEG (a) and PVP (b), and the standard data for bulk monetite phase CaHPO4 works as the reference

Figure 2 XRD patterns of the HAP samples prepared by hydrolysis with PEG (a) and PVP (b) as additives, and the standard data of Ca10(PO4)6(OH)2 as reference

Figure 3 TEM micrographs of HAP samples prepared with different additives by the precipitation-hydrolysis synthetic routes: a bundle of HAP nanorods with PEG (a), one single nanorod (b), and the SAED (c) corresponding to (b), a bundle of HAP nanorods with PVP (d), one single nanorod (e), and the SAED (f) corresponding to (e)

Figure 4 XRD patterns of HAP prepared by direct precipitation method with PEG (a) and PVP (b) as surfactants, and the standard data for bulk Ca10(PO4)6(OH)2 as a reference

Figure 5 TEM micrographs of HAP samples prepared by the direct precipitation synthetic route: the sample prepared with PEG (a), a single particle (b), the sample prepared with PVP (c), and the corresponding SAED (d)
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 327
- HTML全文浏览量: 30
 下载:
下载:
